
Abstract
Discovering novel catalysts for hydrogen evolution reaction (HER) holds the potential to revolutionize the energy chemistry and unlock new tool for synthetic processes. Inspired by hydrogenases, we pair alkali metals with cobalt-Salen catalysts which allow the integration of naked base site into bimetallic HER catalysts. The incorporation of alkali metals (Na, K, Rb, Cs) significantly enhances HER activity. Among these, the [Co/K] system exhibits the highest HER catalytic efficiency (kobs ~ 31.4 s⁻¹), which is 9 times higher than the mononuclear analogue. Remarkably, this HER catalyst is repurposed for the terminal C(sp³)-H functionalization of N-allylimines with imine/aldehyde, a previously inaccessible transformation. Mechanistic studies reveal that the naked base site enables selective C-H activation via proton relay, overriding the inherent preference for Pinacol coupling. The electrochemical protocol features good functional group tolerance, and opens up a streamlined avenue for chiral pyrrolines, key precursors of the anti-cancer medicine Larotrectinib. More importantly, the alkali metal effect is rationalized through structural analysis, density functional theory (DFT) calculations, and control experiments.
Similar content being viewed by others
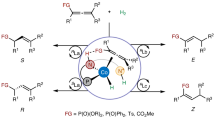

Introduction
Developing highly efficient catalysts for the hydrogen evolution reaction (HER)1,2,3,4 is one of the central topics in energy chemistry, since hydrogen can be considered an ideal surrogate for fossil fuels. Natural [NiFe]- and [FeFe]-hydrogenases5,6 achieve high HER activity through synergistic bimetallic centers and proton-relaying base sites (Fig. 1a). These two factors have been individually leveraged to facilitate the HER catalyst discoveries. The bimetallic7,8,9,10 strategy has been widely explored and well-documented in the hetero-11,12 and homogeneous13,14,15 HER catalyst discovery (Fig. 1b). Meanwhile, the proton relay strategy16,17 has proven highly effective in molecular HER catalysts, as demonstrated in the compelling reports18,19,20,21 (Fig. 1b). Nevertheless, integrating proton relay sites into bimetallic HER catalysts has received far less attention22. With our long-term research interest23,24,25,26 in cobalt-Salen27,28,29,30,31 HER catalysis, we envisaged that its modular structure could serve as an ideal platform for developing such bimetallic HER catalysts (Fig. 1c). The less explored alkali metals (AM)32,33 were selected to combine with cobalt-Salen systems, given their low toxicity and strong hydride affinity, which would lead to more reactive metal hydride for HER catalysis. Moreover, the interaction between alkali metals and oxygen sites can release a naked base site, enabling rapid proton transfer during HER. Guided by this concept, we devised a series of cobalt/alkali-Salen catalysts bearing naked base sites, and potassium ion demonstrated an unmatched enhancement in HER efficiency.

a The structure of [NiFe] and [FeFe] hydrogenases. b Reported strategies for HER catalysts. c The structure of cobalt/alkali-Salen HER catalyst. d The C–H functionalization of N-allylimine. AM alkali metal.
The robust bimetallic HER catalyst offers an unconventional tool34,35,36,37,38,39,40 for electrochemical C–H functionalization23,24,25,26,41,42,43,44,45,46,47, as it can break sp³ C–H bonds into the relative carbanion species while simultaneously releasing H₂. The functionalization of N‑allylimine48,49,50,51,52 with imines provides direct access to various bioactive amines, including both α- and γ-selective products (Fig. 1d). Conventional protocol52 involving excessively strong base nBuLi and the activator N,N,N’,N’-tetramethylethylenediamine (TMEDA) predominantly yields α-selective products. Selectively accessing terminal C–H (γ-C–H) functionalized products remains a formidable challenge, although this unconventional product allows a straightforward pathway to diverse chiral pyrrolines, which are widespread in 40 U.S. FDA-approved drugs53 and amino organocatalysts54. In this context, we envisaged that the developed bimetallic HER catalysts might address the challenge of terminal C–H selectivity in this reaction. However, the electrochemical C–H functionalization of N‑allylimine faces inherent challenges. That is, the N-allylimine substrate preferentially couples with imines or aldehydes to give Pinacol coupling55,56,57 products under electroreduction, owing to its redox-active imine moiety (Fig. 1d). In contrast, C–H bonds of N-allylimine are poor acidic (pKa > 30) and typically considered redox-unactive. This contradiction poses a great obstacle for electrochemical C–H functionalization of N-allylimine.
In this work, we unveil that the bimetallic [Co/K] catalyst58 overcomes this inherent limitation by selectively activating C–H bonds with a naked base site and efficiently breaking C–H bonds with in situ formed Co/K hydrides. This electrocatalytic protocol exhibits several impressive advantages, including good functional group tolerance, high stereoselectivity, mild reaction conditions, and good scalability. The versatility of the [Co/K] catalyst is further extended to the terminal C–H functionalization of N‑allylimine with aldehydes.
Results
The catalysts synthesis and catalytic activity evaluation
We set out to synthesize the bimetallic cobalt/alkali-Salen catalyst via a one-pot procedure (Fig. 2a). The Salen ligand was prepared through the direct condensation between (L)−3-aminoalanine hydrochloride and two equivalents of o-vanillin using various alkali metal hydroxides as base at 50 °C. Further treatment with cobalt acetate and an additional two equivalents of base gave the desired bimetallic complexes as a colored precipitate, which can be directly used for catalysis after a hot filtration. To our delight, this approach is amenable to give a series of alkali metals (Na, K, Rb, Cs) bimetallic catalysts on a gram scale. Nevertheless, lithium hydroxide failed to give the desired complex, presumably due to its mismatched ionic radius. These bimetallic catalysts were unambiguously characterized by nuclear magnetic resonance (NMR) (Figs. S2–S9) and high-resolution mass spectrometry (HRMS). It reveals that the methoxy anion coordinates with the cobalt center as an extra ligand. This structure assignment was also verified by the single-crystal diffraction analysis of cat 2 (Deposition number 2422475 (for cat 2) contains the supplementary crystallographic data for this paper. These data are provided free of charge by Cambridge Crystallographic Data Centre). As depicted in Fig. 2b, the potassium ion is firmly chelated by four oxygen atoms to release a free carboxylate anion. The resulting structure resembles a scorpion, with a potassium ‘head’ and a carboxylate ‘tail’. In the solid state, the bimetallic complexes dimerize readily via a head-to-tail connection.

a Synthetic route of cobalt/alkali-Salen catalysts. b Single-crystal structure of cat 2. c The chemical shifts of the protons in the HER catalysts. d Redox potentials of catalysts. e Cyclic voltammograms of HER catalyst (0.01 mmol) and acetic acid (0.175 mmol) in DMF (3.0 mL) solution with nBuNClO4 as electrolyte. DMF N,N-dimethylformamide, Fc ferrocene.
With the structurally well-defined bimetallic catalysts, we next probed the influence of alkali metals on the cobalt center by conducting a comparative analysis of the chemical shift of surrounding proton Ha and Hb (Fig. 2c). A consistent trend in the variation of chemical shifts was observed for both protons. The stronger the metallic character of the alkali metal (Cs > Rb > K > Na), the more electron-rich the cobalt center became. This increase in electron density is expected to generate more alkaline metal hydride species, thereby enhancing the efficiency of the hydrogen evolution reaction. Subsequently, we evaluated the electrochemical properties of the catalysts using a relevant cobalt-Salen (cat 5) as a reference. The redox potentials corresponding to CoIII/II and CoII/I processes were identified (Figs. S11–S15) and summarized in Fig. 2d. It clearly shows that alkali metals significantly change the electrochemical behaviors of Salen catalysts (cat 1–cat 4) compared to cat 5. The redox couples of CoIII/II uniformly shifted to more negative potentials, which might be ascribed to the steric hindrance and electronic effect of the carboxylate anion. On the other hand, the potential of CoII/I decreased with the increasing metallicity of the alkali metals; potassium, rubidium, and cesium resulted in more negative peaks than that of cat 5. Subsequently, we investigated the effect of alkali metals on the HER catalytic performance using a glassy carbon (GC) electrode in DMF solution with acetic acid as a proton source (Fig. 2e). Upon treating with the acid, the reduction waves associated with CoII/I were enhanced dramatically. We quantified the current increment with the ratio (ic/ip) of the catalytic current and peak current. The result indicates that bimetallic HER catalysts (cat 1–cat 4) exhibited higher efficiency than the reference catalyst, cat 5. Notably, the [Co/K] catalyst (cat 1) was recognized as the most efficient one, displaying the highest ic/ip value (3.56) and a 9-fold higher rate constant (kobs = 31.4 s−¹) compared to cat 5 (kobs = 3.5 s−¹). This result underscores the critical role of the appropriate metallicity and ionic radius of alkali metals in contributing to both the structural stability of the metal framework and the reactivity required for H₂ production. Additionally, the gas chromatography (GC) experiment further verified the hydrogen product during the electrolysis (Fig. S32).
Reaction optimization and substrate scope examination
Encouraged by the promising HER catalytic performance, we sought to access unconventional terminal C–H selectivity in the reaction between N-allylimine (1a) and Ellman imine59 (2a). Initially, we conducted the reaction in the absence of any catalysts using graphite felt anode and nickel cathode with DMF as solvent (Entry 1, Table 1). As expected, the corresponding cross-Pinacol coupling product 3a’ was delivered in 21% yield along with some homo-coupling byproduct. Variations on other electrolysis parameters, such as electrolyte and solvent, failed to afford the desired C–H functionalization product (for details, see Table S2). Gratifyingly, introducing HER catalysts substantially changed the reaction selectivity (Entry 2–6). The desired terminal C–H selective product 3a was exclusively accessed by using cat 5, albeit with only 30% yield (Entry 2). Further modifications on the structure of mononuclear Salen catalyst have a negligible impact on the reaction efficiency (Table S2). In contrast, bimetallic cobalt/alkali-Salen catalysts showed great enhancement in the reaction yield, which is in good consistency with the HER activity evaluation. The highest activity was observed for the [Co/K] complex (cat 2), and the desired product 3a was afforded in 81% yield (Entry 4). The reaction monitoring experiment (Table S3) further demonstrates that the HER catalyst (cat 2) exhibits high initial catalytic efficiency and maintains good durability under progressively basic reaction conditions. The control experiment (Entry 7), removing electricity directly shut down the reaction even with prolonged reaction time (48 h), implying the necessity of electricity. Replacing the graphite felt anode with a sacrificial zinc anode largely maintained the reaction yield (52%). This suggests that the terminal C–H selective product 3a should be generated via cathodic electrolysis (Entry 8).
Following systematic optimization, we next examined the protocol generality with variations on both substrates. A broad range of the Ellman imines was first subjected to the optimal conditions (Fig. 3). The electronic effect investigation revealed that electron-deficient substrates (3h-3m) gave the desired product with slightly higher yields compared to the electron-rich ones (3b-3g). Some useful but conventionally challenging substituents (e.g., thioether, amine, vinyl, iodine, ester) were amenable to give the terminal C–H functionalization product in moderate to good yields (3n-3r). Notably, this method showed excellent tolerance for other fused rings (3s-3u) and medicinally relevant heterocycles, including quinoline (3v), pyridine (3w), thiophene (3x-3y), and imidazole (3z). Moreover, ferrocene (3aa), as a key moiety of functional materials, proved to be suitable in the transformation. The protocol compatibility for substituent patterns (3ab-3am) was also tested, and the reaction efficiency was largely maintained. Specifically, substrates bearing bulky groups, such as 2-diphenylphosphino (3ah), 2-allyloxy (3ai), 2,6-methyl (3aj), and chlorine (3ak) still reacted smoothly, although they resulted in lower diastereoselectivity. Bioactive fluorinated imines furnish the desired products (3al-3am) in 79% and 62% yields, respectively. Aliphatic imine (3an) and diaryl ketimine (3ao) resulted in deteriorated reaction performance, whereas it should be noted that the radical-sensitive cyclopropyl ring was intact after electrolysis, supporting an ionic reaction pathway. Furthermore, the mildness of the method allows a facile platform for the late-stage functionalization of pharmaceutical (Adapalene 3ar) and natural products (Diacetonefructose 3ap, Vitamin E 3aq, Cholesterol 3as) that bear multiple functional groups. Nevertheless, Ellman imines (2at-2au) with acidic α-C–H were found to be unsuccessful in the reaction since it would interfere with the HER of N-allylimine C–H. The scope of N-allylimines was also tested by varying β-substituents. Alkyl groups afforded the corresponding products (3at-3au) in good stereocontrol, while aryl groups (3av-3bc) led to lower E/Z selectivity owing to its similar steric hindrance compared with the sulfinamide moiety in the product. N-homoallylimine 1bd with a poor acidic C–H bond was found to be incompatible in the transformation.

Reaction conditions: 1 (1.5 mmol), 2 (0.5 mmol), cat 2 (5 mol%), nBu4NClO4 (1.0 mmol), DMF (10 mL), graphite felt anode, nickel plate cathode, undivided cell, CCE = 18 mA, 3 h (4.03 F/mol), 0 °C; Yields were based on isolated products; dr > 15/1, unless otherwise noted. aThe reaction is conducted on a 0.3 mmol scale. GF graphite felt, CCE constant current electrolysis, DMF N,N-dimethylformamide.
To further demonstrate the robustness of the [Co/K] catalyst, we extended its application to the terminal C–H functionalization of N-allylimines with aldehydes. As shown in Fig. 4a, high reaction efficiency and excellent functional group tolerance (4a-4t) were readily achieved merely with 2 mol% catalyst loading. Intriguingly, N-allylimine containing diene moiety was competent, and the terminal C–H was selectively functionalized with benzaldehyde to give a new product 4t. We next investigated the scalability of the electrocatalytic approach (Fig. 4b). Under simply modified reaction conditions, 5 mmol scale reactions of both benzaldehyde and Ellman imine (2a) proceed smoothly to couple with 1a, affording respective products 4a and 3a in acceptable yields. Finally, the derivatization of products to chiral pyrrolines, diamine, and amino alcohol was conducted. A sequential procedure involving acidic deprotection and reductive amination allows a rapid route to transform chiral sulfinamide into pyrroline. Fortunately, the good retention of the chiral center is observed; corresponding products 5 and 6 were afforded with 94% and 96% ee values, respectively. Noteworthy, chiral pyrroline 6 is a key intermediate for the anti-cancer medicine Larotrectinib60 with an identical configuration. Furthermore, the direct reduction of corresponding products with excessive sodium borohydride gives direct access to chiral diamine (7) and amino alcohol (8).

a Electrochemical terminal C–H functionalization of N-allylimine with aldehydes. b Gram-scale reaction and product derivatization.
Mechanism investigation
We conducted a series of cyclic voltammetry (CV) experiments to better understand the role of the [Co/K] bimetallic catalyst. Initially, the comparison of substrates' reductive potential illustrates that the electrophilic reactant 2a and benzaldehyde are more susceptible to cathodic reduction than N-allylimine 1a (Fig. 5a). This result verifies that the Pinacol-type coupling between these substrates should be more favored without the intervention of a catalyst. Titrating substrates with cat 2 resulted in contrasting results. For instance, treating 1a with an increasing gradient of cat 2 gives a surge of reductive peak of 1a (Fig. 5b). Conversely, no significant change was detected in the case of 2a (Fig. S25). Mixing benzaldehyde with cat 2 failed to boost the peak current, nor did it (Fig. S26). From these observations, we drew a conclusion that cat 2 could selectively promote the HER of 1a in the presence of more reducible substrates 2a and benzaldehyde. The naked base site of cat 2 might be responsible for the selective interaction with the acidic allylic C–H bond of 1a, thus facilitating the inner-sphere electron transfer for the HER of 1a. Neither varying 1a to the weakly acidic substrate 1bd nor replacing cat 2 with cat 5 resulted in a significant increase in the cathodic peak current of the substrates (Figs. S27–S28), which evidenced the necessity of acid-base interaction. Further CV titration experiment of cat 2 with excessive 1a gave rise of a new noticeable peak at −1.58 V (Fig. 5c), which was tentatively ascribed to the in situ formed species A via deprotonation from 1a. We validated this postulation by using acetic acid to replace 1a, and an analogous peak emerged at the same position (Fig. 5d). Finally, the GC detection of hydrogen byproduct from the reaction supports the hydrogen evolution process of 1a (Fig. S33). Taken together, bimetallic catalyst cat 2 selectively enhanced the HER of 1a by forming the reactive species A.

a Cyclic voltammogram of 1a (0.05 mmol), 2a (0.05 mmol) and benzaldehyde (0.05 mmol) in DMF (3.0 mL) solution with nBuNClO4 as electrolyte. b Cyclic voltammogram of 1a (0.05 mmol) and cat 2 in DMF (3.0 mL) solution with nBuNClO4 as electrolyte. c Cyclic voltammogram of cat 2 (0.01 mmol) and 1a in DMF (3.0 mL) solution with nBuNClO4 as electrolyte. d Cyclic voltammogram of cat 2 (0.01 mmol) and HOAc in DMF (3.0 mL) solution with nBuNClO4 as electrolyte. e Control experiments. Fc ferrocene, DMF N,N-dimethylformamide, TEMPO 2,2,6,6-Tetramethylpiperidinooxy.
To further elucidate the reaction mechanism, various control experiments were carried out (Fig. 5e). We first ruled out the possible radical pathway by radical suppression (Eq. a) and radical clock experiments (Eq. b). Introducing excessive radical scavengers, such as 1,1-diphenylethene, 2,2,6,6-tetramethylperidinooxy (TEMPO), and triethyl phosphite, led to negligible changes in the reaction yield. Probing the reaction with the substrate 1be only afforded the desired product 3be without detecting any radical-initiated ring expansion product. This excludes the reaction mechanism triggered by the single-electron reduction of the imine moiety. We next confirmed the ionic pathway by a deuterium incorporation experiment (Eq. c). Treating the model reaction with 6 equivalents of deuterium oxide (D2O) gave a partially deuterated product 3a in a diminished yield (9%). This result can be interpreted as the deuterium exchange of allyl carbanions during the reaction. Furthermore, the kinetic isotope effect (KIE) was studied to get insight into the rate-determining step (RDS). Mono-deuterated substrate 1a-D was employed in the intramolecular competing experiment (Eq. d). Remarkably, the deuterium atom has been largely retained, and the ratio of kH/kD was determined to be 15.5/1, suggesting the C–H cleavage should be the RDS. Finally, we tried to simulate the in situ formed metal hydride species in the HER catalysis with sodium hydride as a surrogate (Eq. e). To our delight, the multistep procedure involving deprotonation and nucleophilic addition gave a single product 3a, albeit with inferior yield (19%). This comparable selectivity confirms that the metal hydride should be the reactive species in the electrochemical reaction.
On the basis of the experimental observations and related mechanism13 on bimetallic HER, a plausible reaction mechanism is proposed (Fig. 6a). The HER catalytic cycle is initiated by the CoII species (cat 2’) via the cathodic reduction of its precatalyst cat 2. The naked carboxylate site promotes the proton dissociation from substrate 1a to deliver a protonated species A, which leads to a further cathodic reduction and gives CoI species B. With the high nucleophilicity of the CoI center, intramolecular proton transfer occurs rapidly to yield a CoIII hydride C, in which the hydride bridges cobalt and alkali ions owing to its affinity to alkali metals. Subsequently, the single-electron reduction of C delivers more alkaline CoII hydride D, and it facilitates further deprotonation of 1a, releasing hydrogen and resonant carbanions Int-I and Int-II. Finally, the nucleophilic addition between the resulting carbanions and chiral imine 2a affords the result product 3a and a possible branched product 3a-1. Noteworthy, the dimeric form of the catalytic species (cat 2’, C and D) may participate in the catalytic cycle due to strong interactions between the carboxylate moiety and the K⁺ ion of a neighboring molecule. On the anode side, the oxidation of the DMF should be the predominant reaction based on the cyclic voltammetry experiment (Fig. S31).

a Proposed catalytic cycle for the electrochemical functionalization. b DFT calculations for the catalytic species. c DFT calculations for the nucleophilic addition between carbanions and Ellman imine. DFT density functional theory.
We rationalized the effect of alkali metals on the HER performance by density functional theory (DFT) calculations (Fig. 6b). The results illustrate that alkalic metals significantly affect the structure of the CoII catalysts (cat’) and their transformation into the intermediate D. Specifically, Na and K ions result in nearly planar geometry in catalysts cat 1’-cat 2’ with dihedral (K-O-Co-O) ranging from 1.7 to 3.3°. In contrast, the larger Rb and Cs ions lead to bowed structures (dihedrals 15.9–26.3°), which would introduces pronounced steric hindrance for the formation of the hydride species D. Transition-state calculations (Figs. S45–S48) validate that larger alkali metals correspond to higher energy barriers (1.90–5.83 kcal/mol), indicating a more sluggish process for the generation of intermediate D based on the further rate constant calculation61. The alkalinity of the in situ formed metal hydrides is the other factor for the HER efficiency, which has a positive correlation with the alkali metallicity. Consequently, the superior catalytic performance of the potassium-based catalyst (cat 2) can be attributed to its optimal ionic radius and the reasonable alkalinity of its metal hydride intermediate. Moreover, the DFT calculations (Fig. S49) for the reaction between carbanions Int-I/Int-II and imine 2a rationalize that the carbanion adduct IM2(3a-RR) is a kinetic control product with the lowest reaction activation energy of 9.5 kcal/mol (Fig. 6c).
Discussion
In summary, we have developed an array of bimetallic HER catalysts by incorporating redox-active cobalt centers with alkali metal ions. These combinations significantly enhance HER performance, enabling the terminal C–H selectivity in the functionalization of N-allylimines. With a naked base site in the bimetallic catalyst, the HER of N-allylimines is promoted to outcompete the undesired reductive coupling, thereby providing broad access to chiral amine products. DFT calculations and control experiments reveal that the enhancement of HER performance by alkali metals is dictated by their ion radius and metallicity. Our laboratory is actively exploring the repurposing of these bimetallic HER catalysts to solve the challenges in synthetic chemistry.
Methods
General procedure for electrochemical terminal C–H functionalization of N‑Allylimine
An undivided cell was equipped with a magnetic stirrer, nickel plate (1.8*1.5 cm2), graphite felt (1.8*1.5 cm2), as cathode and anode, respectively (the electrolysis setup is shown in Fig. S35). The substrate N‑allylimine 1a (332 mg, 1.5 mmol), Ellman imine 2a (105 mg, 0.5 mmol), cat 2 (12 mg, 0.025 mmol), and nBu4NClO4 (342 mg, 1.0 mmol) were added to the solvent DMF (10 mL). The resulting mixture was allowed to stir and electrolyze under constant current conditions (18 mA, J = 6.7 mA•cm−2) at 0 °C for 3 h. The reaction mixture was subsequently poured into water (200 mL) and extracted with ethyl acetate (40 mL*3). The combined organic phases were washed with saturated brine solution (100 mL). The volatile solvent was then removed with a rotary evaporator, and the residue was purified by column chromatography (PE/EA = 5/1–2/1, v/v basified with 1% triethylamine) on silica gel to afford the desired product 3a (174 mg) in 81% yield.
Data availability
All data supporting the findings of this study, including experimental details, spectroscopic characterization data for all compounds, are available in the text and the Supplementary Information section. Crystallographic data for the structure reported in this article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2422475 (cat 2). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All data are available from the corresponding author upon request. All of the original figure data in this study are provided in the Source Data file. The Cartesian coordinates generated in the DFT calculation are provided in Supplementary Data 1. Source data are provided with this paper.
References
Koper, M. T. M. & Bouwman, E. Electrochemical hydrogen production: bridging homogeneous and heterogeneous catalysis. Angew. Chem. Int. Ed. 49, 3723–3725 (2010).
McKone, J. R., Marinescu, S. C., Brunchwig, B. S., Winkler, J. R. & Gray, H. B. Earth-abundant hydrogen evolution electrocatalysts. Chem. Sci. 5, 865–878 (2014).
Zhu, J., Hu, L., Zhao, P., Lee, L. Y. S. & Wong, K.-Y. Recent advances in electrocatalytic hydrogen evolution using nanoparticles. Chem. Rev. 120, 851–918 (2020).
Zheng, Y., Jiao, Y., Vasileff, A. & Qiao, S.-Z. The hydrogen evolution reaction in alkaline solution: from theory, single crystal models, to practical electrocatalysts. Angew. Chem. Int. Ed. 57, 7568–7579 (2018).
Tard, C. & Pickett, C. J. Structural and functional analogues of the active sites of the [Fe]-, [NiFe]-, and [FeFe]-hydrogenases. Chem. Rev. 109, 2245–2274 (2009).
Tai, H., Hirota, S. & Stripp, S. T. Proton transfer mechanisms in bimetallic hydrogenases. Acc. Chem. Res. 54, 232–241 (2021).
Wang, F. et al. Selective functionalization of alkenes and alkynes by dinuclear manganese catalysts. Acc. Chem. Res. 57, 2985–3006 (2024).
Chen, K. et al. Switch in selectivities by dinuclear nickel catalysis: 1,4-hydroarylation of 1,3-dienes to Z-olefins. J. Am. Chem. Soc. 145, 24877–24888 (2023).
Chen, Q.-F., Xiao, Y., Hua, K., Zhang, H.-T. & Zhang, M.-T. Bimetallic synergy in oxygen reduction: how tailored metal–metal interactions amplify cooperative catalysis. J. Am. Chem. Soc. 147, 14504–14518 (2025).
Peng, P. et al. Unlocking the nucleophilicity of strong alkyl C–H bonds via Cu/Cr catalysis. ACS Cent. Sci. 9, 756–762 (2023).
Wei, Z., Sun, J., Li, Y., Datyte, A. K. & Wang, Y. Bimetallic catalysts for hydrogen generation. Chem. Soc. Rev. 41, 7994–8008 (2012).
Liu, L. & Corma, A. Bimetallic sites for catalysis: from binuclear metal sites to bimetallic nanoclusters and nanoparticles. Chem. Rev. 123, 4855–4933 (2023).
Brazzolotto, D. et al. Nickel-centred proton reduction catalysis in a model of [NiFe] hydrogenase. Nat. Chem. 8, 1054–1060 (2016).
Cloward, I. N. et al. Catalyst self-assembly accelerates bimetallic light-driven electrocatalytic H2 evolution in water. Nat. Chem. 16, 709–716 (2024).
Valdez, C. N., Dempsey, J. L., Brunschwig, B. S., Winkler, J. R. & Gray, H. B. Catalytic hydrogen evolution from a covalently linked dicobaloxime. Proc. Natl. Acad. Sci. USA 109, 15589–15593 (2012).
Bullock, R. M. & Helm, M. L. Molecular electrocatalysts for oxidation of hydrogen using earth-abundant metals: shoving protons around with proton relays. Acc. Chem. Res. 48, 2017–2026 (2015).
Haake, M., Reuillard, B., Chavarot-Kerlidou, M., Costentin, C. & Artero, V. Proton relays in molecular catalysis for hydrogen evolution and oxidation: lessons from the mimicry of hydrogenases and electrochemical kinetic analyses. Angew. Chem. Int. Ed. 63, e202413910 (2024).
Barton, B. E., Olsen, M. T. & Rauchfuss, T. B. Aza- and oxadithiolates are probable proton relays in functional models for the [FeFe]-hydrogenases. J. Am. Chem. Soc. 130, 16834–16835 (2008).
Helm, M. L., Stewart, M. P., Bullock, R. M., DuBois, M. R. & DuBois, D. L. A synthetic nickel electrocatalyst with a turnover frequency above 100,000 s−1 for H2 production. Science 333, 863–866 (2011).
Bediako, D. K. et al. Role of pendant proton relays and proton-coupled electron transfer on the hydrogen evolution reaction by nickel hangman porphyrins. Proc. Natl. Acad. Sci. USA 111, 15001–15006 (2014).
Queyriaux, N. et al. Electrocatalytic hydrogen evolution with a cobalt complex bearing pendant proton relays: acid strength and applied potential govern mechanism and stability. J. Am. Chem. Soc. 142, 274–282 (2020).
Lin, S. et al. Electrochemical strategy for proton relay installation enhances the activity of a hydrogen evolution electrocatalyst. J. Am. Chem. Soc. 144, 20267–20277 (2022).
Zhang, S. et al. Electrochemically generated carbanions enable isomerizing allylation and allenylation of aldehydes with alkenes and alkynes. J. Am. Chem. Soc. 145, 14143–14154 (2023).
Liang, Y. et al. A hydrogen evolution catalyst [Co2O2] metallacycle enables regioselective allene C(sp2)-H functionalization. Angew. Chem. Int. Ed. 63, e202400938 (2024).
Liu, K. et al. Paired electrocatalysis unlocks cross-dehydrogenative coupling of C(sp3)-H bonds using a pentacoordinated cobalt-salen catalyst. Nat. Commun. 15, 2897 (2024).
Feng, J. et al. Electrochemical allene C─H functionalization via carbanion sampling. Angew. Chem. Int. Ed. 64, e202508369 (2025).
Song, L. et al. Dual electrocatalysis enables enantioselective hydrocyanation of conjugated alkenes. Nat. Chem. 12, 747–754 (2020).
Cai, C.-Y. et al. Tailored cobalt-salen complexes enable electrocatalytic intramolecular allylic C–H functionalizations. Nat. Commun. 12, 3745 (2021).
Chen, M., Wu, Z.-J., Song, J. & Xu, H.-C. Electrocatalytic allylic C–H alkylation enabled by a dual-function cobalt catalyst. Angew. Chem. Int. Ed. 61, e202115954 (2022).
Yang, F. et al. Electrocatalytic oxidative hydrofunctionalization reactions of alkenes via Co(II/III/IV) cycle. ACS Catal. 12, 2132–2137 (2022).
Huang, C., Tao, Y., Cao, X., Zhou, C. & Lu, Q. Asymmetric paired electrocatalysis: enantioselective olefin–sulfonylimine coupling. J. Am. Chem. Soc. 146, 1984–1991 (2024).
Shah, A. H. et al. The role of alkali metal cations and platinum-surface hydroxyl in the alkaline hydrogen evolution reaction. Nat. Catal. 5, 923–933 (2022).
Ji, S. G. et al. Alkali metal cations act as homogeneous cocatalysts for the oxygen reduction reaction in aqueous electrolytes. Nat. Catal. 7, 1330–1338 (2024).
Zhang, S. & Li, M.-B. Repurposing HER catalysis toward metal hydride-mediated electro-reductive transformations. Tetrahedron Chem. 11, 100080 (2024).
Gnaim, S. et al. Cobalt-electrocatalytic HAT for functionalization of unsaturated C–C bonds. Nature 605, 687–695 (2022).
Wu, X. et al. Intercepting hydrogen evolution with hydrogen-atom transfer: electron-initiated hydrofunctionalization of alkenes. J. Am. Chem. Soc. 144, 17783–17791 (2022).
Derosa, J., Garrido-Barros, P., Li, M. & Peters, J. C. Use of a PCET mediator enables a Ni-HER electrocatalyst to act as a hydride delivery agent. J. Am. Chem. Soc. 144, 20118–20125 (2022).
Liu, C., Wu, Y., Zhao, B. & Zhang, B. Designed nanomaterials for electrocatalytic organic hydrogenation using water as the hydrogen source. Acc. Chem. Res. 56, 1872–1883 (2023).
Wang, T., He, F., Jiang, W. & Liu, J. Electrohydrogenation of nitriles with amines by cobalt catalysis. Angew. Chem. Int. Ed. 63, e202316140 (2024).
Wang, Y. et al. Electroreduction of unactivated alkenes using water as hydrogen source. Nat. Commun. 15, 2780 (2024).
Yan, M., Kawamata, Y. & Baran, P. S. Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev. 117, 13230–13319 (2017).
Yuan, Y. & Lei, A. Electrochemical oxidative cross-coupling with hydrogen evolution reactions. Acc. Chem. Res. 52, 3309–3324 (2019).
Jiao, K.-J., Xing, Y.-K., Yang, Q.-L., Qiu, H. & Mei, T.-S. Site-selective C–H functionalization via synergistic use of electrochemistry and transition metal catalysis. Acc. Chem. Res. 53, 300–310 (2020).
Li, P., Wang, Y., Zhao, H. & Qiu, Y. Electroreductive cross-coupling reactions: carboxylation, deuteration, and alkylation. Acc. Chem. Res. 58, 113–129 (2025).
Xiong, P. & Xu, H.-C. Molecular photoelectrocatalysis for radical reactions. Acc. Chem. Res. 58, 299–311 (2025).
Zhang, Q., Liang, K. & Guo, C. Enantioselective nickel-catalyzed electrochemical radical allylation. Angew. Chem. Int. Ed. 61, e202210632 (2022).
Shi, A., Xie, P., Wang, Y. & Qiu, Y. Photoelectrocatalytic Cl-mediated C(sp3)–H aminomethylation of hydrocarbons by BiVO4 photoanodes. Nat. Commun. 16, 2322 (2025).
Wang, T.-C. et al. Palladium-catalyzed enantioselective C(sp3)–H/C(sp3)–H umpolung coupling of N-allylimine and α-Aryl ketones. J. Am. Chem. Soc. 143, 20454–20461 (2021).
Fan, X., Gong, X., Ma, M., Wang, R. & Walsh, P. J. Visible light-promoted CO2 fixation with imines to synthesize diaryl α-amino acids. Nat. Commun. 9, 4936 (2018).
Pan, Z.-Z., Li, J.-H., Tian, H. & Yin, L. Copper(I)-catalyzed asymmetric allylation of ketones with 2-aza-1,4-dienes. Angew. Chem. Int. Ed. 63, e202315293 (2024).
Daniel, P. E., Weber, A. E. & Malcolmson, S. J. Umpolung synthesis of 1,3-amino alcohols: stereoselective addition of 2-azaallyl anions to epoxides. Org. Lett. 19, 3490–3493 (2017).
Uphade, M. B., Reddy, A. A., Khandare, S. P. & Prasad, K. R. Stereoselective addition of a lithium anion of 1,1-diphenyl-2-aza-pentadiene to sulfinimines: application to the synthesis of (−)-epiquinamide. Org. Lett. 21, 9109–9113 (2019).
Marshall, C. M., Federice, J. G., Bell, C. N., Cox, P. B. & Njardarson, J. T. An update on the nitrogen heterocycle compositions and properties of U.S. FDA-approved pharmaceuticals (2013–2023). J. Med. Chem. 67, 11622–11655 (2024).
Jensen, K. L., Dickmeiss, G., Jiang, H., Albrecht, Ł & Jørgensen, K. A. The diarylprolinol silyl ether system: a general organocatalyst. Acc. Chem. Res. 45, 248–264 (2012).
Zhang, S. et al. Catalyst-dependent direct and deoxygenative coupling of alcohols by convergent paired electrolysis. CCS Chem. 4, 1938–1948 (2022).
Wang, L.-J., Ye, P., Tan, N. & Zhang, B. Electroreductive cross-coupling between aldehydes and ketones or imines via cathodically generated dianions. Green. Chem. 24, 8386–8392 (2022).
Kwak, D. et al. Electroreductive access to 1,2-aminoalcohols via cross aza-pinacol coupling of N‑acyl diarylketimines and aldehydes. Org. Lett. 26, 2733–2738 (2024).
Deacy, A. C., Moreby, E., Phanopoulos, A. & Willams, C. K. Co(III)/alkali-metal(i) heterodinuclear catalysts for the ring-opening copolymerization of CO2 and propylene oxide. J. Am. Chem. Soc. 142, 19150–19160 (2020).
Ellman, J. A., Owens, T. D. & Tang, T. P. N-tert-butanesulfinyl imines: versatile intermediates for the asymmetric synthesis of amines. Acc. Chem. Res. 35, 984–995 (2002).
Bernhard, L. M., McLachlan, J. & Gröger, H. Process development of enantioselective imine reductase-catalyzed syntheses of pharmaceutically relevant pyrrolidines. Org. Process Res. Dev. 26, 2067–2074 (2022).
Eyring, H. The activated complex in chemical reactions. J. Chem. Phys. 3, 107–115 (1935).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21702113, 22571003 to S.Z., and 22422101, 22371002, 92061110 to M.-B.L.), Anhui University (S020318006/069 to S.Z.), and the Anhui Provincial Natural Science Foundation (2308085Y14 to S.Z.).
Author information
Authors and Affiliations
Contributions
S.Z. and M.-B.L. conceived the project, designed the experiments, and wrote the manuscript. S.Z., L.H., and M.W. performed the experimental work. J.F. and J.H. carried out a density functional theory calculation. Refinement and analysis of single-crystal data was conducted by Y.Z. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Silvia Bordoni, who co-reviewed with Federico Moro; Praveena Gopalan and Hai-Chao Xu for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, S., Hong, L., Feng, J. et al. Bimetallic [Co/K] hydrogen evolution catalyst for electrochemical terminal C-H functionalization. Nat Commun 16, 8435 (2025). https://doi.org/10.1038/s41467-025-63914-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-63914-0
