
Abstract
The increasing demand for higher operating speeds and greater integration densities in electronic devices has made heat dissipation one of the most critical challenges for next-generation technologies. This challenge has driven extensive efforts aimed at achieving a giant electrocaloric effect in ferroelectrics for high-efficiency cooling. Here, we propose a defect dipole engineering strategy to manipulate the polarization behavior of ferroelectric ceramics, leading to superior electrocaloric effect. By incorporating Sm and Li ions, the (SmBȧ-LiBaʹ) defect dipoles enhance the polarizability of BaTiO3. Simultaneously, these dipole defects increase the carrier activation energy, effectively mitigating the inherent trade-off between high breakdown strength and high polarization, thereby allowing the application of a high electric field to fully activate the electrocaloric potential. As a result, defect dipole engineering enables BaTiO3 to achieve a remarkable electrocaloric effect over a wide temperature range, achieving a high temperature change of 2.7 K at 70 °C— typical for integrated circuits.
Similar content being viewed by others

Introduction
The rapid advancement of artificial intelligence technology relies heavily on the increasing operation speed, integration, and power density of electronic circuits, devices, and systems, which in turn present unprecedented challenges in thermal management1,2,3. Research indicates that for every 10 °C increase in the chip temperature, the probability of system failure increases by an order of magnitude, with over 55% failure of integrated circuit (IC) being attributed to excessive heat4,5. Caloric effects, including mechanocaloric (mC), magnetocaloric (MC), and electrocaloric (EC) effects, offer promising technical routes to achieve high-efficiency solid-state refrigeration6,7,8,9,10,11,12. Among these, ECE has received significant attention, as applying electric fields to align electric dipoles and driving entropy change in ferroelectrics is a more straightforward approach than providing mechanical power or magnetic fields for mCE and MCE. Moreover, EC refrigerators are compact and lightweight13,14,15,16,17,18, making them one of the most promising candidates for cooling electronic devices and systems (Supplementary Note 1).
Both ferroelectric ceramics and polymers possess ECE, which involves changes in temperature (∆T) and entropy (∆S). However, the energy barrier for switching dipoles in inorganic displacive ferroelectrics, such as ceramics and single crystals, is lower than that for flipping the polar molecular chains in organic ferroelectrics (e.g., P(VDF-TrFE) and P(VDF-TrFE-CFE)), meaning that the required electric field for ECE in ferroelectric ceramics is lower than that in polymers19,20,21,22,23. Generally, a high ECE is observed near the phase transition region, such as at the Curie temperature, where the degree of polarization (or electric dipole entropy) undergoes a dramatic change due to the phase transition induced by variations in the electric field24,25,26. Therefore, a significant ECE can be achieved by combining competing multiphases with distinct symmetries at the invariant critical point (ICP), as the dipole entropy change associated with multi-conformation transitions is greater than that in a single-phase transition27,28,29. Within the theoretical framework of ICP, improvements in ECE have been demonstrated in ferroelectric ceramics through careful composition and microstructure designs30,31.
The high ECE at ICP arises from the increased change in the order degree of intrinsic dipoles, where the dipole moment (polarizability) is determined by the relative off-center displacement of B-site ions. The number and polarizability of these intrinsic dipoles govern the entropy change that can be induced by applied electric fields in a ferroelectric material at a specific ICP. This suggests that further enhancement of ECE can depend heavily on the introduction of extrinsic polarization factors (Supplementary Note 2). Unlike intrinsic dipoles, doping ions with different valences, whether as acceptors or donors, tend to form polar defect dipoles due to charge neutrality and Coulomb interaction32,33, potentially adding extra polarization to the materials. We hypothesize that the incorporation of polar defect dipoles represents an extrinsic mechanism for ECE promotion.
To explore the effect of defect dipoles on the polarization behavior and ECE of ferroelectric ceramics, in this work, we introduce A-site polar defect complexes (SmBȧ-LiBaʹ) in BaTiO3, a typical ferroelectric material with a simple composition. The incorporated defect dipoles with high polarizability significantly enhance the polarization of BaTiO3. Meanwhile, density functional theory (DFT) calculations suggest that these extrinsic defect dipoles interact with the intrinsic dipoles via the covalent effect, facilitating domain switching in BaTiO3, which in turn enhances electrocaloric strength (the ECE relative to the applied electric fields, ΔT/E and ΔS/E). Additionally, the incorporation of defect dipoles raises the carrier activation energy and suppresses charge transport, thereby mitigating the inherent trade-off between high breakdown strength and high polarization in ferroelectrics. This enables the application of a high electric field to fully exploit the EC potential of BaTiO3. As a result, BaTiO3 with defect dipole engineering showcases an exceptional ECE over a wide temperature range, for example, a high ∆T of 2.7 K and ∆S of 3.1 J kg−1 K−1 has been achieved at elevated temperatures, such as 70 °C—a typical operating temperature for integrated circuits4. These findings establish a defect dipole engineering paradigm for designing ferroelectric ceramics with giant ECE for thermal management.
Results
Construction of defect dipoles in BaTiO3
Ba1-2xSmxLixTiO3 (abbreviated as BT-x(Sm,Li)) ceramics, with varying (Sm,Li) defect complex contents (x = 0.01 to 0.05 with an interval of 0.01), were synthesized using a conventional solid-state method (see Methods). Additionally, to confirm that the enhancement of the ECE originates from the contribution of (SmBȧ-LiBaʹ) defect dipoles formed by the co-doped Sm and Li ions rather than from a simple doping effect, BaTiO3 singly doped with Sm and Li were prepared for comparisons. As observed in the scanning electron microscope (SEM) images, all samples exhibit a dense microstructure (Supplementary Fig. 1 and Supplementary Table 1). The X-ray diffraction (XRD, Fig. 1a and Supplementary Fig. 2) patterns denote that the BaTiO3, BT-0.03Li, BT-0.03Sm, and BT-x(Sm,Li) ceramics possess a pure perovskite structure. The observed splitting (200) peak at ~45o suggests the coexistence of the tetragonal (T) phase (P4mm) and the orthorhombic (O) phase (Amm2) in BT-x(Sm,Li) (Supplementary Fig. 2), which is further confirmed by the scanning transmission electron microscopy (STEM) at the atomic scale (Fig. 1b, c). The T and O phase proportions vary with the concentration of Sm and Li doped ions. As shown in Supplementary Fig. 3, the intensity of the Raman characteristic peaks at around 314 cm−1 corresponding to the tetragonal phase decreases as x in Ba1-2xSmxLixTiO3 increases. Calculated with the XRD Rietveld refinement, the orthorhombic phase fraction increases from 36.9% at x = 0.01 to 54.1% at x = 0.05 (Supplementary Table 2). Moreover, as revealed by the refinement, the unit cell volume of both the orthorhombic and tetragonal phases decreases with increasing x in BT-x(Sm,Li) due to the small ionic radius of Sm3+ and Li+ (Supplementary Table 3).

a XRD patterns of BaTiO3, BT-0.03Li, BT-0.03Sm, and BT-0.03(Sm,Li). b Bright-field STEM image and selected area electron diffraction (SAED) pattern of BT-0.03(Sm,Li). c Mapping of the atomic displacement vector embedded in the atomic-scale HAADF-STEM image of BT-0.03(Sm,Li). The arrows, denoting the orientation of the collective B-site cation displacement, display the B-site polar vectors in the unit cells, and the orange/blue arrows mark the tetragonal and orthorhombic phase regions, respectively. d–f Atomic-resolution HAADF-STEM image and EDS mappings of Ba, Sm, and Ti elements of BT-0.03(Sm,Li). Lattice distortion (c/a ratio) mapping of g BT-0.03Sm and h BT-0.03(Sm,Li) extracted from the atomic-scale HAADF-STEM, respectively. i Lattice c/a ratio distribution of BT-0.03Sm and BT-0.03(Sm,Li), respectively.
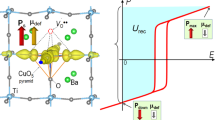
To investigate the substitution of Sm3+ and Li+ ions for the component ions in BaTiO3, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) imaging and corresponding atomic-resolution energy-dispersive X-ray spectroscopy (EDS) mappings of BT-0.03(Sm,Li) were captured. As shown in Fig. 1d, the large and small dots represent the A-site and B-site ions in the perovskite lattice, respectively. By comparing the EDS mapping of the Sm element with the Ba element mappings (Fig. 1e,f), it is evident that the doped Sm3+ ions replace the A-site Ba2+ ions. While it is difficult to directly trace Li+ ions via EDS due to the small atomic number, we hypothesize that the doped Li+ ions also occupy the A site rather than the B site, considering their electronegativity is similar to that of the A site Ba2+ ions. According to the electroneutrality principle, as an acceptor, the equivalently doped Li+ ions tend to replace Ba2+ ions at the A site to neutralize the positive charges of SmBȧ. Otherwise, additional oxygen vacancies \(({V}_{o}^{\cdot \cdot })\) must form to compensate for the negative charges if Li+ ions replace Ti4+ ions at the B site in the lattices. This is confirmed by the high-resolution X-ray photoelectron spectroscopy (XPS), which shows no increase in the \({V}_{o}^{\cdot \cdot }\) concentration in BT-0.03(Sm,Li) compared to that of pristine BaTiO3, further indicating that the Li+ ions substitute at the A-site (Supplementary Fig. 4). We also postulate that Li+ ions tend to locate adjacent to the Sm3+ ions, forming defect dipoles within the BaTiO3 lattice. The statistics in Fig. 1g,h show the lattice c/a ratio distributions extracted from the atomic-scale HAADF-STEM mappings of Ba0.97Sm0.03TiO3 (BT-0.03Sm) and Ba0.94Sm0.03Li0.03TiO3 (BT-0.03(Sm,Li)), respectively. Since the ionic radii of both Sm3+ and Li+ are smaller than that of Ba2+, if the doped Li+ ions substitute randomly at the A site, rather than forming defect dipoles with Sm3+, both the a and c axis of the lattices would decrease synchronously, resulting in an approximately constant c/a ratio (Supplementary Fig. 5)33,34,35,36. However, as shown in Fig. 1i, the average c/a ratio of BT-0.03(Sm,Li) is lower than that of BT-0.03Sm, signifying that the Sm3+ and Li+ ions occupying the A-site form defect dipoles (SmBȧ-LiBaʹ). An important point is that these defect dipoles, which occupy neighboring A sites in the lattices along the [001]c direction, align with the spontaneous polarization direction of the intrinsic Ti⁴⁺-O²⁻ dipoles in BaTiO3. As a result, they are expected to contribute to additional polarization.
Dielectric property and polarization behavior
The temperature-dependent dielectric constants and losses for the samples are shown in Fig. 2a and Supplementary Figs. 6,7. These measurements reveal the impact of Sm³⁺ and Li⁺ substitutions on the material’s dielectric behavior. As indicated by the decrease in Raman peak at ~314 cm−1 (Supplementary Fig. 3b), the substitution of Sm3+ for Ba2+ reduces the tetragonal symmetry of the lattice, leading to a decrease in the Curie temperature and a dispersion of the ferroelectric-paraelectric phase transition (Supplementary Fig. 8), which is evidenced by the low-temperature shift and broadening of the dielectric constant peak observed in BT-0.03Sm. In contrast, the incorporation of Li⁺ induces a marginal change in the dielectric constant. Owing to the lattice distortion caused by the introduction of the defect dipoles (SmBȧ-LiBaʹ) (Supplementary Table 2), the Curie temperature decreases from 123 °C to 60 °C as the x increases from 0.01 to 0.05 in BT-x(Sm,Li). Interestingly, while individual doping of Sm and Li reduces the dielectric constant peak of BaTiO3, the incorporation of (SmBȧ-LiBaʹ) defect dipoles enhances the dielectric anomaly. As shown in Fig. 2a, the dielectric constant peak of BT-0.03(Sm,Li) exceeds 14,000, which is significantly higher than those of BaTiO3, BT-0.03Li, and BT-0.03Sm. This increase in dielectric constant can be attributed to the electron-pinned defect-dipoles (EPDD) effect37,38. Furthermore, the BT-0.03(Sm,Li) composition maintains the characteristic diffused phase transition, as indicated by the broadened dielectric peak, which closely resembles that of BT-0.03Sm. The peak magnitude of BaTiO3 with defect dipoles decreases with increasing frequency, further confirming the dispersed phase transition behavior. Additionally, all samples exhibit low dielectric losses at low temperatures, remaining below 0.03 across below 200 °C (Supplementary Fig. 6). The dielectric loss increases markedly as the temperature is higher than 240 °C (Supplementary Fig. 7).

a Temperature dependence of dielectric constant and loss of BaTiO3, BT-0.03Li, BT-0.03Sm, and BT-0.03(Sm,Li) at 1 kHz. b Polarization-Electric field (P-E) loops and c polarization of BaTiO3, BT-0.03Li, BT-0.03Sm, and BT-0.03(Sm,Li) under 80 kV cm−1 at room temperature, respectively. d Temperature-dependent polarization of BaTiO3, BT-0.03Li, BT-0.03Sm, and BT-0.03(Sm,Li) under the electric fields close to their breakdown strengths. e Remanent polarization of the samples measured at different temperatures. f Breakdown strengths (Eb) of BaTiO3, BT-0.03Li, BT-0.03Sm, and BT-0.03(Sm,Li) determined by the Weibull distribution, respectively. g Experimental and fitting results of Impedance spectra of ln(R) vs. 1000/T. Scatter points represent experimental values, while solid lines represent fitted values. h Electric resistivities of BaTiO3, BT-0.03Li, BT-0.03Sm, and BT-0.03(Sm,Li) under 80 kV cm−1 at room temperature, respectively.
In addition to the dielectric behavior, the concentration of (SmBȧ-LiBaʹ) defect dipoles also plays a significant role in the polarization of BT-xSL. As shown in Fig. 2b,c and Supplementary Fig. 9, the polarization increases substantially from 18.9 μC cm−2 in BaTiO3 to 28.2 μC cm−2 in BT-0.03(Sm,Li) under 80 kV cm−1, and then decreases with further increasing defect dipole concentration (Supplementary Fig. 9). The larger polarization of BT-0.03(Sm,Li) suggests an enhanced ECE14,39, indicating its superior performance in solid-state cooling (Supplementary Note 3). It is important to emphasize that the observed polarization increase originates from the presence of (SmBȧ-LiBaʹ) defect dipoles, rather than the individual contributions from Sm or Li doping. As shown in Fig. 2b, Sm doping narrows the polarization-electric field (P-E) loop by disrupting the long-range ordering of the lattice, which is confirmed by the diffused phase transition (Supplementary Fig. 8). However, this effect has minimal influence on the polarization at elevated electric fields40,41. In contrast, the introduction of Li+ induces a stronger local ferroelectric distortion (Supplementary Table 5), leading to an increase in remanent polarization42. Despite this, the polarization at high fields only shows a moderate increase with Li doping, as displayed in Fig. 2c. In addition, due to the pyroelectric effect43, both maximum polarization and remanent polarization of the samples decrease with increasing temperature (Fig. 2d, e and Supplementary Fig. 10).
Meanwhile, the (SmBȧ-LiBaʹ) defect dipoles were found to greatly enhance the breakdown strength of BaTiO3, as shown in Fig. 2f, where BT-0.03(Sm,Li) exhibits a high breakdown strength of 152 kV cm−1, surpassing that of both pure and Li-doped BaTiO3 (Supplementary Figs. 11,12). Density functional theory (DFT) calculations reveal that Sm doping increases the bandgap of BaTiO₃ from 1.486 eV to 1.925 eV, as shown in Supplementary Fig. 13, and this enlarged bandgap is retained in BT-0.03(Sm,Li). However, Li doping has minimal influence on the bandgap (Supplementary Fig. 14). As illustrated in Fig. 2g and Supplementary Fig. 15, the wide bandgap of 1.829 eV provides BT-0.03(Sm,Li) a higher carrier activation energy compared to pure BaTiO3, effectively preventing charge migration44,45. This enables BaTiO3 doped with an optimal concentration of defect dipoles (i.e., 3 mol% in this study) to achieve a high resistivity exceeding 1011 Ω m (Fig. 2h and Supplementary Fig. 16), which is responsible for the greatly improved breakdown strength. The enhanced breakdown strength allows higher electric field to be applied on the samples, thereby inducing larger polarization. For example, at room temperature, the polarization of BT-0.03Sm and BT-0.03(Sm,Li) increase to 24.3 μC cm−2 and 34.2 μC cm−2, respectively, as the electric field is raised to 150 kV cm−1 (Fig. 2d,e and Supplementary Fig. 10). Moreover, BT-0.03(Sm,Li) exhibits uniform and stable insulating performance (Supplementary Figs. 25 and 26).
To understand the underlying mechanism of the (SmBȧ-LiBaʹ) defect dipoles contributing to the enhanced polarization, we performed theoretical calculations, as shown in Fig. 3. The polarizability of the extrinsic defect dipoles, bound by Coulomb forces, can be assessed as \({{\alpha }}_{{{{\rm{u}}}}}={{{{\rm{q}}}}}^{2}\times {l}^{2} / 3{kT}\), where \({{{\rm{q}}}}\) is the equivalent charges, l = c is the distance of the defect dipole ions (c is the lattice parameter obtained by XRD Rietveld refinement), k is the Boltzmann constant, and T is the temperature, respectively46,47. For the defect dipoles (SmBȧ-LiBaʹ), the calculated polarizability reaches 3.44 × 10−37 F m2, which is over four orders of magnitude higher than that of the intrinsic dipoles (Ti4+-O2-) in BaTiO3 (0.92 × 10−41 F m2). The polarizability of the intrinsic dipoles is calculated by \({\alpha }_{i}={4\pi }_{\varepsilon 0}\times ({3r}^{3}){{{\rm{ / }}}}M(n-1)\) (ɛ0 is the vacuum dielectric constant, r = c/2 is the distance between the centers of Ti4+ ions and O2- ions, Mperovskite = 24.755 is the Madelung constant, and n = 12 is the exponent repulsion potential term in Lennard-Jones potential)48,49, as shown in Fig. 3a. Therefore, even with a low loading of 3 mol.%, the high polarizability of the defect dipoles could increase the polarization of the BaTiO3 ceramic substantially.

a, b Schematic illustration of (SmBȧ-LiBaʹ) defect dipole and the interaction of the defect dipole with the intrinsic (Ti4+-O2-) dipoles. Charge density of c BaTiO3 and d BT-(Sm,Li) calculated by DFT, respectively. Here, δ represents the average displacement of the Ti4+ ion from the center of the oxygen octahedra. e Total DOS and partial DOS of O, Ti for BaTiO3 and BT-(Sm,Li), respectively. The overlap degree of the Ti 3 d state with the primary O 2p valence bands (from the upper valence bands of −7 eV~0 eV to the bottom conduction bands of 2 eV~7 eV) represents the hybridization intensity of Ti-O bonds.
Additionally, the defect dipoles (SmBȧ-LiBaʹ) interact with the intrinsic dipoles in BaTiO3, and the coupling between them further contributes to the enhanced polarization (Fig. 3b). As compared in Fig. 3c1, d1, the covalent bond strength of Sm-O in the lattice is greater than that of Ba-O, owing to the higher electron charge density around Sm (hybrid Sm-O) compared to Ba (which undergoes complete ionization), as revealed by the DFT calculations. Furthermore, the (SmBȧ-LiBaʹ) defect dipoles induce significant lattice distortions, including a pronounced oxygen octahedron rotation away from the [001]c direction (θ = 5.28°) and a large displacement of the Ti4+ ion from the center of the oxygen octahedron (δ = 0.41 Å). These distortions are substantially greater than those observed in pure BaTiO3 lattices or BaTiO3 doped individually with Sm or Li (Fig. 3c2,d2, Supplementary Fig. 17, and Supplementary Table 5), which can be attributed to the smaller ionic radii of Sm3+ and Li+ compared to Ba2+. The hybridization of the Sm-O bond, along with the lattice distortion induced by the defect dipoles, further strengthens the hybridization of the Ti-O bonds in the intrinsic dipoles. DFT calculations reveal a significantly larger overlap of the Ti 3 d partial density of states (DOS) with the primarily O 2p valence bands in lattices containing (SmBȧ-LiBaʹ) defect dipoles, when compared to the pristine BaTiO3 (Fig. 3e) or those doped solely with Sm or Li (Supplementary Fig. 18). This confirms the enhanced Ti-O hybridization and reduced B site-O repulsions in the BT-(Sm,Li) lattices50,51,52,53. Consequently, the intrinsic dipole polarization is further enhanced, and the interaction between the defect dipoles and the (Ti4+-O2-) intrinsic dipoles becomes another contributing factor to the increased macroscopic electric polarization of BT-0.03(Sm,Li), in addition to the high polarizability of the defect dipoles themselves.
Contrary to earlier beliefs that defect dipoles clamp the domain wall motion, the introduced (SmBȧ-LiBaʹ) defect dipoles in BaTiO3 were found to play an important role in facilitating domain switching under applied electric fields, as illustrated in Supplementary Fig. 19. PFM phase images reveal that the domains in BT-0.03(Sm,Li) begin to switch at a low voltage of 2 V and become fully aligned at a moderate voltage of 6 V. In contrast, pure BaTiO3 shows only partial domain switching even at a higher voltage of 8 V. These findings demonstrate that the defect dipoles (SmBȧ-LiBaʹ) lower the activation energy (electric field) required for domain switching in BaTiO3, which is expected to enhance the EC strength of the ceramic. It is suggested that these donor-acceptor defect complexes can serve as nucleation centers, which enhance the domain wall mobility and facilitate domain switching54. Additionally, XPS analysis (Supplementary Fig. 4) reveals that the introduction of (SmBȧ-LiBaʹ) defect dipoles reduces the concentration of oxygen vacancies, which further contributes to domain switching55,56. The additional polarization induced by the defect dipoles57, as evidenced by the asymmetric amplitude-voltage butterfly curves (Supplementary Fig. 20), helps restore the domains to their original state after the removal of the electric field (Supplementary Fig. 19c3,d3), resulting in a significant entropy change between the original and polarized states in BT-0.03(Sm,Li).
Electrocaloric effect
Figure 4 and Supplementary Figs. 21–24,26 summarize the ECE of BaTiO3, BT-0.03Li, BT-0.03Sm, and BT-x(Sm,Li). The temperature and entropy changes of the samples increase with the applied electric field at room temperature, as shown in Fig. 4a,b. For example, ΔT and ΔS of BaTiO3 increase from 0.27 K and 0.33 J kg−1 K−1 to 0.47 K and 0.57 J kg−1 K−1 as the electric field increases from 30 kV cm−1 to 80 kV cm−1, respectively. Due to the high polarizability of the defect dipoles (SmBȧ-LiBaʹ), BT-0.03(Sm,Li) exhibits an improved EC strength in comparison to that of pure BaTiO3 and the samples with single Sm or Li doping (Supplementary Fig. 21), enabling a preferred ECE. Under an electric field of 80 kV cm−1, BT-0.03(Sm,Li) shows ΔT and ΔS of 0.98 K and 1.27 J kg−1 K−1, approximately 2 times higher than those of pure BaTiO3, and 1.4 folds those of BT-0.03Li and BT-0.03Sm, respectively. The results calculated using Maxwell relations agree well with that experimentally evaluated with a heat flux sensor (Supplementary Fig. 22). However, further increase of defect dipole concentration leads to a decay in ECE, in line with the trend observed in polarization (Supplementary Fig. 23). We then increase the applied electric field to fully activate the EC potential of BT-0.03(Sm,Li). Under a high electric field of 150 kV cm−1 (before its breakdown strength), a desirable ECE with a ΔT of 1.54 K and a ΔS of 2.00 J kg−1 K−1 is achieved in BT-0.03(Sm,Li) at room temperature. Although the same high electric field can be applied to BT-0.03Sm due to its high breakdown strength, its ECE is inferior to that of BT-0.03(Sm,Li) because of its lower polarizability (Fig. 4c). It should be noted that the sample thickness in this study was kept at 100 µm, considering the breakdown strength is inversely proportional to the sample thickness58, it is expected that a higher breakdown strength, thus higher ECE will be achieved in a prototypic multilayer ceramic capacitor.

a ΔT and b ΔS of BaTiO3, BT-0.03Li, BT-0.03Sm, and BT-0.03(Sm,Li) under different applied electric fields at room temperature. c A comparison of ΔTmax of BaTiO3, BT-0.03Li, BT-0.03Sm, and BT-0.03(Sm, Li) at room temperature. d ΔT and e ΔS in the temperature range of 25 °C ~ 70 °C close to their corresponding breakdown electric fields (80 kV cm−1, 80 kV cm−1, 150 kV cm−1, and 150 kV cm−1 for BaTiO3, BT-0.03Li, BT-0.03Sm, and BT-0.03(Sm,Li), respectively. f Comparison of ΔT of BT-0.03(Sm,Li) ceramic with other representative BaTiO3 based ceramics. The detailed information is listed in Supplementary Table 6.
Of particular interest is that the ECE of BT-0.03(Sm,Li) continues to increase as the ambient temperature approaches its Curie point (96 °C, as indicated by the dielectric constant peak in Fig. 2a). As plotted in Fig. 4d,e, at an elevated temperature of 70 °C—a critical temperature for typical electronic devices4,59, BT-0.03(Sm,Li) exhibits a ΔT and ΔS reaching 2.76 K and 3.10 J kg−1 K−1, representing one of the highest ECE among BaTiO3-based bulk materials (Fig. 4f and Supplementary Table 6). In addition, BT-0.03(Sm,Li) maintains a high ECE, with ΔT ≥ 1.5 K and ΔS ≥ 2.0 J kg−1 K−1, across a wide temperature range from 25 °C to 70 °C. The results agree well with that calculated using Maxwell relations (Supplementary Fig. 24). This exceptional EC performance, especially at elevated temperatures, highlights the potential of defect-engineered ferroelectric ceramics for effective thermal management in electronics.
Discussion
In summary, this study demonstrates the significant potential of defect dipole engineering to enhance the ECE in ferroelectric ceramics. By introducing an optimized amount of heterovalent ions (Sm3+ and Li+ ions) to form defect dipoles in BaTiO3, we successfully form (SmBȧ-LiBaʹ) defect dipoles aligning along the spontaneous polarization. This, in turn, facilitates a strong coupling effect between the defect dipoles and the intrinsic (Ti4+-O2-) dipoles, leading to a much-improved polarization and a substantial enhancement in the ECE of BT-0.03(Sm,Li). Simultaneously, the defect dipoles widen the bandgap and increase the carrier activation energy, enabling the application of large electric fields to fully activate the EC potential over a broad temperature range. These findings not only underscore the effectiveness of defect dipole engineering in enhancing the ECE but also suggest its broader applicability in various ferroelectric ceramic systems, including lead zirconate titanate, potassium sodium niobate, and sodium bismuth titanate.
Methods
Material preparation
Ba1-2xSmxLixTiO3 (BT-x(Sm,Li), x = 0.01, 0.02, 0.03, 0.04, and 0.05), BaTiO3, Ba0.97Sm0.03TiO3 and Ba0.97Li0.03TiO3 ceramics were fabricated through the conventional solid-state reactions. All raw materials, including BaCO3 (AR), Sm2O3 (4 N), Li2CO3 (4 N), and TiO2 (4 N), were weighed according to the stoichiometric ratio and then planetary milled for 6 h with alcohol media. The mass ratio of alcohol to raw materials was 1.2:1, and the rotation speed of the planetary milling was 400 r min−1. After drying at 70 °C, the mixed raw material powders were calcined at 1000 °C for 4 h with a heating rate of 3 °C min−1. Then the calcined powders were dried and mixed with a 4 wt.% solution of polyvinyl alcohol after high-energy ball milling for 6 h. The mixtures were uniaxially pressed into disks with a diameter of 10 mm. To improve the densification of the samples, a cold isostatic pressing (CIP) with a pressure of 200 MPa was applied to the disk samples for 300 seconds. Finally, the samples were sintered at 1240 °C ~ 1350 °C for 3 h with a heating rate of 3 °C min−1.
Material characterization
The surface morphologies of BaTiO3, BT-0.03Li, BT-0.03Sm, and BT-x(Sm,Li) were observed using the field emission scanning electron microscope (SEM) (Zeiss GeminiSEM300, Germany). The phase structure characteristics of all samples were determined by the X-ray diffractometer (XRD, 7000 S/L, Shimadzu Corp., Japan) with a Cu Kα rotating anode from 10° to 80° at a scanning speed of 0.3° min−1 ~ 7.0° min−1. To analyze the specific phase composition and lattice parameters of BT-x(Sm,Li), the XRD data were refined through the Rietveld method using the software GSAS-EXPGUI. HAADF-STEM images were captured by a double spherical aberration corrected scanning transmission electron microscope (STEM, Spectra 300, ThermoFisher Scientific) equipped with probe and image correctors and operated at 300 kV. The atomic-scale elemental analysis was performed using an energy-dispersive X-ray spectroscopy (EDS) detector attached to the STEM. TEM samples were prepared by a dual-beam Focused Ion Beam (FIB, Helios 5 UC, ThermoFisher Scientific). The final thicknesses of TEM lamellas were about 30 ~ 40 nm. A Raman spectrometer (LabRAM HR800, Horiba Jobin Yvon, France) was used to further verify the crystal structure of the samples. High-resolution X-ray photoelectron spectroscopy (XPS) spectra of O 1 s and Ti 2p were measured through an X-ray photoelectron spectrometer (AXIS-ULTRA DLD-600W). The observation of the oriented domain and the local poling experiments were presented by a piezoresponse force microscope (PFM) (Bruker Dimension Icon, USA). The dielectric constant and loss under different temperatures and frequencies, as well as the impedance spectra, were measured by the dielectric properties test system (DPTS-3000, Wuhan Yanhe Technology Co., Ltd., China). The polarization-electric field (P-E) loops at room temperature were measured by the ferroelectric tester (FETS-2000, Wuhan Yanhe Technology Co., Ltd., China). The electric conductivity of all samples was measured by a high-voltage leakage current test system (Keithley 6517B, Tektronix Company, America). The bulk densities of the samples were measured through the Archimedes method. The specific heat capacities of all samples were measured by the differential scanning calorimeter (DSC) instruments (Q2000, TA Instruments-Waters LLC). The ECE testing experiment with the isothermal heat flux measurement (Supplementary Note 4) was performed using the electrocaloric tester (ECTS-2000, Wuhan Yanhe Technology Co., Ltd., China)14,60.
First principles calculations
The first principles calculations were carried out through the density functional theory (DFT) using the Vienna Ab-initio Simulation Package (VASP) with the projector-augmented wave (PAW) method. The generalized gradient approximation (GGA) with the Perdew Burke Ernzerhof (PBE) function was adopted to describe the electron exchange and correlation effects61. Uniform Monkhorst Pack k-points meshes with a resolution of 2π×0.03 Å−1. The simulations were run at a cutoff energy of 500 eV throughout the entire calculation process, and the total energy converged stays within 10−6 eV per atom, and all forces on per atom were less than 0.005 eV Å−1. In the calculations, the lattice parameters of bulk BaTiO3, BT-Li, BT-Sm, and BT-(Sm,Li) were identified from the Rietveld refinements of the XRD patterns. The 1 × 2 × 2 supercells with diverse polarization orientations were built for BaTiO3, BT-Li, BT-Sm, and BT-(Sm,Li), respectively. In the supercell of BT-(Sm,Li), two adjacent A-site Ba ions along the c-axis are replaced by one Sm ion and one Li ion. The calculated lattice parameters of bulk BaTiO3, BT-Li, BT-Sm, and BT-(Sm,Li) are a = 4.006 Å, b = 4.006 Å, c = 4.197 Å; a = 3.933 Å, b = 7.853 Å, c = 8.750 Å; a = 4.005 Å, b = 8.010 Å, c = 8.015 Å; and a = 3.918 Å, b = 7.787 Å, c = 8.521 Å, respectively. Then, based on the full relaxed supercells, electronic structures were calculated until the total energy converged smaller than 10−8 eV.
Data availability
Data supporting the figures in this paper and the other findings of this study are either available in the article and its Supplementary Information, or are available from the corresponding author upon request. Source data are provided with this paper.
References
Li, J. et al. High cooling performance in a double-loop electrocaloric heat pump. Science 382, 801–805 (2023).
Yuan, M., Pu, J. & Pei, Q. Electrocaloric cooling over high device temperature span. Joule 5, 780–793 (2021).
Du, F. et al. Giant electrocaloric effect in high-polar-entropy perovskite oxides. Nature 640, 924–930 (2025).
Garimella, S. V., Persoons, T., Weibel, J. A. & Gektin, V. Electronics thermal management in information and communications technologies: challenges and future directions. IEEE Trans. Compon. Packag. Manuf. Technol. 7, 1191–1205 (2017).
Yan, Z. et al. On-chip high speed localized cooling using superlattice microrefrigerators. IEEE Trans. Compon. Packag. Technol. 29, 395–401 (2006).
Franco, V. et al. Magnetocaloric effect: from materials research to refrigeration devices. Prog. Mater. Sci. 93, 112–232 (2018).
Tušek, J. et al. The elastocaloric effect: a way to cool efficiently. Adv. Energy Mater. 5, 1500361 (2015).
Zhang, Z. et al. Thermal batteries based on inverse barocaloric effects. Sci. Adv. 9, eadd0374 (2023).
Zou, K. et al. Electronic cooling and energy harvesting using ferroelectric polymer composites. Nat. Commun. 15, 6670 (2024).
Nouchokgwe, Y. et al. Giant electrocaloric materials energy efficiency in highly ordered lead scandium tantalate. Nat. Commun. 12, 3298 (2021).
Li, Q. et al. Low-k nano-dielectrics facilitate electric-field induced phase transition in high-k ferroelectric polymers for sustainable electrocaloric refrigeration. Nat. Commun. 15, 702 (2024).
Son, Y. et al. Polarization stability and its influence on electrocaloric effects of high entropy perovskite oxide films. Acta Mater. 283, 120576 (2025).
Son, Y., Zhu, W. & Trolier-McKinstry, S. E. Electrocaloric effect of perovskite high entropy oxide films. Adv. Electron. Mater. 8, 2200352 (2022).
Liu, Y., Scott, J. F. & Dkhil, B. Direct and indirect measurements on electrocaloric effect: Recent developments and perspectives. Appl. Phys. Rev. 3, 031102 (2016).
Plaznik, U. et al. Bulk relaxor ferroelectric ceramics as a working body for an electrocaloric cooling device. Appl. Phys. Lett. 106, 043903 (2015).
Bai, Y. et al. The giant electrocaloric effect and high effective cooling power near room temperature for BaTiO3 thick film. J. Appl. Phys. 110, 094103 (2011).
Shirsath, S. E. et al. Interface-charge induced giant electrocaloric effect in lead free ferroelectric thin-film bilayers. Nano Lett. 20, 1262–1271 (2020).
Liu, X. Q., Chen, T. T., Wu, Y. J. & Chen, X. M. Enhanced electrocaloric effects in spark plasma-sintered Ba0.65Sr0.35TiO3-based ceramics at room temperature. J. Am. Ceram. Soc. 96, 1021–1023 (2013).
Zheng, S. et al. Colossal electrocaloric effect in an interface-augmented ferroelectric polymer. Science 382, 1020–1026 (2023).
Chen, Y. Q. et al. An all-scale hierarchical architecture induces colossal room-temperature electrocaloric effect at ultralow electric field in polymer nanocomposites. Adv. Mater. 32, 1907927 (2020).
Nair, B. et al. Large electrocaloric effects in oxide multilayer capacitors over a wide temperature range. Nature 575, 468–472 (2019).
Achkar, M. et al. Electrocaloric characterization of samarium doped barium titanate ceramics synthesized by sol-gel process. Ceram. Int. 50, 13794–13801 (2024).
Feng, H. et al. Low-electric-field-induced high electrocaloric properties via defect regulation in samarium-doped BaTiO3 ceramics. J. Alloy. Compd. 989, 174362 (2024).
Moya, X. et al. Giant electrocaloric strength in single-crystal BaTiO3. Adv. Mater. 25, 1360–1365 (2013).
Yan, Y. et al. Correlation between cation order/disorder and the electrocaloric effect in the MLCCs of complex perovskite ferroelectrics. Acta Mater. 254, 118990 (2023).
Yan, G. et al. Enhanced energy storage property and dielectric breakdown strength in Li+ doped BaTiO3 ceramics. J. Alloy. Compd. 857, 158021 (2021).
Liu, Z. K., Li, X. & Zhang, Q. M. Maximizing the number of coexisting phases near invariant critical points for giant electrocaloric and electromechanical responses in ferroelectrics. Appl. Phys. Lett. 101, 082904 (2012).
Zhao, C., Yang, J., Huang, Y., Hao, X. & Wu, J. Broad-temperature-span and large electrocaloric effect in lead-free ceramics utilizing successive and metastable phase transitions. J. Mater. Chem. A 7, 25526–25536 (2019).
Qian, X. S. et al. Giant electrocaloric response over a broad temperature range in modified BaTiO3 ceramics. Adv. Funct. Mater. 24, 1300–1305 (2014).
Zhao, L. et al. Large electrocaloric effect over a wide temperature range in BaTiO3-modified lead-free ceramics. J. Mater. Chem. C. 7, 1353–1358 (2019).
Li, J. et al. Large room-temperature electrocaloric effect in lead-free BaHfxTi1−xO3 ceramics under low electric field. Acta Mater. 115, 58–67 (2016).
Feng, Y. et al. Defects and aliovalent doping engineering in electroceramics. Chem. Rev. 120, 1710–1787 (2020).
Feng, Y. et al. Defect engineering of lead-free piezoelectrics with high piezoelectric properties and temperature-stability. ACS Appl. Mater. Interfaces 8, 9231–9241 (2016).
Ren, X. Large electric-field-induced strain in ferroelectric crystals by point-defect-mediated reversible domain switching. Nat. Mater. 3, 91–94 (2004).
Ren, X. & Kazuhiro, O. Universal symmetry property of point defects in crystals. Phys. Rev. Lett. 85, 1016–1019 (2000).
Lee, D. et al. Active control of ferroelectric switching using defect-dipole engineering. Adv. Mater. 24, 6490–6495 (2012).
Hu, W. et al. Electron-pinned defect-dipoles for high-performance colossal permittivity materials. Nat. Mater. 12, 821–826 (2013).
Meng, Y. et al. Defect engineering in rare-earth-doped BaTiO3 ceramics: Route to high-temperature stability of colossal permittivity. J. Am. Ceram. Soc. 105, 5725–5737 (2022).
Vales-Castro, P. et al. Origin of large negative electrocaloric effect in antiferroelectric PbZrO3. Phys. Rev. B 103, 054112 (2021).
Li, F. et al. Giant piezoelectricity of Sm-doped Pb(Mg1/3Nb2/3)O3-PbTiO3 single crystals. Science 364, 264–268 (2019).
Pan, H. et al. Ultrahigh energy storage in superparaelectric relaxor ferroelectrics. Science 374, 100–104 (2021).
Zou, J. et al. Enhancing piezoelectric coefficient and thermal stability in lead-free piezoceramics: insights at the atomic-scale. Nat. Commun. 15, 8591 (2024).
Shen, M. et al. High room-temperature pyroelectric property in lead-free BNT-BZT ferroelectric ceramics for thermal energy harvesting. J. Eur. Ceram. Soc. 39, 1810–1818 (2019).
Lee, H., Lee, K., Schunke, J. & Burton, L. Leakage currents in multilayer ceramic capacitors. IEEE Trans. Compon. Hybrids Manuf. Technol. 7, 443–453 (1984).
Gao, Y. et al. Ultrahigh energy storage in tungsten bronze dielectric ceramics through a weakly coupled relaxor design. Adv. Mater. 36, 2310559 (2024).
Liu, Y. X. et al. Isolated-oxygen-vacancy hardening in lead-free piezoelectrics. Adv. Mater. 34, 2202558 (2022).
Lai, L. et al. Giant electrostrain in lead-free textured piezoceramics by defect dipole design. Adv. Mater. 35, 2300519 (2023).
Zhao, Z., Lv, Y., Dai, Y. & Zhang, S. Ultrahigh electro-strain in acceptor-doped KNN lead-free piezoelectric ceramics via defect engineering. Acta Mater. 200, 35–41 (2020).
Johnson, Q. C. & Templeton, D. H. Madelung constants for several structures. J. Chem. Phys. 34, 2004 (1961).
Cohen, R. E. Origin of ferroelectricity in perovskite oxides. Nature 358, 136–138 (1992).
Shi, T., Xie, L., Gu, L. & Zhu, J. Why Sn doping significantly enhances the dielectric properties of Ba(Ti1-xSnx)O3. Sci. Rep. 5, 8606 (2015).
Cohen, R. E. & Krakauer, H. Electronic structure studies of the differences in ferroelectric behavior of batio3 and PbTiO3. Ferroelectrics 136, 65–83 (1992).
Lu, N. & Zhu, J. Electron low energy-loss functions of Pb(Mg1/3Nb2/3)O3: Theory and experiment. J. Appl. Phys. 104, 034109 (2008).
Khachaturyan, R., Dimou, A. & Grünebohm, A. Domain wall acceleration by ultrafast field application: An ab initio-based molecular dynamics study. Phys. Status Solidi RRL 16, 2200038 (2022).
Li, X. Y. et al. Domain wall motion in perovskite ferroelectrics studied by the nudged elastic band method. J. Phys. Chem. C122, 3091–3100 (2018).
Li, Z., Yu, J., Hao, S. & Janolin, P. Enhancing properties of lead-free ferroelectric BaTiO3 through doping. J. Eur. Ceram. Soc. 42, 4693–4701 (2022).
Geng, H. et al. Deciphering the effect of defect dipoles on the polarization and electrostrain behavior in perovskite ferroelectrics. Nano Lett. 24, 12148–12155 (2024).
Yang, L. et al. Perovskite lead-free dielectrics for energy storage applications. Prog. Mater. Sci. 102, 72–108 (2019).
Bo, Y. et al. Electrostatic actuating double-unit electrocaloric cooling device with high efficiency. Adv. Energy Mater. 11, 2003771 (2021).
Xiao, W. et al. Significant enhancement of electrocaloric effect in ferroelectric polycrystalline ceramics through grain boundary barrier engineering. Adv. Funct. Mater. 34, 2405241 (2024).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (52272110, 62105110, 52202134, 52372108, 52172114, 52001117, and 523B2011) (S.J., K.L., W.D., G.Z., J.G., and F.L.), the National Key Research and Development Program of China (2022YFA1204603) (G.Z., and K.L.), and Hubei Provincial Natural Science Foundation of China (2024AFA012) (K.L.). We would also like to thank the Analytical and Testing Center of HUST.
Author information
Authors and Affiliations
Contributions
G.Z. and W.X. conceived the idea. G.Z., S.J., S.Z., K.L., J.G., X.W., and W.D. supervised the project. W.X. designed the experiments. W.X. and X.Z. fabricated the samples. W.X., Y.L., B.Y., Z.Z., X.G., F.L., L.L., and J.G. performed the characterizations. Y.W., W.X., S.L., and W.D. carried out the DFT calculation. G.Z., S.Z., W.X., X.W., J.L. and K.L. prepared the manuscript, with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiao, W., Wu, Y., Liu, Y. et al. Enhanced electrocaloric effect in ferroelectric ceramics via defect dipole engineering. Nat Commun 16, 8909 (2025). https://doi.org/10.1038/s41467-025-63963-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-63963-5
