
Abstract
Metastatic castration-resistant prostate cancer (mCRPC) is a lethal disease requiring additional therapeutic strategies. MCL1, an anti-apoptotic BCL2 family member, promotes cancer-cell survival, but its role in mCRPC remains poorly understood. Here, we characterise MCL1 in multiple mCRPC biopsy cohorts and patient-derived models, assessing responses to MCL1 inhibition. MCL1 copy number gain (14%–34%) correlates with increased MCL1 expression and worse outcomes. MCL1 inhibition exhibits anti-tumour effects in MCL1-gained mCRPC models. Co-inhibition of MCL1 and AKT induces cancer-specific cell death in PTEN-loss/PI3K-activated models in vitro and in vivo, modulating BAD-BCLXL and BIM-MCL1 interactions, with durable anti-tumour activity in models with AKT inhibitor acquired resistance. Finally, CDK9-mediated MCL1 downregulation combined with AKT inhibition recapitulates these findings, providing further opportunities for clinical translation. These data support early phase clinical trials targeting MCL1, both as monotherapy for MCL1-gained mCRPC, and in combination with AKT inhibition for PTEN-loss/PI3K-activated mCRPC.
Similar content being viewed by others


Introduction
Despite significant progress in the treatment of metastatic castration-resistant prostate cancer (mCRPC), the disease remains lethal, with a median overall survival (OS) ranging from 2 to 3 years1,2,3. Additional therapeutic strategies are urgently required to improve the outcome for patients with advanced disease. Cell death escape and circumvention of apoptosis is a hallmark of cancer, driving tumourigenesis and therapeutic resistance4. Intrinsic apoptosis is tightly regulated by a multitude of pro- and anti-apoptotic B-cell lymphoma 2 (BCL2) proteins, which interact on the mitochondrial outer membrane. Among the anti-apoptotic members, Myeloid Cell Leukaemia 1 (MCL1) has been shown to promote cancer cell survival in various malignancies, including prostate cancer (PC)5,6. Moreover, MCL1 is one of the most frequently amplified genes in human malignancies, with amplification found in 10.9% of cancers across multiple tumour types7.
Beyond its anti-apoptotic function, it is postulated that MCL1 is involved in other key cancer cell processes such as cell cycle regulation, DNA repair, metabolism and calcium homoeostasis8. In addition, MCL1 may play a role in the development of resistance to androgen deprivation therapy (ADT) and DNA damage-inducing chemotherapies in PC9,10, although further studies are required in this space. Moreover, MCL1 is thought to promote cell survival in therapy-induced senescent PC cells, a phenotype that contributes to tumour proliferation and metastasis via the Senescence-Associated Secretory Phenotype and has recently been reported to play a key role in endocrine treatment resistance11,12. Taken together, MCL1 holds promise as a therapeutic target to induce apoptotic cell death, rather than growth inhibition in mCRPC, potentially mitigating the risk of acquired therapeutic resistance.
Importantly, small molecule BH3 mimetics have been developed to inhibit the anti-apoptotic BCL2 proteins, by binding to their hydrophobic groove5. Although developing high-affinity MCL1-specific BH3 mimetics has been challenging, there are now multiple agents under investigation in first-in-human clinical trials, with a focus on hematopoietic cancers. Despite these advances, preclinical studies have shown limited anti-tumour activity for single protein targeting, potentially due to functional redundancies among the anti-apoptotic BCL2 family members, including BCLXL13,14,15. Indeed, it has been shown that PC is co-dependent on MCL1 and BCLXL and that both proteins must be inhibited to induce apoptosis16,17, which suggests that targeting pathways regulating the expression or activity of BCLXL, in combination with an MCL1 inhibitor, may drive apoptotic cell death. Taken together, the identification of predictive biomarkers that enrich for response to single agent MCL1 inhibition in PC, and the discovery of rational MCL1 inhibitor combination strategies that drive apoptosis in PC, would address an urgent unmet clinical need.
Here, we show that MCL1 copy number gain and amplification occur early in lethal PC evolution and associate with worse clinical outcomes. We demonstrate that single agent MCL1 inhibition has marked anti-tumour activity in PC models with MCL1 copy number gain, and that MCL1 targeting (directly through BH3 mimetics or indirectly through CDK9 inhibition) synergises with AKT inhibition to deliver cancer-specific killing in PTEN-loss/PI3K-activated PC models, with ongoing anti-cancer activity in PC models with acquired AKT resistance. These data support the evaluation of therapeutic strategies targeting MCL1, as a single agent for PC with MCL1 copy number gains, and in combination with AKT inhibition in PTEN loss/PI3K-activated PC, within ‘proof of concept, proof of mechanism’ early phase clinical trials.
Results
MCL1 copy number gains are common in lethal prostate cancer, occur early in tumour evolution, and increase with castration-resistance
To investigate the genomic status of MCL1 in PC, we examined gene copy number alteration data from multiple PC biopsy cohorts. MCL1 is located on chromosome 1q21 and falls within a commonly amplified multi-gene cassette (Fig. 1A, B). MCL1 amplification (high level copy number gain) was rarely seen in primary PC from the The Cancer Genome Atlas (TCGA) cohort (1%), the majority of which are cured after radical treatment (Fig. 1A, B). In advanced CRPC biopsies from the Stand Up To Cancer/Prostate Cancer Foundation (SU2C/PCF) cohort, MCL1 amplification was more common (14%) (Fig. 1A, B). Analysis of matched same-patient CSPC and CRPC biopsies from the Royal Marsden Hospital (RMH) cohort showed a similar frequency of MCL1 amplification (CSPC 11.36%, CRPC 13.64%), suggesting this event occurs early in tumour evolution and associates with CRPC development (Fig. 1A, B). Despite this, a higher proportion of CRPC biopsies (34.09%) exhibited MCL1 copy number gain when compared to matched, same patient, CSPC biopsies (15.91%) (Fig. 1A, B), suggesting this might be part of an adaptive response emerging with treatment resistance. In addition, consistent with treatment resistance emergence, androgen receptor (AR) locus (Xq12) amplification was almost exclusively observed in CRPC biopsies (36%) when compared to CSPC biopsies from the same patient (Fig. 1A, B).

A, B Copy number alterations in TCGA (primary PC), SU2C/PCF (CRPC), and RMH (matched HSPC and CRPC) cohorts. A Chromosomes 1 (MCL1) and X (AR) are highlighted; B MCL1 locus (1q21) marked with an asterisk. CNA copy number alterations. Gain: copy number gain; Amp amplification. C Kaplan–Meier progression-free interval curves in TCGA cohort (n = 492) comparing patients with MCL1 gain/amplification vs those without. Hazard ratio (HR) with 95% confidence intervals and p-value from log-rank test shown. D Comparison of Gleason score, American Joint Committee on Cancer (AJCC) pathological tumour (T) and node (N) stages between patients with MCL1 gain/amplification vs those without in TCGA (n = 492). E, F Residual cancer burden in patients from the NCI neoadjuvant cohort (n = 37), treated with 6 months ADT and enzalutamide, stratified by MCL1 copy number (gain/amplification vs no gain/amplification). F Two-tailed Mann-Whitney U test; median indicated. INR Incomplete/non-responder, ER exceptional responder. G Kaplan–Meier overall survival for patients from FIRSTANA and PROSELICA trials (n = 152), stratified by MCL1 gain/amplification on baseline ctDNA. HR with 95% confidence intervals and p-values from log-rank test shown. H Baseline PSA and Hb in patients from FIRSTANA and PROSELICA trials (n = 152), stratified by MCL1 gain/amplification on baseline ctDNA; Two-sided Mann-Whitney U test used. Box shows the interquartile range (IQR), line indicates the median, and whiskers the min/max values. I MCL1 RNA expression by copy number status across TCGA (n = 491), SU2C/PCF (n = 106), RMH (n = 86), and UW/FH (n = 151) cohorts. Box (IQR), line (median), whiskers (min/max). Two-tailed Mann-Whitney U test. J MCL1 RNA expression in UW/FH cohort split by homogeneous (with and without MCL1 copy number gain/amplification) and heterogeneous copy number (n = 115 tumours from 50 patients with >1 tumour). Mean with min–max floating bars shown. NA Not available. K MCL1 IHC in 30 mCRPC biopsies from RMH cohort with RNA-seq data. Spearman correlation between MCL1 RNA and protein (OD) levels. L Representative IHC images from the 30-biopsy set. Scale bar = 100 µm. Source data are provided as a Source Data file.
MCL1 copy number gains associate with worse clinical outcome
Having shown that genomic gains of MCL1 are common in mCRPC, we evaluated their association with prognostic markers and clinical outcome. In primary PC (TCGA cohort), MCL1 copy number gain/amplification was associated with significantly shorter progression-free interval (PFI) (HR 1.93, CI 1.05-3.53, p = 0.03) (Fig. 1C), a higher Gleason score and pathological tumour stage, and an increased chance of lymph node involvement at diagnosis (Fig. 1D). Furthermore, in a phase II trial evaluating 6 months of neoadjuvant ADT and enzalutamide prior to radical prostatectomy in men with localised intermediate- or high-risk PC18, MCL1 somatic copy number gains at baseline were associated with an inferior pathologic response to therapy. Four out of 37 tumours (11%) had MCL1 copy number gain/amplification, all of which were classified as pathologic incomplete or non-responders, providing evidence that gains in MCL1 may be associated with worse clinical outcome (Fig. 1E, F). In mCRPC, analysis of plasma cell-free DNA from patients enrolled in the FIRSTANA and PROSELICA clinical trials showed that subjects whose tumours have MCL1 copy number gain/amplification have shorter overall survival (HR 1.35, CI 0.95–1.90, p = 0.09) (Fig. 1G). MCL1 copy number gain/amplification was also associated with prior exposure to second-generation hormonal therapies (abiraterone and/or enzalutamide), whereas no significant differences were observed in terms of prior radical prostatectomy or radiotherapy (Supplementary Fig. 4A). MCL1 copy number gain/amplification did not, however, associate with taxane response highlighting its potential role as a prognostic, rather than predictive biomarker, in this specific context (Supplementary Fig. 4B). When considering prognostic markers in CRPC, MCL1 copy number gain/amplification associated with higher PSA and lower haemoglobin (Fig. 1H), but not with the other putative prognostic biomarkers analysed herein (Supplementary Fig. 4C–E). Overall, these data suggest that MCL1 copy number changes as PC progresses to mCRPC and is associated with worse clinical outcome.
MCL1 RNA and protein are highly expressed in mCRPC
Transcriptomic analysis revealed an association between MCL1 copy number gain/amplification and increased RNA expression in the TCGA and SU2C/PCF cohorts, as well as in the rapid autopsy University of Washington/Fred Hutchinson (UW/FH) CRPC cohort which also demonstrated highly concordant intra-patient, inter-tumour, MCL1 expression19 (Fig. 1I, J). The same trend was observed in the RMH cohort although not statistically significant (p = 0.301) (Fig. 1I). These data underline the functional significance of MCL1 copy number gain/amplification. However, a substantial number of tumours with unaltered MCL1 copy number exhibit high RNA expression, suggesting that additional mechanisms can also drive MCL1 overexpression (Fig. 1I, J). To investigate the correlation between MCL1 RNA and protein levels, Immunohistochemistry (IHC) was performed on 30 mCRPC biopsies from the RMH mCRPC cohort (Fig. 1K). MCL1 staining exhibited a cytoplasmic and granular pattern, which is consistent with its mitochondrial localisation (Fig. 1L). MCL1 RNA expression was found to be positively correlated with MCL1 protein cytoplasmic optical density values (n = 30, Spearman r = 0.482, p = 0.007) (Fig. 1K, L), suggesting that elevated RNA levels led to functionally relevant protein expression. While a range of expression levels was observed, none of the tumours exhibited loss of MCL1 protein or RNA (Supplementary Fig. 5A). Cytoplasmic MCL1 H-score was found to be correlated with MCL1 cytoplasmic optical density levels (Supplementary Fig. 5B). In addition, higher tumour MCL1 expressions (H-Score ≥100) was associated with shorter overall survival in CRPC patients (HR 2.09, 95% CI 0.85–5.12, p = 0.11) (Supplementary Fig. 6). These data confirm that MCL1 copy number changes associate with MCL1 RNA and protein, which are broadly expressed in advanced PC, further underlying its potential importance in PC biology.
Response to MCL1 inhibition is enriched in PC models with MCL1 copy number gain
Following the demonstration that MCL1 copy number was gained in a subset of mCRPC patients and linked to poor prognosis, we explored MCL1 targeting as a therapeutic strategy, hypothesising that MCL1 copy number gain would indicate dependency and enrich response to MCL1 inhibition, as suggested in other tumour types7,20,21. We first characterised seven prostate-derived cell lines and found that 22Rv1 and DU145 harbour MCL1 copy number gain; however, only 22Rv1 exhibited markedly elevated MCL1 protein levels, suggesting additional mechanisms regulate MCL1 protein levels in DU145 (Fig. 2A–C). Among the tested cell lines, 22Rv1 showed the highest sensitivity to MCL1 inhibition, as evidenced by increased caspase 3/7 activity and reduced cell viability at 24 h and 6 days (Fig. 2D–F). LNCaP95 displayed a comparable response, indicating that sensitivity to MCL1 inhibition can be influenced by factors beyond basal MCL1 copy number or protein levels (Fig. 2D-F). The lack of response in DU145 may stem from its reported apoptosis resistance due to BAX deficiency22 (Fig. 2D–F). Next, we evaluated the apoptotic response to MCL1 inhibition of five CRPC PDX-O models with varying MCL1 copy numbers and protein levels (Fig. 2G–K). Both AZD5991 and S63845 increased caspase activity (6 h) and reduced organoid viability (at 96 h) in the MCL1 copy number gain models CP50c and CP267c, which exhibited high MCL1 protein expression (Fig. 2G–K and Supplementary Fig. 7). In contrast, models with unaltered MCL1 copy number and lower protein levels (CP142c, CP253c and CP336c) were minimally impacted (Fig. 2G–K and Supplementary Fig. 7). The effects of MCL1 inhibition demonstrated herein reflect short-term apoptosis; thus, the potential impact on PDX-O proliferation may require extended incubation times. These data suggest that, at least a subset of CRPC cells with high MCL1 levels may be sensitive to single agent MCL1 inhibition.

A Violin plot of MCL1:Centromere (1p12) ratio in PNT2, LNCaP, C4-2, LNCaP95, 22Rv1, PC-3, and DU145 cell lines (n = 100 cells each). B Representative MCL1 FISH images (1 biological replicate; n = 100 cells). Scale bar = 10 µm. C MCL1, BCLXL, and BCL2 protein levels assessed by western blot in the same cell lines. GAPDH used as loading control. D Caspase 3/7 activity following AZD5991 (1 µM, left) or S63845 (1 µM, right) for 6 h using Caspase-Glo 2D. Mean ± Standard error (SEM) from three biological replicates, each with three technical replicates. Cell viability assessed by CellTiter-Glo 3D after 24 h (E) and 6 days (F) following treatment with six concentrations (1 µM highest, two-fold dilutions) of AZD5991 (left) or S63845 (right). Mean ± SEM from three biological replicates, each with three technical replicates is shown. Data depicted as fold change relative to vehicle (DMSO). G Workflow for mCRPC patient-derived xenograft (PDX) organoids (PDX-Os). Created in BioRender. Jimenez Vacas (2025) https://BioRender.com/7s2gcp5. H Violin plot of MCL1:Centromere (1p12) ratios in CP253c, CP336c, CP50c, CP267c (n = 100 cells each) and CP142c (n = 50). Median and IQR shown. One-way ANOVA with Tukeyʼs post-hoc test was performed. I Representative MCL1 FISH images of single PDX-O cells (1 biological replicate). Median MCL1:Centromere ratio shown. Scale bar = 5 µm. J Representative MCL1 IHC images from PDX tissues (single passage). Cytoplasmic H-score shown. Scale bar = 100 µm. K Caspase 3/7 activity (6 h) and organoid viability (24 and 96 h) in PDX-Os treated with AZD5991 or S63845 (1 µM), measured with Caspase-Glo 3D and CellTiter-Glo 3D. Mean ± SEM from three biological replicates, each with five technical replicates. Data shown as fold change relative to vehicle (DMSO). One-way ANOVA with Dunnett post-hoc test was performed. Source data are provided as a Source Data file.
MCL1 inhibition synergises with PI3K inhibition to trigger apoptosis in CRPC cells
In order to enhance and/or broaden the effect of MCL1 inhibition, we conducted a screen testing 166 FDA-approved drugs (at 5 µM), which included AR inhibitors (apalutamide, darolutamide and enzalutamide), as single agents and in combination with AZD5991 (1 µM). We evaluated the impact on apoptosis induction (6 h) and cell viability (24 h) in the C4-2 and LNCaP95 CRPC models, as well as in the immortalised prostate epithelium cell line PNT2. Specifically, PI3K pathway inhibitors (copanlisib, idelalisib, duvelisib and alpelisib) showed synergistic effects, decreasing cell viability, and inducing apoptosis in C4-2 and LNCaP95, but not in PNT2, suggesting that this combination selectively kills tumour cells (Fig. 3A, B and Supplementary Fig. 8). These results were validated using copanlisib and buparlisib at four different concentrations, revealing a dose-dependent induction of caspase 3/7 activity (6 h), accompanied by a dose-dependent decrease in cell viability (24 h) when combined with AZD5991 (Supplementary Fig. 9A, B). Taken together, these data demonstrate that MCL1 and PI3K pathway inhibitors synergise to induce cancer cell specific death in PC models.

A FDA-approved drug screen (n = 166 drugs) in LNCaP95, C4-2, and PNT2 cells, in the presence and absence of AZD5991 (1 µM). FDA approved drugs used at 5 µM. Caspase 3/7 activity (6 h; Caspase-Glo 2D) and cell viability (24 h; CellTiter-Glo 2D) were assessed. The x-axis shows the effect of each FDA drug alone (FDA/DMSO), and the y-axis shows the added effect of combining AZD5991 with each drug (FDA + AZD5991/FDA). Experiment performed as a single biological replicate in technical singlets. PI3K inhibitors are highlighted in red. B Heatmaps showing the combined effect of AZD5991 with FDA drugs on caspase activation (6 h) and cell viability (24 h) in LNCaP95 and C4-2, normalised to the effect on the normal-like prostate cell line PNT2. Blue and red indicate increased or decreased caspase 3/7 activity or cell viability, respectively. C Zero Interaction Potency (ZIP) synergy scores comparing combinations of AZD5991 with PI3K inhibitors (copanlisib, buparlisib) versus AKT inhibitors (capivasertib, ipatasertib) in LNCaP95 cells. Data are mean ± SEM from three biological replicates (each with three technical replicates). One-way ANOVA with Tukey’s post-hoc test. D ZIP synergy surface plots for AZD5991 combined with PI3K (copanlisib, buparlisib; left) or AKT (capivasertib, ipatasertib; right) inhibitors in LNCaP95. Caspase 3/7 activity (6 h), cell viability (24 h), and protein expression of cleaved PARP, cleaved caspase 3, and phosphorylated AKT (Ser473) following treatment with DMSO, AZD5991 (1 µM), capivasertib (1 µM), ipatasertib (1 µM), or combinations in LNCaP95 (E) and C4-2 (F). Bars show mean ± SEM of three biological replicates (three technical replicates). Statistical comparisons performed using one-way ANOVA with Tukey’s post-hoc test. Asterisks indicate statistical significance between groups (***p < 0.001). Source data and exact p values are provided are provided as a Source Data file.
MCL1 inhibition synergises with AKT inhibition to trigger apoptosis in CRPC cells
Given that AKT inhibition has shown anticancer activity in mCRPC clinical trials, we next tested whether direct inhibition of AKT could also elicit a robust synergistic effect when combined with MCL1 inhibition23,24. The ZIP synergy scores obtained in response to the co-inhibition of AKT (capivasertib and ipatasertib) surpassed those observed with PI3K inhibitors (copanlisib and buparlisib) when combined with the MCL1 inhibitor AZD5991 in LNCaP95 and C4-2 cells (Fig. 3C, D and Supplementary Fig. 10). These results were further validated independently in an all vs all screen of 42 agents with 30 distinct mechanisms of action (861 different combinations), carried out at the National Cancer Institute (NCI). Specifically, the MCL1 inhibitor S63845 synergised with the PI3K inhibitors copanlisib and paxalisib, as well as with the AKT inhibitors capivasertib and ipatasertib to decrease cell viability at 48 h in the LNCaP95 cells (Supplementary Fig. 11A, B). The induction of apoptosis in response to the combination of AZD5991 with capivasertib and ipatasertib was further validated by protein analyses at 6 h, with cleavage of PARP, caspase 7 (determined by western blot) and caspase 3 (determined by both western blot and IHC) in LNCaP95 and C4-2 cells (Fig. 3E, F and Supplementary Fig. 12). These results were orthogonally validated using siRNAs to target AKT1/2/3 and MCL1, confirming “on-target” pharmacological mechanism of action (Supplementary Fig. 13). Overall, these findings demonstrate that, beyond the synergy observed with MCL1 and PI3K inhibition, the combined impact of MCL1 and AKT inhibition is more pronounced, leading to a heightened induction of apoptosis in PC models.
The synergistic anti-tumour activity of AKT and MCL1 inhibition is mediated through alterations in BAD-BCLXL and BIM-MCL1 interactions
Given the essential role of BH3-only proteins in the control of the intrinsic apoptosis pathway, through the regulation of the activity of anti-apoptotic BCL2 family members25, we conducted a pro-apoptotic BCL2 family siRNA screen in C4-2 and LNCaP95 cells in response to capivasertib, ipatasertib, AZD5991, and their combinations. Specifically, individual silencing of the BH3-only proteins BIM and BAD, and the mitochondrial pore forming effector BAK partially prevented the induction of apoptosis (assessed by caspase 3/7 assay, and cleaved caspase 3 and cleaved PARP protein levels) and the reduction in cell viability (assessed by CellTiter-Glo) induced by the combined treatments in LNCaP95 and C4-2 cells (Supplementary Fig. 14 and Supplementary Fig. 15). When silencing LNCaP95 and C4-2 cells with a combination of BIM, BAD and BAK siRNAs, a near-complete rescue was achieved (Fig. 4A, B), suggesting that BIM, BAD and BAK are co-operative in the molecular mechanism underlying the synergistic effects driven by AKT and MCL1 co-inhibition. Furthermore, western blot analyses showed no significant changes in BAK, BAD, or BIM protein levels in response to capivasertib, ipatasertib, AZD5991, or their combinations (Fig. 4C). However, phosphorylation of BAD at Ser136 decreased with AKT inhibitors (capivasertib and ipatasertib) and combined treatments (Fig. 4C). Co-immunoprecipitation experiments revealed that ipatasertib increased BAD-BCLXL and BIM-MCL1 binding, with the latter being more evident when BIM was pulled down, in both LNCaP95 and C4-2 lines (Fig. 4D). Additionally, AZD5991 (as a single treatment or combined with ipatasertib) disrupted BIM-MCL1 and BAK-MCL1 binding (Fig. 4D), in keeping with its mechanism of action26. These results were orthogonally validated by PLA in C4-2 cells (Supplementary Fig. 16). Taken together, these data suggest that the induction of apoptosis by MCL1 and AKT inhibition is mediated by BAD-driven sequestration of BCLXL and the displacement of BIM from MCL1.

Impact of combined treatment 1 [CMB1; capivasertib (1 µM) and AZD5991 (1 µM)], and combined treatment 2 [CMB2; ipatasertib (1 µM) and AZD5991 (1 µM)] upon triple knockdown of BAK, BAD, and BIM (using specific OnTarget siRNAs at 25 nM for 72 h) or OnTarget Control siRNA (75 nM, 72 h) on caspase 3/7 (6 h; left panel), cell viability (24 h; middle panel) and protein expression (6 h; right panel) in LNCaP95 (A) and C4-2 (B). Caspase 3/7 activity, cell viability, and protein expression were measured by Caspase-Glo 3/7 2D, CellTiter-Glo 2D and western blot, respectively. GAPDH was used as a housekeeping. Data represent three biological replicates, each with three technical replicates. Bars indicate the mean ± SEM. Statistical significance was assessed using one-way ANOVA with Tukey’s post-hoc test. Asterisks indicate statistical significance between groups (**p < 0.01; ***p < 0.001). C Protein levels of BAK, phosphorylated BAD (Ser136), total BAD and BIM were measured by western blot in LNCaP95 and C4-2 cells treated with DMSO, AZD5991 (1 µM), capivasertib (1 µM), ipatasertib (1 µM), or their combinations. Experiment performed as a single biological replicate. D Immunoprecipitation of BCLXL (top), MCL1 (middle), and BIM (bottom) in LNCaP95 and C4-2 cells treated with DMSO, ipatasertib (1 µM), AZD5991 (1 µM), or the combination. BAD, BCLXL, BIM, BAK and MCL1 were blotted. Input and IgG pulldown controls included. Experiment performed as a single biological replicate. Source data and exact p values are provided as a Source Data file.
AKT and MCL1 co-inhibition triggers BAK-dependent apoptosis in PTEN-loss/PI3K-activated CRPC cells
To identify potential predictive biomarkers, we assessed phospho-BAD, total BAD, BIM and BAK levels and examined responses to AKT and MCL1 co-inhibition in seven cell lines with different molecular backgrounds (Fig. 5A, B and Supplementary Fig. 17A, B). Synergistic caspase 3/7 activation and reduced viability were observed in PTEN loss lines (LNCaP, C4-2, LNCaP95) and the PIK3CA-mutated line 22Rv1, which exhibited higher levels of phospho-BAD, total BAD, BIM, and BAK (Fig. 5A, B and Supplementary Fig. 17A, B). In contrast, no synergy was observed in PNT2, DU145 (both PTEN WT) nor in PC-3 despite harbouring PTEN loss (Fig. 5B and Supplementary Fig. 17A, B). Specifically, PC-3 cells have low levels of phospho-BAD, BIM and BAK, and additionally we showed that they rely on BAX rather than BAK (as opposed to the responder LNCaP95) to undergo apoptosis (Fig. 5B and Supplementary Fig. 17C, D). This suggests that undergoing apoptosis in a BAK-dependent manner is required for PC cells to be sensitive to AKT and MCL1 co-inhibition.

A The protein levels of phosphoBAD (Ser136; pBAD); total BAD, BIM, and BAK in PNT2, LNCaP, C4-2, LNCaP95, 22Rv1, PC3, and DU145 cell lines, with GAPDH as a loading control, were detected by western blot. This experiment was performed as a single biological replicate. B Heatmaps showing the effects of capivasertib (1 µM), ipatasertib (1 µM), AZD5991 (1 µM), S63845 (1 µM), and their combinations on caspase 3/7 activity (left panel, 6 h, measured by Caspase-Glo 3/7 assay) and cell viability (right panel, 24 h, measured by CellTiter-Glo 2D assay) in prostate-derived cell lines. Caspase 3/7 activity and cell viability are expressed as fold changes relative to vehicle (DMSO), with caspase data logarithmically transformed (log₁₀). C PI3K/AKT pathway activity was assessed by calculating the Reactome PI3K/AKT signalling pathway score from RNAseq data in CP50c, CP253c, CP267c, CP336c, and CP142c patient-derived xenograft (PDX) models. Data represent six biological replicates. The box shows the interquartile range, the line indicates the median, and the whiskers represent the minimum and maximum values. Statistical analysis was performed using one-way ANOVA with Tukey’s post-hoc test. D Protein levels of phosphoBAD (Ser136; pBAD), total BAD, BIM, and BAK were detected by western blot, with GAPDH as a loading control, in the PDX models. Caspase 3/7 activity at 6 h (E) and cell viability at 24 and 96 h (F) in response to vehicle (DMSO), ipatasertib (1 µM), AZD5991 (1 µM), S63845 (1 µM), and their combinations in CP50c, CP253c, CP267c, CP336c, and CP142c PDX-derived organoids (PDX-Os). Statistical analysis was performed using one-way ANOVA with Tukey’s post-hoc test. The vehicle, AZD5991, and S63845 arms (represented by dotted-pattern bars) were previously shown in Fig. 2K (same experiment). Asterisks indicate statistical significance between groups (*p < 0.05; **p < 0.01; ***p < 0.001). G Representative microscopy images of the PDX-Os on day 4 after treatment. The scale bar indicates a length of 50 µm. All the experiments were performed in three biological replicates and the standard error of the mean is shown. Source data and exact p values are provided as a Source Data file.
We next analysed CRPC PDX-Os with PI3K/AKT pathway aberrations, including CP50c (AKT1 amplification), which shows the highest PI3K-AKT pathway score among the SU2C/PCF samples27, and CP253c, CP267c, and CP336c (PTEN loss by IHC), as well as the PTEN WT model CP142c (Supplementary Fig. 18 and 19). CP142c has significantly lower PI3K/AKT pathway activity than CP50c, CP253c, CP267c and CP336c, which was inferred by the Reactome PI3K/AKT signalling in cancer score and AKT downstream markers including phospho-GSK3B (Ser9) and phospho-PRAS40 (Thr246) (Fig. 5C and Supplementary Fig. 19). Among the five models, CP50c, CP253c, and CP142c were AR-FL and AR-V7 positive, with CP50c and CP253c also ERG positive, indicating TMPRSS2:ERG fusion28 (Supplementary Fig. 19). Synergistic caspase 3/7 activation and reduced viability were observed in response to ipatasertib combined with MCL1 inhibitors (AZD5991 and S63845) in CP50c, CP253c, CP267c, and CP336c, all of which exhibited higher basal phospho-BAD levels than CP142, where no synergy was observed (Fig. 5D–G). In contrast, BAD, BIM, and BAK levels varied across models (Fig. 5D and Supplementary Fig. 19).
To confirm that PTEN loss is a key determinant of sensitivity to AKT and MCL1 co-inhibition, we treated PC Preclinical Mouse Modelling Platform organoids (ProMPt-Os) with different genomic backgrounds. Only PTEN loss models (PTENKO/TP53WT/MYCOE and PTENKO/TP53KO/MYCOE) responded synergistically to the combination of AKT and MCL1 inhibitors, as evidenced by a more pronounced increase in caspase 3/7 activity and reduced viability compared to single-agent treatments, while no synergy was observed in the PTEN WT model (PTENWT/TP53KO/MYCOE) (Fig. 6A–D). Notably, both PTEN loss models exhibited higher levels of phospho-BAD (Ser136), BAK, and BIM than the PTEN WT model (Fig. 6E). Basal phosphorylated BAD levels strongly correlated with the degree of response to AKT and MCL1 co-inhibition across all models, suggesting its potential as a predictive biomarker for sensitivity to this combination therapy (Fig. 6F, G). These data demonstrate that co-inhibition of AKT and MCL1 is synergistic and induces BAK-dependent cancer-specific killing in PTEN-loss/PI3K-activated PC models, supporting the clinical evaluation of this therapeutic strategy in a molecularly defined patient population.

Prostate Cancer Preclinical Mouse Modelling Platform organoids (ProMPt-Os) were treated with vehicle (DMSO), ipatasertib (1 µM), AZD5991 (1, 5, and 10 µM), S63845 (1, 5, and 10 µM), and their combinations to assess caspase 3/7 activity at 6 h (A), cell viability at 24 h (B), and cell viability at 96 h (C). Caspase 3/7 activity and cell viability were assessed by Caspase-Glo 3/7 3D and CellTiter-Glo 3D, respectively. Data represent the mean ± SEM of three independent biological replicates, each performed with three technical replicates. Statistical analysis was performed using one-way ANOVA with Tukey’s post-hoc test. D Representative microscopy images of the ProMPt-Os on day 4 after treatment. This experiment was repeated three times with similar results. The scale bar indicates a length of 200 µm. E Protein levels of MCL1, BCLXL, pBAD (Ser136), BAD, BIM, and BAK were analysed by western blot with GAPDH as the loading control in ProMPt-Os. This experiment was performed as a single biological replicate. Spearman correlation between caspase 3/7 activity (left panels) or cell viability at 24 h (right panels) in response to ipatasertib + AZD5991 (F) or ipatasertib + S63845 (G) and basal pBAD (Ser136) levels in all cell lines, PDX-O, and ProMPt-O models. Simple linear regression is depicted, with the line representing the mean and the shaded area indicating the 95% CI. Caspase 3/7 activity is expressed as log10 fold change relative to vehicle, cell viability as fold change relative to vehicle, and pBAD levels as log10 of pBAD intensity normalised to GAPDH intensity. Spearman correlation coefficients and two-sided p-values are shown. Caspase 3/7 activity was measured using Caspase-Glo assays (2D for cell lines and 3D for PDX-Os/ProMPt-Os), and cell viability was measured using CellTiter-Glo assays (2D for cell lines and 3D for PDX-Os/ProMPt-Os). Source data are provided as a Source Data file.
Synergistic inhibition of tumour growth by AKT and MCL1 co-inhibition in vivo
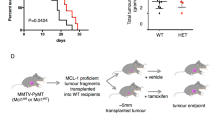
To evaluate AKT and MCL1 co-inhibition in vivo, we assessed CP253c, a PTEN loss, AR-FL/AR-V7 positive and ERG rearranged PDX model derived from a lymph node biopsy of an mCRPC patient previously treated with abiraterone (Supplementary Fig. 3 and Supplementary Fig. 19), in response to ipatasertib [50 mg/kg, p.o., 5 days/week (Monday–Friday)], S63845 [25 mg/kg, i.v., 3 days/week (Monday, Wednesday, and Friday)], combined treatment, or vehicle. The combined treatment led to a significant suppression of tumour growth, while single-agent treatments showed no statistically significant effect, indicating a synergistic interaction between the two drugs in vivo (Fig. 7A–C). No mice exhibited weight loss exceeding 20% of their baseline or showed any signs of discomfort or distress Supplementary Fig. 20). However, this should be taken with caution as S63845 (and other MCL1 inhibitors) show less affinity for mouse MCL1 than human MCL129. Histopathological examination of terminal tumours revealed a significant increase in cleaved caspase 3 in response to the combined treatment, and a decrease in KI67 with ipatasertib treatment, suggesting that the combined treatment inhibited the growth of CP253c by triggering apoptosis (Fig. 7D). Furthermore, downstream markers of the PI3K/AKT pathway (i.e., pGSK3B and pPRAS40) were downregulated in response to both ipatasertib single treatment and the combined treatment with S63845 (Fig. 7D). These data orthogonally validate our in vitro studies, confirming that AKT and MCL1 co-inhibition demonstrates marked anti-tumour activity in a PTEN loss patient-derived model in vivo, with associated pharmacodynamic biomarker modulation.

A Individual tumour volumes of CP253c treated with ipatasertib (50 mg/kg; n = 5), S63845 (25 mg/kg; n = 4), combined treatment (n = 5) and vehicle (n = 5). B Predicted tumour growth using a linear mixed-effect model of CP253c treated with ipatasertib (50 mg/kg; n = 5), S63845 (25 mg/kg; n = 4), combined treatment (n = 5) and vehicle (n = 5). C Forest plots showing the results from the longitudinal mixed effect model for log-transformed tumour volume. Estimates and p-values refer to the interaction term of treatment arm and time indicating the difference in tumour volume growth rate between each treatment arm and the vehicle arm. Points represent the random effect coefficients for each drug group and error bars the 95% confidence intervals. The black dotted line indicates the vehicle (reference level). D Comparison of protein expression for cleaved caspase 3 (% positive cells), Ki67 (% positive cells) and pGSK3B, pPRAS40 and MCL1 H-score for the different treatment arms. The box shows the interquartile range, the line indicates the mean, and the whiskers represent the minimum and maximum values. One-way ANOVA with post-hoc Fisher’s LSD test was performed. Representative IHC micrographs for end-of-treatment CP253c tumours are also shown (bottom panel). The scale bar indicates a length of 100 µm. Source data are provided as a Source Data file.
CRPC cells with acquired resistance to capivasertib remain sensitive to AKT and MCL1 co-inhibition, unlike those with acquired resistance to MCL1 inhibitors
To mimic acquired resistance to AKT inhibition seen in mCRPC patients during clinical trials23,24, we generated capivasertib-resistant ‘Capi-R’ C4-2 and LNCaP95 cell lines. We first validated the models, demonstrating a >10-fold higher capivasertib IC50 in Capi-R compared with parental cells (Fig. 8A, B). Both C4-2 and LNCaP95 Capi-R cells exhibited a slower growth rate compared to their parental counterparts (Fig. 8A, B). The IC50 of capivasertib significantly decreased in the presence of AZD5991 in both LNCaP95 and C4-2 Capi-R cells (Fig. 8C, D), suggesting sensitivity to this combined treatment. Moreover, a synergistic increase in caspase 3/7 activity was observed in response to the combination of capivasertib and AZD5991 in LNCaP95 and C4-2 Capi-R cells (Fig. 8E, F). Furthermore, western blot analysis of cleaved PARP and caspase 3 orthogonally validated apoptosis induction (Fig. 8G, H). Apoptosis induction was more pronounced in LNCaP95 Capi-R compared to C4-2 Capi-R cells (Fig. 8E, F, G, H). BAK, BAD, and BIM were downregulated in Capi-R cells, with a more pronounced decrease in C4-2 compared to LNCaP95 Capi-R cells (Fig. 8I, J and Supplementary Fig. 21A, B), which may contribute to their reduced sensitivity to the combined treatment. Noteworthy, phosphorylation levels of BAD (Ser136) decreased with capivasertib in both C4-2 and LNCaP95 Capi-R cells (Fig. 8K, L and Supplementary Fig. 21C, D).

A, B Comparison of capivasertib dose-response curves (left panel), capivasertib IC50 (central panel) and basal cell growth (fold change to day 0; right panel) between Capi-R and parental LNCaP95 (A) and C4-2 (B) cells. A two-sided unpaired t-test assuming normal distribution was performed. Comparison of capivasertib dose-response curves (left panel) and capivasertib IC50 (right panel) in presence and absence of AZD5991 (1 µM, 24 h) between Capi-R and parental cells in LNCaP95 (C) and C4-2 (D). Floating bars (min to max) with line at mean are depicted. One-way ANOVA with Tukey’s post-hoc test was performed. Caspase 3/7 activity at 6 h (left panel) and cell viability at 24 h (right panel) in parental and Capi-R LNCaP95 (E) and C4-2 (F) cells treated with vehicle (DMSO), capivasertib (1 µM), AZD5991 (1 µM) and combined treatment. Two-way ANOVA with Tukey’s post-hoc test was performed. Cell viability and caspase 3/7 activity were determined using Caspase-Glo 3/7 2D assay and CellTiter-Glo 2D, respectively. Protein expression of cleaved PARP and cleaved caspase 3 (at 6 h; determined by western blot) in LNCaP95 (G) and C4-2 (H) treated with vehicle (DMSO), capivasertib (1 µM), AZD5991 (1 µM) and combined treatment (combo). Comparison of protein levels of BIM, BAD, BAK, total and p-AKTSer473 between parental and Capi-R cells in LNCaP95 (I) and C4-2 (J). Protein levels of total and p-BADSer136, total BAD, and p-AKTSer473 in response to vehicle (DMSO) and varying capivasertib concentrations (0.1 µM 0.5 µM, 1 µM, 5 µM) between parental and Capi-R cells in LNCaP95 (K) and C4-2 (L). Vinculin was used as a housekeeping protein. All the experiments were performed in three biological triplicates (apart from the western Blot shown in (H and J); n = 1) and technical singlets. The standard error of the mean is shown. Asterisks (*p < 0.05; **p < 0.01; ***p < 0.001) indicate statistically significant differences between groups. Source data and exact p values are provided as a Source Data file.
We next generated AZD5991- and S63845-resistant LNCaP95 cells and assessed their response (Supplementary Fig. 22A–C). While AKT inhibition alone continued to reduce cell viability, the combination of AKT and MCL1 inhibitors no longer increased caspase 3/7 activity or synergistically decreased viability (Supplementary Fig. 22D–F). The MCL1 inhibitor-resistant cell lines exhibited lower levels of phospho-BAD, BIM, and BAK compared to parental LNCaP95 cells (Supplementary Fig. 22G).
These results suggest that co-inhibition of AKT and MCL1 induces apoptosis, retaining efficacy in PC cells with acquired resistance to AKT inhibitors (as long as AKT inhibition continues to dephosphorylate BAD), while this synergy is lost in cells with acquired resistance to MCL1 inhibitors.
Targeting MCL1 through CDK9 inhibition recapitulates the phenotype of MCL1 inhibition in combination with AKT blockade in CRPC cells
CDK9 inhibition rapidly downregulates MCL1 protein levels in several tumour-types30,31,32. We thus hypothesised that CDK9 inhibitors could also trigger acute apoptosis when combined with AKT inhibitors, in CRPC cells with PI3K/AKT hyperactivation. First, fadraciclib significantly decreased MCL1 protein levels after 6-h of incubation in LNCaP95 and C4-2 cells (Fig. 9A, B). The co-inhibition of AKT (using capivasertib and ipatasertib) and CDK9 (using fadraciclib and AZD4573), synergistically induced apoptosis and reduced cell viability (24 h) (Fig. 9A, B and Supplementary Fig. 23–24). Consistent with the mechanism underlying the synergistic effects of AKT and MCL1 co-inhibition, the caspase 3/7 activity induction and cell viability reduction observed in response to the combination of fadraciclib with AKT inhibitors, were prevented when cells were pre-treated with a combination of BIM, BAD, and BAK siRNAs (Fig. 9C). Furthermore, the response profile to AKT inhibitors and fadraciclib combinations mirrored that of AKT and MCL1 co-inhibition, with the same cell lines, PDX-Os and ProMPt-Os exhibiting sensitivity to both strategies (Fig. 9D–F and Supplementary Fig. 25). In vivo analysis of CP253c PDX showed that the combination of ipatasertib with fadraciclib significantly reduced tumour growth, whereas single treatments showed no significant impact (Fig. 10A). No mice exhibited a weight loss exceeding 20% of their baseline or showed any signs of discomfort or distress (Supplementary Fig. 26). Subsequent examination of terminal tumours by IHC revealed that cleaved caspase 3 induction was observed only in response to the combined treatment, while both single treatments decreased KI67, suggesting that single treatments impacted cell growth, whereas combined treatment triggered apoptosis in vivo (Fig. 10D). Additionally, downstream markers of AKT activity (pGSK3B and pPRAS40) were decreased in response to both ipatasertib alone and when combined with fadraciclib, and MCL1 protein levels were decreased by fadraciclib (Fig. 10D). Finally, while AKT inhibitors combined with fadraciclib continued to synergistically induce apoptosis and cell death in C4-2 and LNCaP95 Capi-R cells, this effect was no longer observed in LNCaP95 cells with acquired resistance to MCL1 inhibitors (Supplementary Fig. 27–28).

Caspase 3/7 activity (6 h), cell viability (24 h), and protein expression (cleaved PARP, cleaved caspase 3, MCL1, total and phospho-AKT Ser473; 6 h) were assessed in LNCaP95 (A) and C4-2 (B) cells treated with vehicle (DMSO), capivasertib (1 µM), ipatasertib (1 µM), fadraciclib (1 µM), and their combinations. GAPDH was used as a loading control. C Caspase 3/7 activity (6 h), cell viability (24 h), and protein expression (6 h) were evaluated in LNCaP95 (left) and C4-2 (right) cells following treatment with either capivasertib + AZD5991 or ipatasertib + fadraciclib, in the context of triple knockdown of BAK, BAD, and BIM (OnTarget siRNAs, 25 nM each for 72 h) or control siRNA (75 nM, 72 h). D Heatmaps show the effects of capivasertib, ipatasertib, fadraciclib, and their combinations on caspase 3/7 activity (6 h) and cell viability (24 h) in PNT2, LNCaP, C4-2, LNCaP95, 22Rv1, PC3, and DU145 cells, shown as fold change relative to vehicle. E Caspase 3/7 activity (6 h) and organoid viability (24 h and 96 h) were assessed in CP50c, CP253c, CP267c, CP336c, and CP142c PDX-derived organoids (PDX-Os) treated with vehicle, ipatasertib (1 µM), fadraciclib (1 µM), or the combination. Vehicle and ipatasertib data (dotted bars/symbols) are from the same experiment shown in Fig. 4D. F Caspase 3/7 activity (6 h) and cell viability (24 h and 96 h) were measured in Prostate Cancer Preclinical Mouse Modelling Platform organoids (ProMPt-Os) treated with vehicle, ipatasertib (1 µM), fadraciclib (1 µM), or the combination. All data represent three biological replicates with three (cell lines, ProMPt-Os) or five (PDX-Os) technical replicates. Bars show mean ± SEM. Statistical analysis was performed using one-way ANOVA with Tukey’s post-hoc test. Caspase 3/7 activity and viability were assessed using Caspase-Glo 3/7 and CellTiter-Glo in 2D (cell lines) or 3D (PDX-Os, ProMPt-Os) formats. Asterisks indicate statistical significance between groups (*p < 0.05; **p < 0.01; ***p < 0.001). Source data and exact p values are provided as a Source Data file.

A Individual tumour volumes of CP253c treated with ipatasertib (50 mg/kg; n = 5), fadraciclib (40 mg/kg; n = 6), combined treatment (n = 6) and vehicle (n = 5). B Predicted tumour growth using a linear mixed-effect model of CP253c treated with ipatasertib (50 mg/kg; n = 5), fadraciclib (40 mg/kg; n = 5), combined treatment (n = 5) and vehicle (n = 5). C Forest plots showing the results from the longitudinal mixed effect model for log-transformed tumour volume. Estimates and p-values refer to the interaction term of treatment arm and time indicating the difference in tumour volume growth rate between each treatment arm and the vehicle arm. Points represent the random effect coefficients for each drug group and error bars the 95% confidence intervals. The black dotted line indicates the vehicle (reference level). D Comparison of protein expression for cleaved caspase 3 (% positive cells), Ki67 (% positive cells) and pGSK3B, pPRAS40 and MCL1 H-score for the different treatment arms. Necrotic samples were not analysed. The box shows the interquartile range, the line indicates the mean, and the whiskers represent the minimum and maximum values. One-way ANOVA with post-hoc Fisher’s LSD test was performed. Representative IHC micrograph for end-of-treatment CP253c tumours are also shown (bottom panel). The scale bar indicates a length of 100 µm. Source data are provided as a Source Data file.
These data demonstrate that CDK9-mediated MCL1 downregulation, combined with AKT inhibition, replicated the in vitro and in vivo findings of direct MCL1 inhibition, providing further opportunities to clinically translate this therapeutic strategy.
Discussion
MCL1 supports cell survival by sequestering pro-apoptotic BCL2 proteins, preventing mitochondrial outer membrane permeabilization and the initiation of the intrinsic apoptosis pathway. Herein, we characterise MCL1 status at genomic, RNA and protein level in multiple independent mCRPC cohorts, including in matched same-patient CSPC and CRPC biopsies. We show that MCL1 amplification is a truncal event occurring early in lethal PC evolution, whilst MCL1 copy number gain occurs at diagnosis but also emerges with treatment resistance, probably representing an adaptive response. Interestingly, these genomic changes associate with worse clinical outcomes and inferior responses to neoadjuvant ADT and enzalutamide. Indeed, previous studies have shown MCL1 upregulation confers resistance to ADT in PC cells9, therefore we suggest that the selection of subclones harbouring MCL1 copy number gains or amplifications is a potential mechanism contributing to tumour progression and therapy resistance. However, no association was observed between MCL1 gain/amplification and taxane response, which could potentially be explained by the ability of taxanes to induce MCL1-independent mitotic catastrophe in these tumours33,34. These data support the urgent development of molecularly stratified therapeutic strategies in MCL1 overexpressing mCRPC to improve the outcome for patients with this lethal disease.
We show that CRPC models with MCL1 copy number gain respond to MCL1-targeting BH3 mimetics, likely due to their increase reliance on MCL1 for survival, with inhibition inducing apoptosis, as previously observed in other tumour-types7,20,21. A limitation of our study is the lack of in vivo validation testing the anti-tumour effects of MCL1 inhibition in models with MCL1 copy number gain. Despite this, we believe that our data support single-agent MCL1 inhibition as an effective anti-cancer strategy for CRPC with MCL1 copy number gains in a molecularly stratified approach. Previous studies have reported limited anti-tumour activity for single agent MCL1 inhibitors in PC models, but this is likely due to the selection of models with unaltered MCL1 copy number status as well as redundancy with other BCL2 family members including BCLXL15,16. Therefore, the major challenge for drug development in this space, is to identify therapeutic strategies that can inhibit both MCL1 and BCLXL to drive cancer cell specific cell death, whilst maintaining tolerability.
Taking this important point into consideration, and to broaden the effect of MCL1 inhibition in CRPC without MCL1 copy number gains, we explored the efficacy of MCL1 inhibition in combination with FDA-approved oncological drugs with a view to discover synergistic combinations that could be rapidly translated to the clinic. This led to the discovery that MCL1 inhibition synergises with AKT blockade to induce BAK-dependent apoptosis by modulating BAD-BCLXL and BIM-MCL1 interactions. This therapeutic combination has been investigated in other tumour types, including myeloma35, leukaemia36, and breast cancer37. However, our study highlights its therapeutic potential in CRPC and offers significant advancements in understanding the underlying molecular mechanisms. Specifically, our study demonstrates that AKT inhibition de-phosphorylates, and subsequently activates the BH3-only protein BAD, as previously reported38. This activation leads to increased binding of BAD to BCLXL, inhibiting its anti-apoptotic function, and therefore triggering apoptosis when combined with MCL1 inhibition39. We showed that AKT inhibition also increases BIM-MCL1 binding, likely in response to the displacement of BIM from BCLXL due to the competitive binding of BAD. This might contribute to increasing dependency on MCL1 in the context of AKT inhibition (and subsequent BCLXL blockade), following the direct activation model40. Altogether, these mechanistic data are consistent with previous findings showing that PC models are co-dependent on MCL1 and BCLXL, with dual blockade needed to induce apoptosis16.
One potential challenge linked to MCL1 and BCLXL dual inhibition is the anticipated clinical toxicity41,42. Critically, this is less likely to be the case for co-inhibition of MCL1 and AKT, as we observed cancer cell killing only in models that harbour PI3K/AKT pathway activating genomic aberrations (such as PTEN loss and AKT1 amplification), which is likely attributed to their significant dependency on phosphorylated BAD for survival38,39,43. Indeed, our data suggest that high basal levels of phosphorylated BAD (Ser136) may be a more sensitive predictive biomarker for this combination therapy than genomic PI3K/AKT pathway aberrations (e.g., PTEN loss). Although single agent MCL1 demonstrated promising activity in PC with MCL1 copy number gains, this remains a small subset when compared with activation of the PI3K/AKT pathway, which accounts for around 50% of mCRPC, and is associated with worse clinical outcome44,45. Taken together, these data demonstrate that co-inhibition of MCL1 and AKT deliver tumour specific killing in PTEN-loss/PI3K-activated lethal PC with high levels of phospho-BAD, providing a molecularly driven therapeutic strategy for a common subtype of lethal PC with poor prognosis45.
AKT inhibitors have shown promise in clinical trials for patients with mCRPC, but have not yet met the threshold for regulatory approval23,24. As such, the development of drug combination strategies to enhance anti-tumour activity or reverse resistance to AKT inhibition is an unmet clinical need. In this space, it should be noted that the co-inhibition of MCL1 and AKT not only drove apoptotic cell death in PC cells sensitive to AKT inhibition but also exerted anti-cancer activity in PC models that had acquired resistance to single agent AKT blockade. This is consistent with the reported sensitivity of AKT inhibition resistant breast cancer cell lines (developed by CRISPR-Cas9 KO techniques, as opposed to acquired resistance) to MCL1 blockade37. Furthermore, we show that dual blockade of AKT and MCL1 is effective in mCRPC models characterised by biomarkers associated with resistance to AR signalling inhibitors and PC progression, including AR negativity, AR-V7 expression and TMPRSS2:ERG fusion46,47,48,49,50,51,52. In conclusion, AKT and MCL1 co-inhibition demonstrates efficacy in overcoming resistance to AKT blockade and provides a targeted therapeutic strategy for common mCRPC lethal subtypes with potential for significant clinical impact. Importantly, we observed that cells with acquired resistance to MCL1 inhibitors no longer responded to dual AKT/MCL1 inhibition. This suggests that combining AKT and MCL1 blockade upfront, rather than after progression on MCL1 inhibitors, may be a more effective treatment strategy.
CDK9 inhibitors rapidly downregulate MCL1 protein levels and have shown anti-tumour activity with manageable toxicity in early-phase clinical trials for haematological malignancies30,31,32,53,54,55. Here, we demonstrate that co-inhibition of CDK9 and AKT mirrors the effects of MCL1 and AKT co-inhibition, inducing cell death in PTEN-loss/PI3K-activated PC cells with high phospho-BAD levels. While CDK9 is a broad transcriptional regulator affecting multiple genes beyond MCL1, some studies suggest that MCL1 depletion is a key driver of the apoptosis-related anti-tumour effects of CDK9 inhibitors30,56,57. Our data support that CDK9-dependent acute MCL1 downregulation contributes, at least in part, to the synergy observed with CDK9 and AKT co-inhibition. Specifically, the synergistic effects observed with both CDK9/AKT and MCL1/AKT co-inhibition strategies require the same BH3-only proteins (BIM, BAD, and BAK), and cells with acquired resistance to MCL1 inhibitors exhibit resistance to both combinations. However, we acknowledge that additional factors acutely regulated by CDK9 inhibition may also contribute to this synergy. Despite this, our findings support CDK9 inhibition combined with AKT blockade as an alternative, promising therapeutic strategy for PTEN-loss/PI3K-activated PC. It is important to acknowledge the challenges associated with direct MCL1 inhibition, particularly due to on-target cardiotoxicity58,59. In this context, future strategies, including antibody-drug conjugates and proteolysis-targeting chimeras (PROTACs), similar to those developed for other BCL2 family members, could enable PC-specific MCL1 targeting while minimising the risk of cardiotoxicity60,61.
Critically, our data support the urgent evaluation of therapeutic strategies targeting MCL1, directly or indirectly, as a single agent for PC harbouring MCL1 copy number gains, and in combination with AKT blockade in PTEN-loss/PI3K-activated lethal PC, within ‘proof of concept, proof of mechanism’ early phase clinical trials.
Methods
Patients and sample collection
Royal Marsden Hospital cohorts
All patients had mCRPC, were treated at the RMH, provided written informed consent, and were enroled in protocols approved by the RMH ethics review committee. For the matched same-patient cohort, treatment-naïve diagnostic samples were retrieved from referring hospitals. CRPC tissue was collected from mCRPC patients. Clinical data and demographics were collected retrospectively from the RMH electronic patient record system. Clinical characteristics and sample disposition are as previously described [RMH matched same-patient cohort62, RMH CRPC cohort63]. For the RMH matched same-patient cohort62, 44 patients were included with copy number analysis data by low pass whole genome sequencing of tumour biopsy DNA. For the RMH CRPC cohort, 86/100 patients had RNA-sequencing data available and were included for this study.
FIRSTANA and PROSELICA cohort
All patients had mCRPC and participated in the FIRSTANA or PROSELICA prospective, randomised, open-label, international phase III trials. In FIRSTANA, patients were randomised to receive cabazitaxel (20 mg or 25 mg/m2) or docetaxel (75 mg/m2), all with prednisone (10 mg/day) as first-line chemotherapy for mCRPC64. In PROSELICA, patients were randomised to receive either cabazitaxel 20 mg/m2 or cabazitaxel 25 mg/m2, each with prednisone 10 mg/day as second-line chemotherapy after docetaxel. All patients included in our study consented to allow for the collection of plasma. Plasma cell-free DNA low-pass whole-genome sequencing data were obtained from baseline samples. Samples with a tumour fraction >5% were assessed for genomic copy number burden (FIRSTANA n = 79, PROSELICA n = 73). OS was defined as the time from randomisation to death of any cause. Clinical characteristics are presented in Supplementary Table 1.
Stand Up To Cancer/Prostate Cancer Foundation cohort
Copy number variation (n = 336) and transcriptome (n = 159) data from mCRPC biopsies, generated by the International SU2C/PCF, were downloaded and re-analysed45,65.
The Cancer Genome Atlas cohort
Clinical data and associated genomic data (n = 494) were downloaded from the TCGA-Clinical Data Resource (CDR) and Progression Free Interval (PFI) was used to evaluate outcome66. PFI was defined as the time from the date of diagnosis until the date of the first occurrence of a new tumour event, which includes progression of the disease, locoregional recurrence, distant metastasis, new primary tumour, or death with tumour.
National Cancer Institute neoadjuvant cohort
For the NCI neoadjuvant cohort, tissue collection and analysis from patients with intermediate- and high-risk locally advanced PC treated with neoadjuvant ADT and enzalutamide prior to radical prostatectomy. This trial was approved by the Institutional Review Board of the Center for Cancer Research, NCI, Bethesda, MD (ClinicalTrials.gov identifier: NCT02430480).
University of Washington/Fred Hutchinson (UW/FH) cohort
For UW/FH cohort, tissue was obtained from patients who had died of mCRPC and had signed written informed consent for a rapid autopsy performed within 6 h of death, under the aegis of the PC Donor Programme at the University of Washington. All procedures involving human subjects in the rapid autopsy programme were approved by the Institutional Review Board of the University of Washington and of the Fred Hutchinson Cancer Center.
Bioinformatic analyses
Copy number analysis
Copy number variations data were processed and analysed as previously described in biopsies from RMH62, SU2C/PCF45,65. In short, exome-sequencing reads were aligned to GRCh37 using BWA MEM. GATK realignment and recalibrate base scores were applied. Germline variance and somatic variance were called using GATK and Mutect2 by comparing tumour-normal paired comparison. ASCAT were used for copy number analysis. TCGA CNA segment data was download from cBioportal (n = 489). Copy number analysis for FIRSTANA/PROSELICA64 cohorts were performed using low pass whole genome sequencing from plasma DNA. Sequencing reads were aligned to GRCh37 using BWA MEM. Aligned reads were quantified by HMMcopy readCounter, and copy number and the tumour fraction were calculated using ichorCNA. For the National Cancer Institute neoadjuvant cohort, exome sequencing of tumour foci and processing of data to derive gene-level somatic copy number estimates was described previously67.
Transcriptome analysis
TCGA transcriptomic data were downloaded from cBioPortal66,68. RMH and SU2C/PCF RNA sequencing dataset analysis (TopHat2 pipeline): CRPC transcriptomes generated by the SU2C/PCF Dream Team, were downloaded and reanalysed45. Paired-end transcriptome sequencing reads for each of the SU2C/PCF (n = 106) and RMH (n = 86) cohorts, comprising samples with matched RNA and copy number data, were aligned to the human reference genome (GRCh37/hg19) using Tophat2 (v2.0.7)63. Gene expression, Fragments Per Kilobase of transcript per Million mapped reads (FPKM), was calculated using Cufflinks. The top expressed genes (n = 15000) were analysed for each cohort respectively. The Spearman’s rank correlation coefficient (Spearman’s rho) between each gene’s expression (FPKM) and MCL1 expression (FPKM) was calculated, and subsequently used for pathway analysis. Pathway analysis was performed using the Gene Set Enrichment Analysis (GSEA) Pre-Ranked algorithm from GSEA software (v4.1.0). GSEA Pre-Ranked results were obtained using the H collection of Hallmark gene sets (MSigDB v7.0), with default parameters. UW/FH mCRPC RNA sequencing: RNA isolation and sequencing of mCRPC tumours were performed as described previously. Sequencing reads were mapped to the hg38 human genome using STAR.v2.7.3a. All subsequent analyses were performed in R. Gene level abundance was quantitated using GenomicAlignments and transformed to log2 FPKM using edgeR. Boxplots by patient were filtered to include only patients with at least two tumours profiled and matched copy number data (n = 115 tumours from 50 patients) and are ordered by per-patient median log2 FPKM gene expression. PI3K/AKT pathway score was calculated by accumulated z-score using genes of the REACTOME_PI3K_AKT_SIGNALING_IN_CANCER pathway obtained from the molecular signature database (MSigDB)69.
Immunohistochemistry
IHC was performed on freshly sectioned tissue blocks where adequate material was present (≥100 tumour cells, reviewed by histopathologist B.G). BIM, BAD, and BAK antibodies were validated for specificity and optimised for IHC (Supplementary Figs. 1 and 2), while MCL1 antibody validation was reported previously70. Details on the IHC assays are available in Supplementary Table 2. Protein quantification was determined for by a histopathologist (B.G) blinded to clinical data using H score method; [(% of absent staining x 0) + (% of weak staining x 1) + (% of moderate staining x 2) + (% of strong staining x 3)], resulting in a score between 0 and 30071. In addition, a pathologist-supervised machine learning algorithm (HALO AI, Indica Labs) was trained to calculate cytoplasmic MCL1 protein levels in patient samples. First, PC cells were differentiated from benign stroma. Colour deconvolution for DAB and haematoxylin staining was performed, and cell recognition and nuclear segmentation optimised. The analysis algorithm was then adjusted to provide continuous data on the intensity of MCL1 cytoplasmic signal intensity normalised per cell (accounting for cell size), averaged across the number of tumour cells identified per sample, providing a value between 0 (no staining) and 1 (black) called optical density (OD).
Fluorescence in situ hybridisation
Fluorescence In Situ Hybridisation (FISH) was performed in cell lines and patient-derived xenograft-organoids (PDX-Os) as previously described72. Briefly, fresh cell lines and organoids were resuspended in 10 mL of phosphate-buffered saline and treated with a fixative solution (FS) containing a 3:1 mixture of methanol and acetic acid (600 μL). The suspension was vigorously vortexed and subsequently centrifuged at 500 × g for 5-min. The supernatant was then carefully decanted, and the cell pellet was resuspended in 15 mL of FS. Following another round of centrifugation and removal of the supernatant, this process was repeated to ensure thorough fixation. A final 2 mL of FS was added to resuspend the cells. To facilitate optimal cell spreading for subsequent analysis, cells were released from a height of 20 cm onto positively charged slides. A dual-colour FISH assay was optimised using a cocktail of two FISH probes commercially available (ZytoLight SPEC MCL1/1p12 Dual Colour Probe, Z-2173-200, Bio SB). The MCL1/1p12 ratio ≥2.0 as previously reported21.
Drug compounds
AZD4573 and CYC065 (fadraciclib) were kind gifts from AstraZeneca and Cyclacel respectively. AZD5363 (capivasertib) and AZD5991 were (in part) a kind gift from AstraZeneca, with further stocks purchased from MedChemExpress. GDC-0068 (ipatasertib), ABT-263 (navitoclax), S63845, copanlisib and buparlisib were purchased from MedChemExpress. AZD5991 and S63845 specificity and selectivity are reported elsewhere26,29. All drugs were prepared as 10 mM stock solution in DMSO and subsequently diluted in the appropriate media to achieve a final DMSO assay concentration of ≤0.1%.
Cell lines
PC cell lines (LNCaP, C4-2, 22Rv1, PC-3, and DU145) were purchased from American Type Culture Collection (ATCC) and normal-like prostate cell line PNT2 from Sigma-Aldrich. LNCaP95 cells were generously provided by Drs Alan K. Meeker and Jun Luo (Johns Hopkins University, Baltimore, MD). The cell culture conditions are depicted in Supplementary Table 3. Cell lines underwent regular Short Tandem Repeat (STR) profiling and mycoplasma testing.
Patient-derived xenograft (PDX) organoids (PDX-Os)
PDX-Os were generated from PDXs derived from human CRPC biopsies using methods described previously27,73,74. The patient treatment histories are detailed in Supplementary Fig. 3. Organoids were formed in Matrigel® matrix (356231, Corning) domes in 24-well plates and then re-seeded into 96-well plates for drug experiments.
Prostate Cancer Preclinical Mouse Modeling Platform (ProMPt) organoids (ProMPt-Os)
The mouse organoids utilised in this study are part of the ProMPt. The mouse strains include PtenLoxP75; p53LoxP (JAX stock: #008462)76; MYCStopFL (JAX stock #020458)77; and the UBC-Cre-ERT2 (JAX stock #007001)78. Organoids were derived from mouse prostate tissue as previously described79. Briefly, mouse prostate organoids were established from prostates isolated from 3-month-old mice. Luminal and basal cells from mouse prostate tissue were separated by double antibody staining for CD24 and CD49f markers. Sorted luminal cells were plated in 30 µl Matrigel drops on 6-well plates and covered with complete mouse prostate organoid medium (1x B27, 1.25 mM N-acetyl-l-cysteine, 50 ng/ml EGF, 200 nM A83-01, 100 ng/ml Noggin, 500 ng/ml R-spondin, 1 nM dihydrotestosterone, 10 µM Y-27632 dihydrochloride in adDMEM/F12 containing penicillin/streptomycin, 10 mM HEPES and 2 mM GlutaMAX). To induce Cre-ERT2-mediated excision of all LoxP exons and STOP cassettes, organoids were treated for two passages with 4-hydroxytamoxifen. Successful CRE-mediated excision was confirmed using quantitative PCR and western blot analysis.
Viability and apoptosis assays
PC cells, PDX-Os and ProMPt-Os were treated two days after seeding, or three days after small interfering RNA (siRNA) transfection, with BH3 mimetics, CDK9 inhibitors, or vehicle. PDX-Os and ProMPt-Os were cultured in full media without Y-27632 (ROCK inhibitor) for 24 h prior to treatment and maintained in the same media throughout the experiment. Cell line viability was evaluated 24 h post-treatment, and PDX-Os/ProMPt-Os viability at both 24 and 96 h after treatment using CellTiter-Glo 2D (G9241; Promega) and CellTiter-Glo3D (G9681; Promega), respectively. The in vitro caspase activity was evaluated 6 h post-treatment using the 2D Caspase-Glo 3/7 Luminescent assay (G8093; Promega) for cell lines and the 3D Caspase-Glo 3/7 Luminescent assay (G8983; Promega) for PDX-Os/ProMPt-Os, in accordance with the manufacturer’s instructions.
Food and drug administration-approved drug screen
The National Cancer Institute/National Institutes of Health (NCI/NIH) Food and Drug Administration (FDA)-approved anti-cancer Oncology Set was obtained from the NCI/NIH DTP Open Chemical Repository. The screen included 166 different compounds, delivered in DMSO at 10 mmol/L (Supplementary Table 4). C4-2, LNCaP95 and PNT2 cells were seeded in 384 well plates at 2000 cells per well. 48 h after seeding, cells were treated with the FDA approved compounds at 5 µM in absence and presence of AZD5991 at 1 µM with a final DMSO concentration in the wells ≤0.01%. Cell viability and apoptosis induction were assessed 24 h and 6 h post-treatment, using CellTiter-Glo 2D and Caspase-Glo 3/7 2D, respectively. The screen was performed as a single biological experiment (n = 1) without technical replicates.
High-throughput combination matrix drug screening
For pairwise drug-combination assessments in matrix format, compounds were acoustically dispensed into a 1536-well white solid bottom tissue culture-treated plate (EWB041000A, Aurora Microplates, Whitefish, MT, USA) with an Echo 550 acoustic liquid handler (Labcyte, San Jose, CA, USA) using previously described methods80,81. A 9-point custom concentration range with 1:2 dilution between points was used for each drug-pair tested. Bortezomib (final concentration 20.3 µM) was used as a positive control for cell cytotoxicity. Cells were seeded into compound-containing plates at a density of 500 cells/well, in a final volume 5 µL of growth media by using a Multidrop Combi dispenser (Thermo Fisher Scientific). Plates were covered by a stainless-steel gasketed lid to prevent evaporation and incubated for 48 h in a humidified CO2 incubator. At the 48 h time point, 3 µL of Cell Titer Glo (Promega) was added to each well using a BioRAPTR® (Beckton Coulter, Brea, CA, USA) and plates were incubated at room temperature for 15 min with the stainless-steel lid in place. Luminescence readings were taken using a Viewlux reader (PerkinElmer) with a 2 s exposure time per plate. Viability of compound treated wells was normalised to DMSO and empty well controls present on each plate, and combination-response plotting was automatically performed for each individual drug+drug combination.
siRNA transfection
Cell lines were transfected with ON-TARGETplus siRNA (SMARTpool, mixture of 4 siRNAs) (Dharmacon, Horizon) using 0.2% Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific) as per manufacturer’s guidelines. For the pro-apoptotic BCL2 family siRNA screen, 3000–5000 cells were seeded in 96-well plates and treated with AZD5991 (1 µM) after 72 h of siRNA exposure. Cell viability (assessed with CellTiter-Glo 2D) and caspase activity (assessed with 2D Caspase-Glo 3/7) were determined 6- and 24 h post-treatment, respectively. Concentration of the siRNAs ranged from 25–75 nM and is specified on figure legends. Detailed information on the siRNAs used in this study is provided in Supplementary Table 5.
Western blot
Cells were plated in 6-well plates at a seeding density of 400,000 cells per well and allowed to attach and grow for 48 h. After a 6-h treatment period, cells were lysed and harvested using Pierce RIPA buffer containing a Pierce Protease and Phosphatase inhibitor tablet (Thermo Fisher Scientific). Following sonication (10 s), the protein lysates were centrifuged for 15-min at 4 °C. Protein concentrations were then determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). A total of 25 μg of protein were loaded on 4–12% NuPAGE Bis-Tris gels (Invitrogen), separated via electrophoresis and subsequently transferred (30 V, overnight) to Immobilon-P PVDF membranes (pore size 0.45 μm). The membranes were incubated with primary and secondary antibodies (Supplementary Table 6) in 5% milk in TBS supplemented with 0.1% Tween20 (Sigma-Aldrich). Chemiluminescence was detected using the ChemiDoc Touch Imaging System (Bio-Rad).
Co-immunoprecipitation
Cells were seeded in 100 mm dishes at a density of 6 million cells, with three dishes per experimental condition. Cells were treated 48 h after seeding. Following a 6-h treatment, cells were washed with cold PBS and lysed in HMKEN buffer (10 mM HEPES, pH 7.2, 5 mM MgCl2, 142 mM KCl, 2 mM EGTA, 0.2% IGEPAL® CA-630, protease, and phosphatase inhibitors). The lysate samples were triturated, and then preclearing was performed by incubating with 1:1 protein A:G Dynabeads (Life Technologies) on a rotator at 4 °C for 1-h. Primary antibodies [BCLXL (#2764, Cell Signaling), MCL1 (16225-1-AP, Proteintech) and BIM (#2933, Cell Signaling Technology)] at a concentration of 0.45 µg were added to the precleared lysates and incubated on a rotator at 4 °C overnight. Subsequently, samples were incubated with 1:1 protein A:G Dynabeads for 2 h at 4 °C. Beads underwent five washes in HMKEN buffer, were resuspended in 50 µL of HMKEN buffer supplemented with reducing and loading buffer. Samples were processed for western blotting.
Proximity ligation assay
Duolink® proximity ligation assay (PLA) was performed on formalin-fixed paraffin-embedded sections from C4-2 cells treated with ipatasertib (1 µM), AZD5991 (1 µM), combined treatment and control (DMSO). Antigen retrieval was performed on a Bond RX autostainer (Leica Biosystems) for 10-min with Bond ER2 solution (AR9640, Leica Biosystems). The following interactions were studied using the red fluorescence assay (DUO92008, Sigma-Aldrich): BAD (1:100, ab32445, abcam)-BCLXL (1:50, ab77571, abcam) and BIM (1:250, #2933, Cell Signaling Technology)-MCL1 (1:100, sc-69838, Santa Cruz). Antibody cocktails were incubated at room temperature for 1-h and the remaining protocol was performed according to manufacturer’s instructions (Sigma–Aldrich). Duolink® in situ mouse PLUS and rabbit MINUS probes were used. The omission of one antibody from an interaction was used as negative control. At the end of the procedure the slides were mounted using Duolink® In Situ Mounting Medium which contains DAPI for nuclear staining. Imaging was performed and PLA dots were counted using pathologist supervised HALOTM image analysis software (Indica Labs).
In vivo analyses with the CP253C CRPC PDX model
All procedures involving mice were conducted in compliance with the Institute of Cancer Research guidelines and approved by the ICR Animal Welfare and Ethical Review Body, adhering to the UK Animals (Scientific Procedures) Act 1986. Animals were housed in a pathogen-free facility in ventilated cages, at 20–24 °C and 40–60% relative humidity, with a 12 h light/dark cycle. CP253c PDX tumours were implanted into the left flank of NSG mice (7 weeks old at implantation; n = 31). Sex was considered in the study design and only male mice were used in the experiment given the disease aetiology. Upon reaching a tumour size of 50–300 mm3, mice were randomly assigned to one of six treatment arms: arm 1 received the vehicle, arm 2 was administered ipatasertib (50 mg/kg), arm 3 received S63845 (25 mg/kg), arm 4 was treated with fadraciclib (40 mg/kg), arm 5 received a combination of ipatasertib (50 mg/kg) and S63845 (25 mg/kg), and arm 6 received a combination of ipatasertib (50 mg/kg) and fadraciclib (40 mg/kg). Ipatasertib and fadraciclib were reconstituted in a solution of 0.5% methylcellulose and 0.2% Tween 80 in water, while S63845 was reconstituted in a solution containing 25 mM HCl and 20% hydroxypropyl-beta-cyclodextrin in water. Ipatasertib and fadraciclib were administered orally 5 days/week (Monday–Friday), while S63845 was delivered intravenously 3 days/week (Monday, Wednesday, and Friday). Tumour measurements and body weight were taken every 2–4 days, grouped in 3-day intervals. Mice were monitored for potential signs of discomfort or distress, including changes in physical appearance (facial expression, piloerection, hunched posture), behaviour (reduced activity), and physiological signs (abnormal breathing or altered food/water intake). The experiment concluded either 24 days after the initial treatment or earlier if tumours approached 14 mm diameter. None of the tumours exceeded the approved maximal size/burden allowed by our license (17 mm diameter). Mice were euthanized 6-h after the final dose, and tumour samples were collected for IHC analysis.
Development of drug-resistant cell lines
LNCaP95 and C4-2 cells were gradually exposed to increasing concentrations of capivasertib to develop capivasertib-resistant (Capi-R) cells, while LNCaP95 cells were similarly treated with AZD5991 or S63845 to generate AZD5991- and S63845-resistant (AZD5991-R and S63845-R) cells. Parental cell lines were maintained in parallel with 0.01% DMSO as a vehicle control. Resistance was defined after 6 days of drug exposure: Capi-R cells showed a >10-fold increase in capivasertib IC50, while AZD5991-R and S63845-R cells showed no significant change in growth when treated with 1 µM AZD5991 or S63845, respectively. Mycoplasma testing and STR profiling confirmed cell line identity and integrity.
Statistical analyses
All statistical analyses were performed using R Statistical Software version 4.1.3 (R Core Team, Vienna, Austria) or GraphPad Prism version 10 (GraphPad Software Inc., San Diego, California, USA). OS and PFI analyses were performed using Kaplan-Meier methodology and the log-rank test. The Mann-Whitney U test was used to compare pathologic tumour response in the NCI neoadjuvant cohort and to compare RNA expression, between tumours with MCL1 copy number gain/amplification and those without. Correlation analyses were performed with Spearman’s rank correlation coefficient. The linear mixed effects model was employed to compare the growth rate between the different treatment arms, and the vehicle, in the CP253c in vivo experiment and was performed using the R package nlme. Estimates and p-values refer to the interaction term of treatment arm and time indicating the difference in tumour volume growth rate between each treatment arm and the vehicle arm. Zero Interaction Potency (ZIP) synergy score was calculated, and ZIP surface plots were downloaded, using SynergyFinder+ web application82,83. Statistical comparisons of cell viability or caspase 3/7 activity between the control (or scramble) and treatments (or siRNAs) were conducted using one-way ANOVA. Post-hoc Tukey tests were employed for comparisons among all groups, whereas post-hoc Dunnett tests were used for comparisons against the control group. Mean and standard deviation were presented unless specified otherwise. A two-sided p value ≤ 0.05 was deemed to be statistically significant. All experiments were performed in biological triplicates (n = 3) unless stated otherwise.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Source Data files are provided with this paper. All raw data are available in the Source Data. Transcriptomic and copy number variation datasets used in this study have been previously published and made available19,45,62,65,66,67,68. The datasets used in this study are publicly available from the following sources: The TCGA data can be accessed in cBioPortal. The SU2C/PCF cohort data are available in cBioPortal and on GitHub under the code prad_su2c_2019. The RMH cohort data are available in the European Nucleotide Archive under accession number PRJEB32038. Finally, the NCI neoadjuvant cohort data have been deposited in the Database of Genotypes and Phenotypes (dbGaP) under accession number phs001938.v2.p1. No new omics datasets were generated. Further reasonable data access requests can be submitted to the corresponding authors. Source data are provided with this paper.
References
Mehtala, J. et al. Overall survival and second primary malignancies in men with metastatic prostate cancer. PLoS One 15, e0227552 (2020).
Moreira, D. M. et al. Predicting time from metastasis to overall survival in castration-resistant prostate cancer: results from search. Clin. Genitourin. Cancer 15, 60–66.e2 (2017).
Sumanasuriya S., De Bono J. Treatment of advanced prostate cancer-a review of current therapies and future promise. Cold Spring Harb. Perspect. Med. 8, a030635 (2018).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Westaby, D. et al. Targeting the intrinsic apoptosis pathway: a window of opportunity for prostate cancer. Cancers. 14, 51 (2021).
Carneiro, B. A. & El-Deiry, W. S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 17, 395–417 (2020).
Beroukhim, R. et al. The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 (2010).
Widden, H. & Placzek, W. J. The multiple mechanisms of MCL1 in the regulation of cell fate. Commun. Biol. 4, 1029 (2021).
Santer, F. R. et al. Mechanistic rationale for MCL1 inhibition during androgen deprivation therapy. Oncotarget 6, 6105–6122 (2015).
Reiner, T. et al. Mcl-1 protects prostate cancer cells from cell death mediated by chemotherapy-induced DNA damage. Oncoscience 2, 703–715 (2015).
Troiani, M. et al. Single-cell transcriptomics identifies Mcl-1 as a target for senolytic therapy in cancer. Nat. Commun. 13, 2177 (2022).
Guo, C. et al. Targeting myeloid chemotaxis to reverse prostate cancer therapy resistance. Nature 623, 1053–1061 (2023).
Wei, G. et al. Chemical genomics identifies small-molecule MCL1 repressors and BCL-xL as a predictor of MCL1 dependency. Cancer Cell 21, 547–562 (2012).
Goldsmith, K. C. et al. Mitochondrial Bcl-2 family dynamics define therapy response and resistance in neuroblastoma. Cancer Res. 72, 2565–2577 (2012).
Pilling, A. B. & Hwang, C. Targeting prosurvival BCL2 signaling through Akt blockade sensitizes castration-resistant prostate cancer cells to enzalutamide. Prostate 79, 1347–1359 (2019).
Arai, S. et al. Tyrosine Kinase inhibitors increase MCL1 degradation and in combination with BCLXL/BCL2 inhibitors drive prostate cancer apoptosis. Clin. Cancer Res. 24, 5458–5470 (2018).
Brim, B. C. et al. Direct Co-Targeting of Bcl-xL and Mcl-1 Exhibits Synergistic Effects in AR-V7-Expressing CRPC Models. Cancer Res Commun. 5, 1396–1408 (2025).
Karzai, F. et al. Sequential prostate magnetic resonance imaging in newly diagnosed high-risk prostate cancer treated with neoadjuvant enzalutamide is predictive of therapeutic response. Clin. Cancer Res. 27, 429–437 (2021).
Kumar, A. et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 22, 369–378 (2016).
Slomp, A. et al. Multiple myeloma with 1q21 amplification is highly sensitive to MCL-1 targeting. Blood Adv. 3, 4202–4214 (2019).
Munkhbaatar, E. et al. MCL-1 gains occur with high frequency in lung adenocarcinoma and can be targeted therapeutically. Nat. Commun. 11, 4527 (2020).
Chandra, D., Choy, G., Daniel, P. T. & Tang, D. G. Bax-dependent regulation of Bak by voltage-dependent anion channel 2. J. Biol. Chem. 280, 19051–19061 (2005).
de Bono, J. S. et al. Randomized Phase II study evaluating akt blockade with ipatasertib, in combination with abiraterone, in patients with metastatic prostate cancer with and without PTEN Loss. Clin. Cancer Res. 25, 928–936 (2019).
Sweeney, C. et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. Lancet 398, 131–142 (2021).
Kale, J., Osterlund, E. J. & Andrews, D. W. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 25, 65–80 (2018).
Tron, A. E. et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 9, 5341 (2018).
Welti, J. et al. Targeting bromodomain and extra-terminal (BET) family proteins in castration-resistant prostate cancer (CRPC). Clin. Cancer Res. 24, 3149–3162 (2018).
Park, K. et al. Antibody-based detection of ERG rearrangement-positive prostate cancer. Neoplasia 12, 590–598 (2010).
Kotschy, A. et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538, 477–482 (2016).
Cidado, J. et al. AZD4573 Is a highly selective CDK9 inhibitor that suppresses MCL-1 and induces apoptosis in hematologic cancer cells. Clin. Cancer Res. 26, 922–934 (2020).
Frame, S. et al. Fadraciclib (CYC065), a novel CDK inhibitor, targets key pro-survival and oncogenic pathways in cancer. PLoS One 15, e0234103 (2020).
Dey, J. et al. Voruciclib, a clinical stage oral CDK9 inhibitor, represses MCL-1 and sensitizes high-risk diffuse Large B-cell Lymphoma to BCL2 inhibition. Sci. Rep. 7, 18007 (2017).
Fabbri, F. et al. Mitotic catastrophe and apoptosis induced by docetaxel in hormone-refractory prostate cancer cells. J. Cell. Physiol. 217, 494–501 (2008).
Wang, Y. & Poon, R. Y. C. MARCH5 regulates mitotic apoptosis through MCL1-dependent and independent mechanisms. Cell. Death. Differ. 30, 753–765 (2023).
Bloedjes, T. A. et al. AKT signaling restrains tumor suppressive functions of FOXO transcription factors and GSK3 kinase in multiple myeloma. Blood. Adv. 4, 4151–4164 (2020).
Li, Y. et al. AKT inhibition sensitizes acute leukemia cells to S63845-induced apoptosis. Hematology 28, 2214465 (2023).
Dunn, S. et al. AKT-mTORC1 reactivation is the dominant resistance driver for PI3Kbeta/AKT inhibitors in PTEN-null breast cancer and can be overcome by combining with Mcl-1 inhibitors. Oncogene 41, 5046–5060 (2022).
Datta, S. R. et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91, 231–241 (1997).
Zha, J., Harada, H., Yang, E., Jockel, J. & Korsmeyer, S. J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 87, 619–628 (1996).
Kim, H. et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat. Cell Biol. 8, 1348–1358 (2006).
Mason, K. D. et al. Programmed anuclear cell death delimits platelet life span. Cell 128, 1173–1186 (2007).
Weeden, C. E. et al. Dual inhibition of BCL-XL and MCL-1 is required to induce tumour regression in lung squamous cell carcinomas sensitive to FGFR inhibition. Oncogene 37, 4475–4488 (2018).
Yang, E. et al. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 80, 285–291 (1995).
Ferraldeschi, R. et al. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur. Urol. 67, 795–802 (2015).
Abida, W. et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 116, 11428–11436 (2019).
Carver, B. S. et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat. Genet. 41, 619–624 (2009).
King, J. C. et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat. Genet. 41, 524–526 (2009).
Sharp, A. et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Investig. 129, 192–208 (2019).
Attard, G. et al. Characterization of ERG, AR and PTEN gene status in circulating tumor cells from patients with castration-resistant prostate cancer. Cancer Res. 69, 2912–2918 (2009).
Yu, J. et al. An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 17, 443–454 (2010).
Wang, H. T. et al. Neuroendocrine Prostate Cancer (NEPC) progressing from conventional prostatic adenocarcinoma: factors associated with time to development of NEPC and survival from NEPC diagnosis-a systematic review and pooled analysis. J. Clin. Oncol. 32, 3383–3390 (2014).
Vlachostergios, P. J., Puca, L. & Beltran, H. Emerging variants of castration-resistant prostate cancer. Curr. Oncol. Rep. 19, 32 (2017).
Piha-Paul, S. et al. A phase 1/2, open-label, multicenter study to investigate the safety, pharmacokinetics, and efficacy of fadraciclib (CYC065), an oral CDK2/9 inhibitor, in subjects with advanced solid tumors and lymphoma. Eur. J. Cancer 174, S17–S18 (2022).
Strati, P. et al. P1126: phase 1B/2A study of AZD4573 (CDK9i) and acalabrutinib in patients (PTS) with relapsed/refractory diffuse large B-cell lymphoma (R/R DLBCL). Hemasphere 7, e47925e9 (Suppl) (2023).
Do, K. T. et al. Abstract CT037: phase I safety, pharmacokinetic and pharmacodynamic study of CYC065, a cyclin dependent kinase inhibitor, in patients with advanced cancers (NCT02552953). Cancer Res. 78, CT037–CT037 (2018).
Baker, A. et al. The CDK9 Inhibitor dinaciclib exerts potent apoptotic and antitumor effects in preclinical models of Mll-rearranged acute myeloid leukemia. Cancer Res. 76, 1158–1169 (2016).
Polier, G. et al. Wogonin and related natural flavones are inhibitors of CDK9 that induce apoptosis in cancer cells by transcriptional suppression of Mcl-1. Cell Death Dis. 2, e182 (2011).
Wang, X. et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev. 27, 1351–1364 (2013).
Desai, P. et al. A Phase 1 First-in-human study of the Mcl-1 inhibitor Azd5991 in patients with relapsed/refractory hematologic malignancies. Clin. Cancer Res. 30, 4844–4855 (2024).
Tolcher, A. W. et al. A first-in-human study of mirzotamab clezutoclax as monotherapy and in combination with taxane therapy in relapsed/refractory solid tumors: dose escalation results. J. Clin. Oncol. 39, 3015 (2021).
Khan, S. et al. A selective BCL-X(L) PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 25, 1938–1947 (2019).
Mateo, J. et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J. Clin. Investig. 130, 1743–1751 (2020).
Fenor de la Maza, M. D. et al. Immune biomarkers in metastatic castration-resistant prostate cancer. Eur. Urol. Oncol. 5, 659–667 (2022).
Sumanasuriya, S. et al. Elucidating prostate cancer behaviour during treatment via low-pass whole-genome sequencing of circulating tumour DNA. Eur. Urol. 80, 243–253 (2021).
Robinson, D. et al. Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228 (2015).
Liu, J. et al. An Integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173, 400–416.e11 (2018).
Wilkinson, S. et al. Nascent prostate cancer heterogeneity drives evolution and resistance to intense hormonal therapy. Eur. Urol. 80, 746–757 (2021).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Milacic, M. et al. The reactome pathway knowledgebase 2024. Nucleic Acids Res. 52, D672–D678 (2024).
Westaby, D. et al. BCL2 expression is enriched in advanced prostate cancer with features of lineage plasticity. J. Clin. Investig. 134, e179998 (2024).
Detre, S., Saclani Jotti, G. & Dowsett, M. A “quickscore” method for immunohistochemical semiquantitation: validation for oestrogen receptor in breast carcinomas. J. Clin. Pathol. 48, 876–878 (1995).
Reid, A. H. et al. Molecular characterisation of ERG, ETV1 and PTEN gene loci identifies patients at low and high risk of death from prostate cancer. Br. J. Cancer 102, 678–684 (2010).
Welti, J. et al. Targeting the p300/CBP Axis in lethal prostate cancer. Cancer Discov. 11, 1118–1137 (2021).
Gil, V. et al. HER3 Is an actionable target in advanced prostate cancer. Cancer Res 81, 6207–6218 (2021).
Chen, Z. et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436, 725–730 (2005).
Marino, S., Vooijs, M., van Der Gulden, H., Jonkers, J. & Berns, A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 14, 994–1004 (2000).
Calado, D. P. et al. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat. Immunol. 13, 1092–1100 (2012).
Ruzankina, Y. et al. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 1, 113–126 (2007).
Drost, J. et al. Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc. 11, 347–358 (2016).
Mathews Griner, L. A. et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc. Natl. Acad. Sci. USA 111, 2349–2354 (2014).
Lin, G. L. et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci. Transl. Med. 11, eaaw0064 (2019).
Zheng, S. et al. SynergyFinder Plus: toward better interpretation and annotation of drug combination screening datasets. Genom. Proteom. Bioinforma. 20, 587–596 (2022).
Yadav, B., Wennerberg, K., Aittokallio, T. & Tang, J. Searching for drug synergy in complex dose-response landscapes using an interaction potency model. Comput. Struct. Biotechnol. J. 13, 504–513 (2015).
Acknowledgements
The authors gratefully acknowledge the patients and the families of patients who contributed to this study. This work was supported by Prostate Cancer UK (TLD-PF19-006 to J.M.J.-V.; Career Acceleration Fellowship to J.T.; Research Funding to J.S.dB.), the Department of Defence Prostate Cancer Research Programme (Early Investigator Research Award to J.M.J-V. and S.W.; Impact Awards to A.G.S. and S.P.B.; Idea Development Award to A.G.S.), Cancer Research UK (Clinical Research Training Fellowship to D.W. and N.W.; Radiation Research Network Seed Funding to A.Sharp; Convergence Centre Grant funding, Centre Programme and Experimental Cancer Medicine Centre grants to J.S.dB.), MINECO (FPU18/02485 to A.J.M-H.; PID2022-138185OB-I00 to R.M.L), National Cancer Institute/National Institutes of Health (R50CA274336, P50CA0907186, P01CA163227, and R21CA277368 to I.C. and P.S.N.; the Intramural Research Programme of the Center for Cancer Research, Research Funding to W.D.), Royal Society of Medicine (International Exchanges Award IES\R3\213131 to A.Sharp and N.A.L.), Terry Fox Research Institute New Frontiers Programme (F22-00589 to N.A.L.), Prostate Cancer Foundation Canada grant-in-aide (to N.A.L.), American Association for Cancer Research (AACR Clinical/Translational Cancer Research Fellowship 18-40-11-BEZZ to M.B.), Medical Research Council (Research Funding to J.S.d.B.), the Prostate Cancer Foundation (Young Investigator Award to S.W.; Challenge Award to S.P.B., J.S.dB. and A.S.), the Movember Foundation through the London Movember Centre of Excellence (CEO13 2-002 to J.S.d.B.), the National Institute for Health and Care Research Biomedical Research Centre (pump priming award to A.Sharp), the Wellcome Trust (Clinical Research Career Development Fellowship to A.Sharp). Portions of this work was supported by the NCI (HHSN261200800001E), the Intramural Research Programme of the NCI, NIH, and the NIH National Center for Advancing Translational Sciences. Portions of this work utilised the computational resources of the NIH HPC Biowulf cluster. The authors thank the patients, families and investigators, and the Institute for Prostate Cancer Research for supporting the University of Washington Tissue Acquisition Programme. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services or the Department of Defence, nor does mention of trade names, commercial products, or organisation imply endorsement by the U.S. Government. J.S.d.B. is a National Institute for Health Research (NIHR) Senior Investigator. The views expressed in this article are those of the author(s) and not necessarily those of the National Health Service, the NIHR, or the Department of Health, nor does mention of trade names, commercial products, or organisation imply endorsement by the UK Government.
Author information
Authors and Affiliations
Contributions
J.M.J.-V., D.W., J.S.d.B. and A.Sharp. conceived and designed the study. J.M.J.-V., D.W., I.F., A.DH.B., A.P., B.G., C.B., S.M., M.L., A.J.M.-H., I.C., I.P.L.Y., L.B., W.Z., A.J.N., J.W., R.P., F.G., N.P., A.F., M.C., R.R., S.D., J.T., N.W., E.H., M.V., J.N., I.B., K.L., T.P., P.G., S.W., S.Y.T., F.K., C.H.C., E.L.B., X.Z., C.K.-T. and A.V. performed experiments and/or collected data. J.M.J.-V., D.W., W.Y., G.S., D.B. and J.R. analysed the data and/or performed statistical analyses. J.M.J.-V., D.W., R.M.L., A.S., F.R., N.A.L., C.J.T., G.H., W.D.F., M.B., A.G.S., P.S.N., S.C., S.P.B., J.S.d.B. and A.Sharp. interpreted the results and/or contributed to data visualisation. J.M.J.-V., D.W., J.S.d.B. and A.Sharp draughted the manuscript. All authors provided advice on, reviewed, edited and approved the manuscript. J.S.d.B. and A.Sharp served as sponsors and monitored the study.
Corresponding authors
Ethics declarations
Competing interests
J.M.J.-V., D.W., I.F., A.d.H.V., A.P., W.Y., G.S., D.B., B.G., C.B., S.M., L.B., W.Z., A.J.N., J.W., J.R., R.P., A.F., M.C., R.R., S.D., J.T., E.H., M.V., J.N., K.L., A.S., F.R., S.C., J.S.d.B. and A.Sharp are employees of the ICR, which has a commercial interest in abiraterone, PARP inhibition in DNA repair defective cancers, and PI3K/AKT pathway inhibitors (no personal income). A.G.S. reports that the National Cancer Institute (NCI) has a Cooperative Research and Development Agreement (CRADA) with Astellas. Resources are provided by this CRADA to the NCI. A.G.S. gets no personal funding from this CRADA but is the primary investigator of the CRADA. J.S.d.B. has served on advisory boards and received fees from many companies, including Amgen, Astra Zeneca, Bayer, Bioxcel Therapeutics, Daiichi, Genentech/Roche, GSK, Merck Serono, Merck Sharp & Dohme, Pfizer, and Sanofi Aventis. He is an employee of the ICR, which has received funding or other support for his research work from AZ, Astellas, Bayer, Cellcentric, Daiichi, Genentech, Genmab, GSK, Janssen, Merck Serono, MSD, Menarini/Silicon Biosystems, Orion, Sanofi Aventis, Sierra Oncology, Taiho, Pfizer, Vertex. J.S.d.B. was named as an inventor, with no financial interest, for patent 8,822,438, submitted by Janssen, that covers the use of abiraterone acetate with corticosteroids. J.S.d.B. has been the CI/PI of many industry-sponsored clinical trials. A.Sharp has received travel support from Sanofi, Roche-Genentech and Nurix, and speaker honoraria from Astellas Pharma and Merck Sharp & Dohme. He has served as an advisor to DE Shaw Research, CHARM Therapeutics, Ellipses Pharma and Droia Ventures. A.Sharp has been the CI/PI of industry-sponsored clinical trials. The remaining authors declare no conflicts of interest.
Peer review
Peer review information
Nature Communications thanks Andrea Lunardi, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiménez-Vacas, J.M., Westaby, D., Figueiredo, I. et al. Elucidating molecularly stratified single agent, and combination, therapeutic strategies targeting MCL1 for lethal prostate cancer. Nat Commun 16, 8806 (2025). https://doi.org/10.1038/s41467-025-64042-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64042-5
