
Abstract
Heterogeneous catalytic ozonation (HCO) triggered by single-atom catalysts (SACs) is a promising technology for advanced wastewater purification. However, high symmetry of conventional metal-N4 structures limits catalytic performance. Herein, we construct an asymmetrically coordinated Fe-N3P1 moiety in Fe-NPC catalyst, where the short-range coordination effect of P significantly enhances HCO. The Fe-NPC/O3 achieves 100% removal of model pollutant p-hydroxybenzoic acid, with a kinetic constant of 0.123 min−1, and also demonstrates excellent advanced treatment for coal chemical wastewater. The degradation of contaminants is attributed to ozone and nonradical singlet oxygen. Theoretical calculations reveal that the central Fe atom in Fe-N3P1 is the main site for HCO, and the introduction of P primarily modulates the electronic structure of Fe atom by altering its coordination environment. This work provides a short-range coordination strategy for regulating the electronic properties of isolated metal centers and sheds light on the HCO pathways with asymmetrically coordinated SACs.
Similar content being viewed by others

Introduction
The imbalance between the rising demand for clean water and the scarcity of freshwater resources underscores the importance and urgency of wastewater reclamation and reuse1,2. Refractory and hazardous organic pollutants in wastewater pose a serious threat to both human health and the environment, which are often resistant to traditional biological treatment. The advanced purification of these harmful contaminants in wastewater is not only essential for protecting ecosystems but also critical for ensuring the safety of reclaimed water3,4. Therefore, there is a growing need for sustainable and advanced treatment technologies capable of effectively addressing this issue.
Advanced oxidation processes (AOPs) offer appealing options to degrade the organics by generating highly oxidizing reactive oxygen species (ROS)5,6,7. As an offshoot of AOPs, heterogeneous catalytic ozonation (HCO) is a highly efficient and promising technique for the advanced wastewater treatment, where ozone is decomposed on the catalyst surface and converted into radical or nonradical ROS, thereby eliminating the recalcitrant organics from wastewater.8,9,10 Generally, the catalytic activity, stability and selectivity in HCO are mainly dependent on the characteristics of the heterogeneous ozone catalysts11,12,13,14,15. Carbon skeletons are preferred ozone catalyst carriers due to their high specific surface areas, tunable chemical structures and environmental friendliness16,17. Transition metals with abundant 3 d empty orbitals commonly possess superior catalytic ozonation performance, which are effective active components12,18,19. Therefore, the rational design of transition metal forms on the surface of carbon carriers is of great significance for the development of HCO in wastewater purification.
In single-atom catalysts (SACs), the metal species exist as atomically dispersed forms on the surface of the carrier20. SACs integrate the maximum utilization of metal atoms, well-defined active sites and precisely tailorable coordination environments, and have raised great interest in the field of catalytic oxidation for water treatment21,22,23. In particular, carbon-supported transition-metal-based SACs are considered the preferred option for facilitating catalytic ozonation to generate radical and nonradical ROS for the degradation of recalcitrant organic contaminants, and these catalysts typically feature atomically dispersed metals coordinated by N atoms, with the optimal active sites being planar and symmetric M-N4 configurations24,25,26. However, the high symmetry of M-N4 results in a fixed d-orbital energy level splitting pattern for the transition metal center, making it difficult to flexibly adjust the electronic structure, which restricts further improvement in catalytic ozonation performance.
Heteroatoms not only provide anchoring sites for single-atom metals but also effectively regulate the charge distribution and spin state of the isolated metal species27,28. Introducing a second heteroatom to break the symmetry of the single-atom M-N4 structure could optimize the local coordination environment and electronic structure of the atomically dispersed metal atoms29,30,31. Based on the short-range coordination effect32,33, the substitution of the N atom with other atoms that have less electronegativity and larger atomic radii (such as P, S) to design asymmetrically coordinated single-atom sites might be a promising approach to enhance the catalytic ozonation of SACs. However, in HCO, the structure-activity relationship between the symmetry of coordination structure of single atoms and the catalytic ozonation performance is rarely reported owing to the challenge of precise control of the coordination environment. Moreover, the catalytic mechanisms triggered by the asymmetrically coordinated single-atom sites have not been thoroughly investigated.
In this work, the heteroatom P was incorporated to modulate the geometric and electronic structures of single-atom iron sites via a short-range coordination engineering strategy, and nitrogen-phosphorus coordinated single-atom iron catalysts (Fe-NPC) with a precisely controlled atomic structure of Fe-N3P1 were designed for catalytic ozonation and wastewater purification. Fe-N3P1 sites exhibited superior catalytic ozonation performance compared to symmetrical Fe-N4 structures, excelling in both model pollutant degradation and advanced treatment of coal chemical wastewater. In the Fe-NPC/O3 system, ozone and nonradical singlet oxygen (1O2) were identified as the dominant active species for the degradation of organics. In-situ surface-enhanced Raman spectroscopy (SERS) was employed to identify key surface-adsorbed intermediates (*O, *O2), while density functional theory (DFT) calculations and molecular dynamics (MD) simulations provided atomic-level insights into the detailed mechanisms of ozone dissociation and nonradical ROS generation. These findings highlight the potential of short-range coordination engineering in SACs, enabling precise tuning of the local microenvironment to boost catalytic ozonation for wastewater purification and reclamation.
Results
Synthesis and structural properties of Fe-NPC catalysts
The synthesis strategy of Fe-NPC was illustrated in Fig. 1a. Zinc-based zeolite-like imidazolium framework (ZIF-8) was used as the host nanocage. Iron(III) acetylacetonate (Fe(acac)3) and triphenylphosphine (PPh3) with the molecular diameters of about 9.9 Å and 8.9 Å, respectively (Supplementary Fig. 1), were employed as the guest molecules. Through the simple host-guest method34, ZIF-8 precursors simultaneously encapsulating both Fe(acac)3 and PPh3 (marked as FeP@ZIF-8) were prepared at normal pressure and temperature, exhibiting a well-defined rhombic dodecahedral morphology (Supplementary Figs. 2, 3). Subsequently, Fe-NPC catalysts were successfully fabricated by the pyrolysis of FeP@ZIF-8 precursors, effectively incorporating Fe and P into the catalyst framework and paving the way for enhanced catalytic ozonation performance.

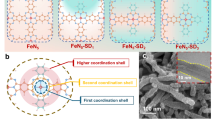
a Synthesis scheme of Fe-NPC catalyst; b SEM image, c TEM image and d EDS mappings of Fe-NPC; e HAADF-STEM image of Fe-NPC (yellow circles indicate single Fe atoms); f three-dimensional Gaussian-function-fitting mapping for the red square region in e.
The iron species in Fe-NPC catalysts exhibited an atomically dispersed state. The SEM and TEM images (Fig. 1b, c and Supplementary Fig. 4) demonstrated that Fe-NPC catalysts still maintained the dodecahedral shape. Supplementary Fig. 5 showed that the introduction of Fe source effectively promoted the catalyst dispersion and improved the degree of graphitization of the carbon substrate. Supplementary Fig. 6 and Supplementary Table 1 depicted that the presence of Fe and P species exerted little influence on the pore structure and specific surface area. The EDS mappings (Fig. 1d) showed that C, N, P, and Fe were uniformly dispersed and highly overlapped. The HAADF-STEM image (Fig. 1e) showed that highly dispersed bright spots (highlighted by yellow circles) were observed on the carbon skeleton, and the three-dimensional Gaussian function fitted mapping of the red square region (Fig. 1f) presented only one distinct peak. The XRD pattern of Fe-NPC (Fig. 2a) also exhibited only two broad diffraction peaks corresponding to graphitized carbon, with no detectable metal peaks. These results indicated that the iron species in Fe-NPC catalysts existed as single atoms.

a XRD patterns, (b) N 1 s XPS spectra, and (c) P 2p XPS spectra of different catalysts. d XANES spectra of Fe-NPC and reference samples; e Linear fitting curve of Fe K-edge energy and Fe valence; f FT-EXAFS spectra; g WT-EXAFS contour plot of Fe-NPC; EXAFS spectra and fitting curves at (h) k space and (i) R space.
Heteroatom P modulated the local geometric and electronic structure of single-atom iron in Fe-NPC catalysts through the short-range coordination effect. The iron content loaded on Fe-NPC was measured to be 0.62 wt.% by ICP-OES. As displayed in Fig. 2b, in addition to pyridinic-N, pyrrolic-N and graphitic-N, the Fe-N peak was also detected in the high-resolution N 1 s XPS spectrum of Fe-NPC, suggesting that N species, especially pyridinic-N with the highest percentage, provided anchoring sites for single-atom iron. The P 2p spectrum of Fe-NPC (Fig. 2c) was deconvolved into three peaks at 128.7, 132.0, and 133.6 eV, which can be assigned P-Fe, P-C, and P-N, respectively28,35. Figure 2d showed that the Fe K-edge XANES spectrum of Fe-NPC lay between those of Fe foil and Fe2O3, demonstrating that single-atom iron in Fe-NPC was positively charged. A linear relationship was established between the valence state and the position of Fe K-edge of the control samples (Fig. 2e), and the valence state of iron of Fe-NPC was calculated to be approximately +2.56.
Furthermore, the precise coordination environment of single-atom iron in Fe-NPC was acquired from the EXAFS. Both Fourier-transform EXAFS spectrum and wavelet transform EXAFS contour map of Fe-NPC (Fig. 2f, g and Supplementary Fig. 7) exhibited one prominent peak, which was attributed to the Fe-N coordination structure. Since Fe-N and Fe-P were detected in Fe-NPC, these two backscattering pathways were used to fit the EXAFS curve. The fitting results (Fig. 2h, i and Supplementary Table 2) displayed that the first-shell coordination environment of single-atom iron in Fe-NPC was three N atoms and one P atom. The formation energy of Fe-N3P1 configuration calculated by DFT was about −13.93 eV (Supplementary Fig. 8), indicating spontaneous generation during calcination. The electrochemical characterizations (Supplementary Fig. 9) displayed that Fe-NPC catalysts possessed the highest current density and the lowest Tafel slope, indicating the best reactivity and the fastest electron transfer capability of Fe-NPC catalysts. On the whole, the local coordination configuration of single-atom iron in Fe-NPC catalysts was atomically dispersed Fe-N3P1 structure, which might present the excellent reactivity in HCO.
Catalytic ozonation performance by Fe-NPC catalysts
Asymmetrically coordinated single-atom iron (Fe-NPC) exhibited superior catalytic ozonation for the degradation of refractory organics in wastewater (Fig. 3). A typical phenolic compound, p-hydroxybenzoic acid (pHBA), was used as the model contaminant. The adsorption of pHBA by different catalysts (Fig. 3a) demonstrated that the simultaneous introduction of Fe and P effectively promoted the adsorption, which was conducive to the diffusion and catalytic oxidation of the model contaminant. Supplementary Fig. 10 displayed that the ozonation and catalytic ozonation profiles of pHBA were well-fitted to the pseudo-first-order kinetics model. As shown in Fig. 3b, electron-rich pHBA could be attacked by ozone molecules with the rate constant of 0.057 min−1. Compared with ozonation, the catalytic ozonation based on NC and NPC showed limited enhancement for pHBA removal. However, the catalytic ozonation was dramatically enhanced after the introduction of Fe species, and the degradation kinetic constant in Fe-NC/O3 system was 0.088 min−1. Furthermore, Fe-NPC presented the best catalytic ozonation activity, achieving complete removal of pHBA with the degradation rate constant of 0.123 min−1. Supplementary Figs. 11, 12 showed that as for the pHBA removal kinetic constant normalized by the specific surface area or Fe-loaded amount, Fe-NPC/O3 still exhibited the superior removal performance. A Fe-NPC-loaded polyvinylidene fluoride (PVDF) catalytic membrane reactor was continuously operated for 48 h. As shown in Supplementary Fig. 13, the water fluxes of the PVDF membrane and the Fe-NPC-PVDF membrane were approximately 245 and 230 LMH, respectively, indicating that the catalyst loading had negligible impact on filtration performance. In the PVDF/O3 system, the removal of pHBA remained around 10%, while in the Fe-NPC-PVDF/O3 system, nearly complete and stable removal of pHBA was achieved. The results demonstrated the excellent catalytic ozonation performance of the Fe-NPC-PVDF membrane and highlighted its potential for application in continuous-flow reactors.

a pHBA degradation in different systems and (b) corresponding removal rate constants; c OA degradation in Fe-NPC/O3 system; d Comparison of mass activity for catalytic ozonation in terms of OA degradation; Effects of (e) NaCl concentration (insert: corresponding kinetic rate constants) and (f) Na2SO4 concentration (insert: corresponding kinetic rate constants); g Advanced treatment of biotreated coal chemical wastewater; h COD and TOC removal after treatment; i Reusability tests of Fe-NPC. The error bars in the figures represent the standard deviations from triplicate tests.
In addition, Fe-NPC/O3 system also demonstrated excellent removal of oxalic acid (OA) with the rate constant of 0.057 min−1 (Fig. 3c). The catalytic activity of Fe-NPC was evaluated against existing catalysts by determining the mass activity for OA degradation. Figure 3d and Supplementary Table 3 showed that Fe-NPC outperformed numerous state-of-the-art ozone catalysts in terms of mass activity for OA degradation, and Fe-NPC-based catalytic ozonation was a promising and advanced technology. A saturated ozone water with a concentration of about 4.0 mg L−1 was prepared to carry out ozone decomposition tests (Supplementary Fig. 14). Fe-NPC also exhibited outstanding capability to decompose ozone, and the dissolved ozone was completely removed in 5 min. The results indicated that single-atom iron in Fe-NPC possessed superior catalytic ozonation, and the presence of P further intensified the reactivity of single-atom iron. Thus, Fe-NPC catalysts containing asymmetric coordinated single-atom Fe-N3P1 active sites presented excellent catalytic ozonation for wastewater decontamination.
The factors influencing pHBA removal in Fe-NPC/O3 system were examined. Increasing the dosage of Fe-NPC catalysts and the ozone concentration at the gas inlet both showed a positive correlation with the rate constant for pHBA removal (Supplementary Figs. 15, 16). With the increase of P doping amount, the rate constant for pHBA removal initially increased and then tended to level off (Supplementary Fig. 17). Moreover, the degradation of pHBA was also accelerated by raising the solution pH (Supplementary Fig. 18). The effects of different concentration gradients of Cl− and SO42− were investigated (Fig. 3e, f). Slight promotion was observed with the presence of NaCl and Na2SO4 concentrations of 0.1 and 1.0 g L−1. It was found that increasing the concentration of NaCl and Na2SO4 to 10.0 g L−1, the removal of pHBA was dramatically enhanced, with the kinetic constants of 0.145 and 0.309 min−1, respectively. Supplementary Figs. 19–22 indicated that the accelerated pHBA degradation kinetics in the Fe-NPC/O3 system with increasing salt concentration can be attributed to the reduced bubble size and enhanced gas-liquid mass transfer (See Supplementary Discussion 1 for detailed discussion).
Advanced treatment of coal chemical wastewater
The catalytic ozonation based on asymmetrically coordinated single-atom iron (Fe-NPC) was also more advantageous in real wastewater treatment. The COD of the biotreated coal chemical wastewater used in this work was about 95 mg L−1. As shown in Fig. 3g, after advanced treatment with O3 alone, NC/O3, NPC/O3, Fe-NC/O3, and Fe-NPC/O3 systems, the effluent COD values were about 78 ± 1, 73 ± 2, 71 ± 1, 56 ± 3, and 48 ± 2 mg L−1, respectively. Among them, Fe-NPC catalysts possessed the best catalytic ozonation for the degradation of refractory organics in coal chemical wastewater, with the COD and TOC removals of 48% and 47%, respectively (Fig. 3h). In the reusability tests (Fig. 3i and Supplementary Fig. 23), the effluent CODs were all lower than 50 mg L−1 and there was almost no iron ion leaching during the treatment, indicating the excellent catalytic stability of Fe-NPC catalysts. The characterization results of XRD and TEM-EDS (Supplementary Figs. 24, 25) demonstrated that the iron species remained uniformly dispersed on the carbon framework, indicating the well structural stability of single-atom Fe. The water quality analysis results (Supplementary Figs. 26–30) showed that in the biotreated coal chemical wastewater, the contents of simple aromatic proteins, fulvic acid-like substances, and aliphatic compounds were relatively higher, and after the advanced treatment by Fe-NPC-based catalytic ozonation process, various organic pollutants were effectively removed (See Supplementary Discussion 2 for detailed discussion). An economic evaluation (Supplementary Discussion 3) was also conducted. Compared to pure ozonation, the Fe-NPC-based catalytic ozonation process could reduce the estimated total cost (primarily arising from ozone consumption and catalyst synthesis) by ~80%, demonstrating its promising cost-effectiveness and engineering potential.
Identification of reactive oxygen species
Catalytic ozonation of organic pollutants was generally dependent on the decomposition and conversion of ozone into ROS. The types of ROS generated on the catalyst surface and in the solution were analyzed. The EPR spectroscopy was implemented to detect the signals of •OH, O2•−, and 1O2 in the solution. The results (Fig. 4a-c) demonstrated that the signal of •OH characteristic peak was not recorded, while both signals of O2•− and 1O2 were identified in Fe-NPC/O3 and compared with the control systems, the introduction of Fe and P effectively gave rise to the peak intensities of DMPO-O2•− and TEMPO. In-situ Raman analysis was conducted to ascertain the surface-adsorbed ROS. As shown in Fig. 4d, after introducing ozone, four peaks were recorded on the Fe-NPC catalyst surface, among which the peak at about 989 cm−1 was surface-adsorbed atomic oxygen species (*O) and the peaks at about 810 cm−1, 897 cm−1, and 1034 cm−1 were assigned to surface-adsorbed peroxide species (*O2). Notably, the *O with the oxidation-reduction potential of 2.43 V can directly attack the surface-adsorbed organics, and the *O2 can convert into other ROS to participate in the removal of contaminants.

EPR spectra of (a) DMPO-•OH, (b) DMPO-O2•− and (c) TEMPO; (d) In-situ Raman spectrum of Fe-NPC/O3 system; (e) Quenching tests by MeOH, DMSO and FFA and (f) corresponding pHBA removal rate constants. The error bars in the figures represent the standard deviations from triplicate tests.
The contributions of different ROS were evaluated by quenching tests (Fig. 4e, f). The direct oxidation by O3 and the indirect oxidation by 1O2 were the predominant removal pathways for electron-rich pHBA in Fe-NPC/O3 system. MeOH is a specific scavenger for •OH both in the aqueous solution and on the catalyst surface, while the inhibitory effect of MeOH was slight for pHBA removal, indicating the limited role of •OH. DMSO has a strong affinity for surface-adsorbed ROS11. However, the removal of pHBA was also slightly inhibited after the introduction of DMSO, with the kinetic constant of 0.080 min−1. The results indicated the •OH oxidation and surface oxidation might not be the predominant removal pathways for pHBA.
The role of 1O2 in pHBA oxidation was verified with furfuryl alcohol (FFA) as the quenching agent. It was found that pHBA degradation was almost terminated in the presence of FFA, and the degradation rate constant was only 0.012 min−1, demonstrating that 1O2 was involved in the catalytic oxidation process. O2•− is a key intermediate in catalytic ozonation, which might be responsible for the generation of 1O2 through single electron transfer process8,36,37. Thus, an important source of 1O2 is the self-disproportionation of O2•− in the Fe-NPC/O3 system. It is worth noting that electron-rich pHBA can also be attacked by the direct oxidation of O3. As a consequence, the removal of pHBA mainly occurred in the aqueous solution, and the O3 and 1O2 were dominated active species.
Theoretical predication of interaction sites
The short-range coordination effect of P could improve the electronic structure of single-atom Fe, thereby promoting the adsorption of ozone and organics. The interaction site of Fe-N3P1 moiety with ozone was predicted by electrostatic potential (ESP), Fukui functions (f + , f − ) and dual descriptor (DD) calculated through DFT. Ozone molecules are polar, and the ESP map of ozone showed that the central O atom was positively charged and the two terminal O atoms were negatively charged. Figure 5a displayed the isosurface maps of f + , f− and DD of ozone. The terminal O atoms covered a larger region than that of the central O atom, indicating a higher reactivity of the terminal O atoms38,39. Condensed DD (CDD) was calculated by Hirshfeld charges to quantitatively compare the DD of each atom. The CDD values of the two terminal O atoms were both −0.02 (Supplementary Table 4), demonstrating that the terminal O atoms in the ozone molecule were more nucleophilic and tended to attract positively charged reagents or those with more empty orbitals.

a Electrostatic potential (ESP) map, Fukui functions (f + , f − ) and dual descriptor (DD) of ozone molecule; b Model and ESP map of Fe-N4 structure; c Model and ESP map of Fe-N3P1 structure; d Maximum ESP of different structures; e Adsorption energies of ozone and pHBA by different structures.
The ESP maps of different catalytic sites were plotted in Fig. 5b, c and Supplementary Fig. 31, and the introduction of Fe into catalytic sites significantly modulated the surface charge distributions. The central Fe atom in Fe-N4 moiety possessed the highest ESP, and the presence of P further enhanced the ESP of the central Fe atom in Fe-N3P1 moiety (Fig. 5d). Since the local coordination environment of single-atom iron was broken after introducing heteroatom P, the graphitic carbon skeleton was locally deformed and stretched. Supplementary Fig. 32 showed the density of states (DOS) of the Fe-N4 and Fe-N3P1 structures, and the results demonstrated that the d-band center of the Fe-N4 structure was located at about 1.76 eV, whereas that of Fe-N3P1 downshifted significantly to 0.93 eV, indicating that P doping effectively modulated the adsorption capability of single-atom Fe toward small molecules. Compared with Fe-N4 structure, the adsorption energies of asymmetrically coordinated single-atom Fe-N3P1 structure for ozone and model contaminant were more negative (Fig. 5e), which indicated that the introduction of P could promote the adsorption and diffusion processes of ozone and organic pollutants. Furthermore, Supplementary Fig. 33 presented that the energy barrier for the reaction between ozone and the Fe-N3P1 site was 1.20 kcal mol−1, which was 30% lower than that of the reaction with the Fe-N4 site (1.71 kcal mol−1). A lower energy barrier allows a greater proportion of molecular collisions to overcome the activation threshold, thereby accelerating the reaction rate. Therefore, the substitution of P for N coordination effectively modulated the electronic structure and spin state of single-atom iron and enhanced catalytic ozonation, and based on the electrostatic effect, the central Fe atom in Fe-N3P1 structure and the terminal O atoms in ozone molecule might be the interaction sites.
Dynamic catalytic ozonation pathways by Fe-N3P1 moiety
The chain dynamic catalytic ozonation pathways by Fe-N3P1 structure were revealed through molecular dynamics (MD) simulations (Supplementary Movies 1–4) and the generation mechanisms of 1O2 were identified by DFT. Figure 6a and Supplementary Fig. 34 showed the first step of catalytic ozonation by Fe-N3P1 structure. In the initial configuration (State 1-1), an ozone molecule was located above the Fe-N3P1 site at a distance of about 5.0 Å. Attracted by the catalyst surface, the ozone molecule gradually moved towards the catalyst surface, and after about 200 fs, the more electrophilic terminal O atoms in ozone were facing downwards (State 1-2). After continuing the reaction for 145 fs, the central Fe atom of Fe-N3P1 site bonded with the terminal oxygen atom O1 of the ozone molecule to form O3-Fe-N3P1 composite structure (State 1-3), which was consistent with the prediction of the DFT calculations. Within the next 75 fs, the O1-O3 bond in the ozone molecule was elongated and then broke, with the result that the terminal oxygen atom O1 bonded with the central Fe atom (O-Fe-N3P1, marked as O species-1) remained on the catalyst surface, while the O2-O3 species (marked as O2 species-1) gradually moved away from the surface and into solution. As demonstrated in Fig. 6b and Eq. 1, combined Mulliken charge and spin population analysis calculated by DFT, the O species-1 was *O with high oxidation ability to attack organics through surface oxidation processes, and the O2 species-1 was triplet oxygen.

a Dynamic interaction process of Fe-N3P1 structure with ozone and (b) Mulliken charges and spin populations of the O species-1 and O2 species-1 in O-Fe-N3P1…O2; c Dynamic interaction process of O-Fe-N3P1 (*O) structure with ozone and (d) Mulliken charges and spin populations of the O2 species-2 and O2 species-3 in Fe-N3P1…O2…O2; e Relative energy changes in the major catalytic processes; f Schematic diagram of catalytic ozonation pathways by Fe-N3P1 structure.
The *O in the O-Fe-N3P1 group was also a key intermediate for further ozone decomposition (Fig. 6c and Supplementary Fig. 35). An ozone molecule was placed above the O-Fe-N3P1 group with a distance of about 4.7 Å, forming the initial configuration of the second step (State 2-1). Influenced by the catalyst surface, the O4-O6 bond in the ozone molecule experienced rapid stretching and broke within 30 fs. In the State 2-3, ozone was decomposed into a lone oxygen atom O4 and a free O5-O6 species (labelled O2 species-2). In the next 20 fs, the lone oxygen atom O4 gradually approached the catalyst surface and then was captured by *O in the O-Fe-N3P1 group, forming a surface-adsorbed O1-O4 species (labelled O2 species-3). After 70 fs, the Fe-O1 bond broke and the O1-O4 species was released into the solution. Figure 6d and Eq. 2 indicated that the O2 species-2 with about 0 charges and 0 spin population might be 1O2, and the O2 species-3 with −0.519 charges and −0.886 spin population might be O2•−. Furthermore, the self-disproportionation reaction of O2•− is another important pathway to generate 1O2 (Eq. 3). Thus, in the second step, 1O2 originated not only from the direct decomposition of ozone, but also from the self-disproportionation of O2•−. The whole catalytic pathways were spontaneous and exothermic (Fig. 6e). The previous experimental results demonstrated that the indirect oxidation pathway of the nonradical 1O2 played a pivotal role in the degradation of organic pollutants.
Although the P atom in the O-Fe-N3P1 structure might serve as a reaction site to decompose ozone (the third step, Supplementary Figs. 36–38 and Eq. 4), in ozone decomposition products, the oxygen atom adsorbed in P site did not have unpaired electrons and presented no oxidizing ability, while the O2 species was 1O2. After the third step of catalytic ozonation, the P site adsorbed an O atom without oxidizing activity, which did not affect the further ozone decomposition by *O adsorbed on Fe site (the fourth step, Supplementary Figs. 39–41). The ozone decomposition products in the fourth step were similar to those in the second step (Eq. 5). Moreover, in the N3P1 /O3 control system (Supplementary Figs. 42-47), although ozone could be decomposed by P site, the resulting O-containing intermediates were chemically inert (See Supplementary Discussion 4 for detailed discussion).
In summary, theoretical calculations revealed that the central Fe atom in Fe-N3P1 moiety served as the primary site to interact with ozone. The short-range coordination effect of P effectively modulated the electronic structure of single-atom Fe, enhancing the adsorption of ozone and organic pollutants. Furthermore, as shown in Fig. 6f, after the first two steps of ozone decomposition, the single-atom Fe-N3P1 catalytic site completed a catalytic ozonation cycle, which was the major ozone decomposition and ROS generation pathways. In the chain catalytic ozonation reactions, 1O2 was generated not only from the decomposition of ozone but also the self-disproportionation of O2•−. The origin of 1O2 was revealed by DFT and MD, consistent with the experimental results.
Discussion
In this work, we successfully synthesized an asymmetrically coordinated single-atom iron (Fe-NPC) catalyst with Fe-N3P1 active sites through a nanocage-based dual-encapsulation strategy, which demonstrated excellent catalytic ozonation performance in wastewater decontamination. The introduction of heteroatom P broke the first-shell coordination environment and effectively regulated the electronic structure of single-atom iron through short-range coordination effect. Fe-NPC exhibited superior catalytic ozonation activity and stability in wastewater treatment. Compared to HCO based on symmetrically coordinated single-atom iron (Fe-NC/O3), the Fe-NPC/O3 system not only achieved complete removal of pHBA but also exhibited a 40% increase in the kinetic rate constant (0.123 min−1). Fe-NPC-based catalytic ozonation also presented excellent advanced treatment of coal chemical wastewater with the COD decreasing from ~95 to ~48 mg L−1. Moreover, the direct oxidation of O3 and the indirect oxidation of 1O2 in the bulk solution were the dominant pathways for organic degradation. Both DFT calculations and MD simulations indicated that the central Fe atom in the Fe-N3P1 moiety was identified as the main catalytic site for ozone decomposition and ROS generation, and the presence of P can effectively modulate the electronic structure of single-atom iron to promote the adsorption of ozone and organic contaminants. The unique design of single-atom iron enabled easy and efficient application in advanced treatment, improving regional water quality and ecological safety. This work provided in-depth insights into the modulation of electronic structure of single-atom metal centers and revealed the catalytic ozonation mechanisms by asymmetrically coordinated SACs at the atomic level, which was of vital significance for the design of advanced heterogenous ozone catalysts.
Methods
Synthesis of Fe-NPC catalysts
Detailed chemicals and reagents are provided in Supplementary Method 1. Single-atom iron catalysts with nitrogen-phosphorus coordination were successfully synthesized by a dual encapsulation strategy using nanocages. Specifically, 10 mmol of Zn(NO3)2•6H2O, 4 mmol of iron(III) acetylacetonate (Fe(acac)3), and 6 mmol of triphenylphosphine (PPh3) were dissolved in 100 mL of MeOH to obtain solution A, and 80 mmol of 2-methylimidazole (2-MI) was dissolved in another 100 mL of MeOH to obtain solution B. At normal temperature and pressure, solution A was quickly poured into solution B and the mixture was stirred continuously for 1 h before standing for 24 h. After centrifugation and washing, FeP@ZIF-8 precursors were fabricated. The precursors were calcined at 1000 °C for 3 h under Ar atmosphere. Zn species evaporated during pyrolysis, resulting in the synthesis of Fe-NPC catalysts. Fe-NPC-x series were obtained by varying the doping amount of P, where x denoted the concentration of PPh3. According to whether Fe or P sources were added, the control precursors were named ZIF-8, P@ZIF-8, and Fe@ZIF-8, and the corresponding catalysts were labelled as NC, NPC, and Fe-NC.
Characterization methods
High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of Fe-NPC was taken on a FEI Titan 80-300 instrument. X-ray absorption fine structure (XAFS) measurements were performed to identify the valence and local coordination environment of single-atom iron in Fe-NPC. Electrochemical tests were performed on a workstation (PGSTAT-128N, Autolab, Switzerland & VersaSTAT, Princeton, USA) with a conventional three-electrode cell system40,41. The signals of •OH, O2•−, and 1O2 were detected by electron paramagnetic resonance (EPR, JEOL FA-200) spectroscopy with DMPO and TEMP as the spin trapping agents42,43. The signals of surface-adsorbed ROS were recorded by an in-situ Raman spectrometer (HORIBA HR) at 532 nm11,44. Other characterization methods were shown in Supplementary Method 2.
Catalytic ozonation experiments
Typically, for the degradation of model contaminant pHBA, 0.20 g L−1 of catalysts were added into 250 mL of simulated wastewater with 50 mg L−1 of pHBA. Then, a gas mixture of ozone and oxygen was immediately introduced to initiate the catalytic ozonation, where the ozone concentration was 5.5 mg L−1 in the mixture gas and the gas flow rate was 0.2 L min−1. A Fe-NPC-PVDF membrane-coupled catalytic oxidation reactor was operated continuously for 48 h. The system was designed with two separate inlet channels: one for a 20 mg L−1 pHBA solution and the other for a saturated ozone solution with a concentration of approximately 5 mg L−1. The two streams were mixed and introduced into the membrane reactor, where ozone activation and pollutant degradation occurred. The effective filtration area of the membrane reactor was approximately 7 cm2, and the catalyst loading was 1.6 mg cm−2. In the OA degradation experiment by Fe-NPC/O3, the initial OA concentration was 100 mg L−1, the catalyst dosage was 0.05 g L−1, the gas flow rate was 0.2 L min−1, the ozone concentration at the inlet was 10 mg L−1, and the pH was about 3.2. Quenching tests were carried out with TBA, MeOH, DMSO, and FFA as the scavengers, and the concentrations were all 2 mM. In the advanced treatment of real wastewater, biotreated coal chemical wastewater taken from a coal chemical wastewater treatment plant in northern China was employed as the refractory organic wastewater. The COD of the wastewater sample was about 95 mg L−1, the conductivity was about 5200 μS cm−1, and the pH was about 8.0. The catalyst dosage was adjusted to 0.50 g L−1 and the ozone concentration in the gas mixture was 10.0 mg L−1 in the coal wastewater treatment. During the treatment of simulated or real wastewater, the samples were taken at certain time intervals and filtered for further detection. The detailed water analysis methods were shown in Supplementary Method 3.
Computational methods
Gaussian 16 software was used to perform density functional theory (DFT) calculations38,45. CP2K v9.1 software with Quickstep module and MOLOPT basis set was used to carry out Ab initio molecular dynamics (AIMD) simulations of catalytic ozonation pathways46,47,48,49. Details were shown in Supplementary Method 4.
Data availability
The source data generated in this study are provided in the Source Data file. Data are available from the corresponding author upon request. Source data are provided with this paper.
Code availability
No custom code was developed for this study.
References
Shannon, M. A. et al. Science and technology for water purification in the coming decades. Nature 452, 301–310 (2008).
Griggs, D. et al. Sustainable development goals for people and planet. Nature 495, 305–307 (2013).
Qu, J. & Chen, J. Pathways toward a pollution-free planet and challenges. Front. Environ. Sci. Eng. 18, 67 (2024).
Huo, Z.-Y., Wang, X., Huang, X. & Elimelech, M. Intensifying electrified flow-through water treatment technologies via local environment modification. Front. Environ. Sci. Eng. 18, 69 (2024).
Miklos, D. B. et al. Evaluation of advanced oxidation processes for water and wastewater treatment – A critical review. Water Res. 139, 118–131 (2018).
Hodges, B. C., Cates, E. L. & Kim, J.-H. Challenges and prospects of advanced oxidation water treatment processes using catalytic nanomaterials. Nat. Nanotechnol. 13, 642–650 (2018).
Huang, H., Ma, R. & Ren, H. Scientific and technological innovations of wastewater treatment in China. Front. Environ. Sci. Eng. 18, 72 (2024).
Yu, G., Wang, Y., Cao, H., Zhao, H. & Xie, Y. Reactive oxygen species and catalytic active sites in heterogeneous catalytic ozonation for water purification. Environ. Sci. Technol. 54, 5931–5946 (2020).
Wei, K. et al. A hybrid fluidized-bed reactor (HFBR) based on arrayed ceramic membranes (ACMs) coupled with powdered activated carbon (PAC) for efficient catalytic ozonation: A comprehensive study on a pilot scale. Water Res. 173, 115536 (2020).
Zhou, Z. et al. Catalytic ozonation in advanced treatment of kitchen wastewater: multi-scale simulation and pilot-scale study. Front. Environ. Sci. Eng. 17, 146 (2023).
Wang, Y. et al. Mechanistic investigations of the pyridinic N–Co structures in Co embedded N-doped carbon nanotubes for catalytic ozonation. ACS EST Eng. 1, 32–45 (2020).
Cao, H. et al. Different roles of Fe atoms and nanoparticles on g-C3N4 in regulating the reductive activation of ozone under visible light. Appl. Catal. B: Environ. 296, 120362 (2021).
Cheng, Y. et al. Surface oxygen vacancies prompted the formation of hydrated hydroxyl groups on ZnO in enhancing interfacial catalytic ozonation. Appl. Catal. B: Environ. 341, 123325 (2024).
Wang, Y. et al. Occurrence of both hydroxyl radical and surface oxidation pathways in N-doped layered nanocarbons for aqueous catalytic ozonation. Appl. Catal. B: Environ. 254, 283–291 (2019).
Zhang, J. et al. Roles of oxygen-containing functional groups of O-doped g-C3N4 in catalytic ozonation: Quantitative relationship and first-principles investigation. Appl. Catal. B: Environ. 292, 120155 (2021).
Wang, Y., Duan, X., Xie, Y., Sun, H. & Wang, S. Nanocarbon-based catalytic ozonation for aqueous oxidation: engineering defects for active sites and tunable reaction pathways. ACS Catal. 10, 13383–13414 (2020).
Liu, Y., Chen, C., Duan, X., Wang, S. & Wang, Y. Carbocatalytic ozonation toward advanced water purification. J. Mater. Chem. A 9, 18994–19024 (2021).
Chen, S. et al. MnNx-Carbon-Silica-Framework for highly efficient heterogeneous catalytic ozonation of electron-rich organics through nonradical pathway. Chem. Eng. J. 466, 143110 (2023).
Wei, K. et al. Zhang, 2D-Like catalyst with a micro-nanolinked functional surface for water purification. Environ. Sci. Technol. 58, 3007–3018 (2024).
Yang, X.-F. et al. Single-atom catalysts: A new frontier in heterogeneous catalysis. Acc. Chem. Res. 46, 1740–1748 (2013).
Wu, X. & Kim, J.-H. Outlook on single atom catalysts for persulfate-based advanced oxidation. ACS EST Eng. 2, 1776–1796 (2022).
Cheng, Y., Chen, Z., Wang, S. & Duan, X. Single atom catalysts for heterogeneous catalytic ozonation. Curr. Opin. Chem. Eng. 41, 100945 (2023).
Chen, F. et al. Embedding electronic perpetual motion into single- atom catalysts for persistent Fenton- like reactions, proceedings of the national academy of sciences of the united states of america, 121 (2024).
Guo, Z. et al. Single-atom Mn–N4 site-catalyzed peroxone reaction for the efficient production of hydroxyl radicals in an acidic solution. J. Am. Chem. Soc. 141, 12005–12010 (2019).
Wang, J. et al. Manipulating selectivity of hydroxyl radical generation by single-atom catalysts in catalytic ozonation: Surface or solution. Environ. Sci. Technol. 56, 17753–17762 (2022).
Ren, T. et al. Single-atom Fe-N4 sites for catalytic ozonation to selectively induce a nonradical pathway toward wastewater purification. Environ. Sci. Technol. 57, 3623–3633 (2023).
Fan, M. et al. Improving the catalytic activity of carbon-supported single atom catalysts by polynary metal or heteroatom doping. Small 16, 1906782 (2020).
Wan, J. et al. In situ phosphatizing of triphenylphosphine encapsulated within metal–organic frameworks to design atomic Co1–P1N3 interfacial structure for promoting catalytic performance. J. Am. Chem. Soc. 142, 8431–8439 (2020).
Xu, W. et al. Research progress of asymmetrically coordinated single-atom catalysts for electrocatalytic reactions. J. Mater. Chem. A 10, 14732–14746 (2022).
Zhou, X. et al. Identification of Fenton-like active Cu sites by heteroatom modulation of electronic density. Proc. Natl Acad. Sci. USA 119, e2119492119 (2022).
Wang, F. et al. Modulating electronic structure of atomically dispersed nickel sites through boron and nitrogen dual coordination boosts oxygen reduction. Adv. Funct. Mater. 33, 2213863 (2023).
Li, R. & Wang, D. Understanding the structure–performance relationship of active sites at atomic scale. Nano Res. 15, 6888–6923 (2022).
Zhang, L. et al. On the coordination environment of single-atom catalysts. Acc. Chem. Res. 58, 1878–1892 (2025).
Li, Z. et al. Iridium single-atom catalyst on nitrogen-doped carbon for formic acid oxidation synthesized using a general host-guest strategy. Nat. Chem. 12, 764–772 (2020).
Li, Z. et al. Robust ring-opening reaction via asymmetrically coordinated Fe single atoms scaffolded by spoke-like mesoporous carbon nanospheres, Proceedings of the National Academy of Sciences of the United States of America, 120 (2023).
Yang, Z., Qian, J., Yu, A. & Pan, B. Singlet oxygen mediated iron-based Fenton-like catalysis under nanoconfinement. Proc. Natl Acad. Sci. USA 116, 6659–6664 (2019).
Zhao, Y. et al. Janus electrocatalytic flow-through membrane enables highly selective singlet oxygen production. Nat. Commun. 11, 6228 (2020).
Ouyang, C., Wei, K., Huang, X., El-Din, M. G. & Zhang, X. Bifunctional Fe for induced graphitization and catalytic ozonation based on a Fe/N-doped carbon-Al2O3 framework: Theoretical calculations guided catalyst design and optimization. Environ. Sci. Technol. 55, 11236–11244 (2021).
Zhang, P., Yang, Y., Duan, X., Liu, Y. & Wang, S. Density functional theory calculations for insight into the heterocatalyst reactivity and mechanism in persulfate-based advanced oxidation reactions. ACS Catal. 11, 11129–11159 (2021).
Zhang, X. et al. Iron-based clusters embedded in nitrogen doped activated carbon catalysts with superior cathodic activity in microbial fuel cells. J. Mater. Chem. A 8, 10772–10778 (2020).
Yin, M. et al. Boosting wastewater bioelectricity recovery via solvent mediation and zinc fencing: Dual regulation for catalyst spatial structure and active sites. Chem. Eng. J. 453, 139276 (2023).
Finkelstein, E., Rosen, G. M. & Rauckman, E. J. Spin trapping: Kinetics of the reaction of superoxide and hydroxyl radicals with nitrones. J. Am. Chem. Soc. 102, 4994–4999 (1980).
Shi, H. L. et al. Evaluation of spin trapping agents and trapping conditions for detection of cell-generated reactive oxygen species. Arch. Biochem. Biophysics 437, 59–68 (2005).
Zhang, T., Li, W. & Croué, J.-P. Catalytic ozonation of oxalate with a cerium supported palladium oxide: An efficient degradation not relying on hydroxyl radical oxidation. Environ. Sci. Technol. 45, 9339–9346 (2011).
Yu, G. et al. Insights into the mechanism of ozone activation and singlet oxygen generation on N-doped defective nanocarbons: A DFT and machine learning study. Environ. Sci. Technol. 56, 7853–7863 (2022).
Kuehne, T. D. et al. CP2K: An electronic structure and molecular dynamics software package - Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 152, 194103 (2020).
Wang, Y.-G., Mei, D., Glezakou, V.-A., Li, J. & Rousseau, R. Dynamic formation of single-atom catalytic active sites on ceria-supported gold nanoparticles. Nat. Commun. 6, 6511 (2015).
Liu, J.-C., Tang, Y., Chang, C.-R., Wang, Y.-G. & Li, J. Mechanistic insights into propene epoxidation with O2–H2O mixture on Au7/α-Al2O3: A hydroproxyl pathway from ab initio molecular dynamics simulations. ACS Catal. 6, 2525–2535 (2016).
Ren, T. et al. Mn-doped carbon-Al2SiO5 fibers enable catalytic ozonation for wastewater treatment: Interface modulation and mass transfer enhancement. J. Hazard. Mater. 460, 132307 (2023).
Acknowledgements
This research was supported by the Jing-Jin-Ji Regional Integrated Environmental Improvement-National Science and Technology Major Project (2024ZD1200503) awarded to X.Z., National Natural Science Foundation of China (52470082) awarded to X.Z., and Project of Establishing the National Independent Innovation Demonstration of Hohhot, Baotou and Ordos (2023XM07) awarded to X.Z. This research was supported by “Explorer 1000” cluster system of Tsinghua HPC Platform.
Author information
Authors and Affiliations
Contributions
T.R. and X.Z. conceived the project design. T.R., K.L. and T.F. performed the experiments. T.R., H.R. and N.Y. conducted theoretical simulations and analyzed data. J.M. performed the In-situ Raman characterization. X.H. and X.Z. supervised the study and acquired funding. T.R. and X.Z. prepared the paper with critical revisions from all co-authors.
Corresponding author
Ethics declarations
Competing interests
A related patent (pending) about the catalyst design has been applied on behalf of Tsinghua University. X. Zhang, T. Ren, and X. Huang are listed as inventors. Other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ren, T., Lu, K., Tao, F. et al. Phosphorus-induced single-atom iron coordination symmetry disruption for superior catalytic ozonation. Nat Commun 16, 9037 (2025). https://doi.org/10.1038/s41467-025-64099-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64099-2
