
Abstract
Ethylene (C2H4), a cornerstone of the chemical industry, is produced predominantly via fossil-intensive high-temperature processes that contribute significantly to global energy consumption and CO2 emissions. Here, we report an ambient bipolar C2H4 electrosynthesis system that concurrently decarboxylates propanoic acid, a prevalent biorefinery waste, at nanoporous Pt microparticles-coated anode and reduces CO2 at W-doped CuOx-loaded cathode. Physicochemical and operando spectroscopy characterizations, along with theoretical modeling reveal that the polarized Pt-PtO2 interface formed in situ downshifts the d-band relative to Fermi level which favors the desorption of *CH2CH2 intermediate to promote selective propanoic acid decarboxylation toward C2H4. Remarkably, the resulting electrocatalyst couple delivers an unprecedented C2H4 faradaic efficiency (FEC2H4) of 118.7% and a large current density of 1000 mA cm−2, and sustains a FEC2H4 exceeding 103.4% for over 265 h at an industrial current density of 400 mA cm−2, offering a promising pathway to carbon-neutral C2H4 production from waste feedstocks.
Similar content being viewed by others

Introduction
Ethylene (C2H4), the simplest olefin, is a fundamental building block in the chemical industry for producing plastics, resins, fibers, rubbers, and packaging materials, with a global production capacity of 227.6 million metric tons (MMTs) in 20231,2,3,4,5,6,7,8. In fact, annual C2H4 production has been used as a metric to measure the economic activity of a country in petrochemical industry8. Currently, C2H4 is produced primarily by high-temperature steam cracking (>750 °C), and (oxidative) dehydrogenation (300–500 °C) of hydrocarbons from nonrenewable fossil fuels2,3,4,5,6,7,8, leading to high energy consumption and increased global CO2 emissions of >366 MMTs per year. Recently, catalytic dehydration of ethanol has been put forward for C2H4 generation because of the relatively low reaction temperature (200–250 °C) and the availability of bioethanol derived from sustainable biomass8. Although such a scenario can alleviate our dependence on fossil fuels and promote industrial decarbonization partly, the bioethanol production via biomass refinery faces new challenges of food safety, circumstance, and economy, like high cost, intensive energy input, and massive biowastes (e.g, carboxylic acids) discharge9.
Against this backdrop, renewable electricity-driven ambient CO2 reduction offers a green route for C2H4 generation, and a myriad of multiscale studies have focused on developing novel electrocatalysts, electrodes, electrolytes, and electrolyzers to improve the selectivity and faradaic efficiency of C2H4 at cathode10,11,12,13,14,15,16. In most cases, however, the oxidation reactions at anode yield low-value byproducts (e.g., oxygen for water oxidation, and water for hydrogen oxidation) or chemicals (e.g., formate for glycerol oxidation) other than C2H417,18,19,20. All the reported CO2 electrolysis systems can produce C2H4 at only the cathode side, and their faradaic efficiencies (FEC2H4) are mostly low to 85% under industrial current densities (>200 mA cm−2)10,11,12,13,14,15,16, provoking a low electron economy toward C2H4 production. In this context, innovative electrochemistry paradigms are required to effectively produce C2H4, especially using only wastes, at both cathode and anode.
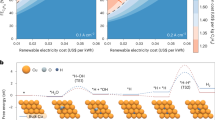
Herein, we report an unconventional bipolar C2H4 electrosynthesis system that couples oxidative decarboxylation of propanoic acid (PA), a typical biowaste acid in biomass refinery9, with CO2 reduction (Fig. 1a). This electrosynthesis system, which operates under ambient conditions, can convert two wastes of PA and CO2 to the same C2H4 (Fig. 1a) with an unprecedented total FEC2H4 of 118.7%, a large current density of 1000 mA cm−2, and robust stability over 265 h at an industrial current density of 400 mA cm−2, using the screened nanoporous Pt microparticles-coated anode and W-doped CuOx-loaded cathode. Combined operando characterizations and theoretical calculations reveal that the polarized Pt-PtO2 interface formed in situ and W-stabilized Cu+ account for efficient PA decarboxylation and CO2 reduction toward C2H4, respectively. This electrosynthesis paradigm for sustainable C2H4 production from both wastes not only maximizes the electron economy, but also gain both economic and ecological benefits.

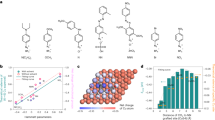
a Schematic of a renewable energy-driven bipolar C2H4 electrosynthesis system from two wastes, CO2 and PA, by pairing electrocatalytic CO2 reduction and PA decarboxylation. b Possible reaction pathways of PA decarboxylation in alkaline solution. c Anode electrocatalyst screening for PA decarboxylation toward C2H4. Source data are provided as a Source data file.
Results
Anode electrocatalysts screening
In view of multiple oxidative pathways of carboxylic acids including dimerization (Kolbe route), disproportionation, deprotonation (non-Kolbe route), Hofer–Moest route, esterification and others (Fig. 1b)21,22,23,24,25, and promising activities of typical transition metal-based materials for oxidation reactions25, we systematically screened eight commercial metal foils (Pt, Ir, Ru, Au, Pd, Fe, Co, Ni) to identify optimal anodes for electrochemical propionic acid (PA) decarboxylation toward C2H4. This screening was conducted in an H-cell containing 2.0 M PA (pH 10.0 ± 0.2) with a mixed electrolyte of KOH (1.5 M) and K3PO4 (0.5 M), using a typical three-electrode configuration (see “Methods” for details). After galvanostatic electrolysis at different current densities, the resulting gas and liquid products were analyzed via gas chromatography and 1H NMR, respectively (Supplementary Fig. 1). As shown in Fig. 1c, noble metal foils commonly exhibit higher FEC2H4 than non-noble metal ones at all the investigated current densities, and Pt foil specifically achieves the highest FEC2H4 among the eight screened metals. Typically, the FEC2H4 for Pt foil is high to 53.7% at a current density of 100 mA cm−2, and the total FEs of C2 products, such as C2H5OH, CH3COOH, and C2H4 reach 79.4% (Supplementary Table 1). Although such a low current density cannot fully fulfill industrial requirements, the decent FEC2H4 inspired us to further fabricate Pt nanomaterials with highly accessible surface areas via nanostructure engineering.
We then used a facile template-free electrodeposition method to synthesize nanoporous Pt microspheres on Ti foil (np-Pt). Scanning electron microscopy (SEM) images of np-Pt reveal a sphere-like morphology with particle sizes of approximately 1.5–2.3 μm and abundant macropores with diameters of about 90 nm on the surface (Supplementary Fig. 2). Such plentiful porosity can facilitate Pt active sites exposure and mass transfer. X-ray diffraction (XRD) pattern of np-Pt indicates the metallic Pt phase (PDF: 04-0802, Supplementary Fig. 3). Transmission electron microscopy (TEM) image also confirms the sphere-like morphology of np-Pt (Supplementary Fig. 4a). High-resolution TEM (HR-TEM) image of np-Pt shows a lattice fringe of 0.222 nm, assignable to the Pt(111) facet (Supplementary Fig. 4b). Analyzing the electrochemically active surface area (ECSA), derived from double-layer capacitance (Cdl), reveals a 3.7-fold higher Cdl of np-Pt than commercial Pt foil (Supplementary Fig. 5). This comparison signifies the enlarged accessible surface area of np-Pt, beneficial to active sites exposure and mass transfer for improved PA decarboxylation. Additionally, the smaller semicircular diameter in the electrochemical impedance spectrum (Supplementary Fig. 6) for np-Pt relative to Pt foil uncovers smaller charge transfer resistance and thus improved PA decarboxylation kinetics. To obtain optimal C2H4 production from PA over np-Pt, electrochemical parameters, including PA concentration and pH, were also screened. The heatmaps intuitively demonstrate that a PA concentration of 2.0 M and an electrolyte pH of 10.0 ± 0.2 are most favorable for C2H4 generation over np-Pt in an H-cell under the investigated current density range (Fig. 2a, b), with the highest FEC2H4 of 60.6% at 200 mA cm−2 (Supplementary Table 2). It is anticipated that initial increase in PA concentration from 0.5 to 2.0 M promotes the coverage of PA molecules over active sites to accelerate the decarboxylation kinetics, leading to higher FEC2H4. While further increasing PA concentration to 3.0 M results in high solution viscosity and thus deteriorated mass transfer, so that lower FEC2H4 is obtained. As for the pH effect, increasing pH over the pKa (4.87) of PA leads to its deprotonation to form propionate anions (CH3CH2COO−), which favors adsorption over active sites, and moderate alkalinity further promotes carbocation formation via the second electron transfer in non-Kolbe reactions25, collectively improving FEC2H4. However, when pH exceeds 10.0 ± 0.2 to 12.0 ± 0.2, excessive OH− competes with propionate anions over active sites to boost oxygen evolution side-reaction and suppress PA decarboxylation as well, leading to reduced FEC2H4. Significantly, the resulting np-Pt shows much higher FEC2H4 under all the applied current densities than commercial Pt foil (Fig. 2c). The superior PA-to-C2H4 activity of np-Pt over Pt foil is also ascertained by the larger partial current density of C2H4 (JC2H4) even under lower applied potentials (Fig. 2d). For example, the np-Pt electrocatalyst achieves a JC2H4 of 179.1 mA cm−2 at ~2.7 V vs the reversible hydrogen electrode (RHE). In contrast, the Pt foil affords a much lower JC2H4 of <80.3 mA cm−2 at a similar potential. These comparisons underscore the positive role of high porosity in np-Pt.

FEC2H4 heatmaps of a PA concentration and b electrolyte pH for anodic PA decarboxylation over np-Pt electrocatalyst in an H-cell. c FEC2H4 under different potentials over np-Pt and Pt foil in an H-cell. d JC2H4 over np-Pt and Pt foil under different potentials with 85% iR correction in an H-cell. e JC2H4 over np-Pt under different potentials with 85% iR correction in a continuous-flow cell. f FEC2H4 over np-Pt under different current densities in a continuous-flow cell. Error bars represent the standard deviation of three independent samples. Data are presented as mean values ± standard deviation. Source data are provided as a Source data file.
To further accelerate mass transfer, a continuous-flow cell instead of an H-cell was utilized to evaluate the PA-to-C2H4 performance of np-Pt with an electrode area of 1.0 cm−2. Figure 2e shows the JC2H4 at different potentials. The np-Pt can afford a JC2H4 of 527.9 mA cm−2 at ~3.8 V vs RHE. Moreover, the FEC2H4-current density plot reveals that np-Pt achieves a remarkable FEC2H4 of 58.8% at a large current density of 400 mA cm−2 (Fig. 2f and Supplementary Table 3). Even at a large current density of up to 1000 mA cm−2, a high FEC2H4 of 52.8% can still be achieved for np-Pt. Notably, such a high FEC2H4 with a large current density is suitable for industrial electrosynthesis of C2H4 from PA waste.
Oxidative decarboxylation mechanism
To understand the superior performance of np-Pt for PA decarboxylation to C2H4, we performed a series of physiochemical characterizations and density functional theory (DFT) calculations. Although the overall XRD pattern of np-Pt after PA decarboxylation at 100 mA cm−2 for 1 h (denoted as post np-Pt) indicates the inheritance of the metallic Pt phase (Fig. 3a), the enlarged pattern exhibits a slight shift of Pt(111) facet toward small diffraction angle, indicating the increased interplanar spacing. This shift suggests the possible surface oxidation of np-Pt during PA decarboxylation, which induces the incorporation of oxygen atoms into interstitial sites within the Pt lattice. Furthermore, high-resolution Pt 4f XPS spectrum of post np-Pt exhibits significant blueshift in the binding energy compared with that of the pristine counterpart (Fig. 3b), indicative of the higher valance state of the former26. Moreover, synchrotron X-ray absorption near-edge structure (XANES) analysis at the Pt L3-edge indicates that the white line peak for post np-Pt is located between those for Pt foil and PtO2 references (Fig. 3c)27, suggesting the oxidative valence state of Pt in post np-Pt as well. HR-TEM image of post np-Pt shows a lattice fringe of 0.250 nm ascribed to (101) plane of PtO2 (PDF: 37-1087), in addition to the interplanar spacing of 0.228 nm assignable to Pt(111) plane (PDF: 04-0802) (Fig. 3d). Elemental mapping images of post np-Pt reveal the presence of appreciable O with a surface-enriched configuration (Fig. 3e). These ex-situ characterizations clearly demonstrate the partial self-oxidation of pristine np-Pt during PA decarboxylation, which is further corroborated by quasi-in situ electrochemical Raman spectroscopy. Figure 3f and Supplementary Fig. 7 show the Raman spectra evolution of pristine np-Pt during PA decarboxylation at 100 mA cm−2 from 0 to 60 min. The characteristic Raman peak at 567.9 cm−1 corresponding to α-PtO2 gradually appears28,29, confirming the oxidation of np-Pt again. Taken together, these results suggest that the in situ-formed Pt-PtO2 interface species serve as real electrocatalytic active sites for PA decarboxylation.

a XRD patterns of post np-Pt and fresh np-Pt. b High-resolution Pt 4 f XPS spectra of fresh np-Pt and post np-Pt. c XANES spectra of post np-Pt, and Pt foil and PtO2 references at Pt L3-edge. d HR-TEM image of post np-Pt. e HAADF-STEM and the corresponding elemental (Pt, O) mapping images of post-np-Pt. f Quasi-in situ Raman spectra of np-Pt after electrolysis at 100 mA cm−2 in 2.0 M PA electrolyte (pH = 10.0 ± 0.2) for different time. Source data are provided as a Source data file.
We then carried out operando electrochemical attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS, Fig. 4a and Supplementary Fig. 8a) to detect the reactive intermediates during PA decarboxylation over np-Pt by varying the applied potential from 1.1 to 2.9 V vs RHE30,31,32. Along with positive potential scanning, an upward absorption band at 1048 cm−1 attributed to the typical C−O vibration of alcohol molecules becomes visible, confirming the formation of ethanol. The band at 1306 cm−1 assignable to the typical C−O stretching of aldehyde or carboxyl groups, along with two bands corresponding to symmetrical (1414 cm−1) and asymmetric (1578 cm−1) stretching of O−C−O collectively prove the formation of acetic acid (CH3COOH). Additionally, the in-plane bending vibration of C−H in methyl (CH3) at 1378 cm−1 and methylene (CH2) at 1460 cm−1 can be also observed. In particular, the band at 1442 cm−1 belonging to the adsorbed *CH2CH2 intermediate is observable, suggestive of the C2H4 formation. We notice that the band at 1714 cm−1 attributable to stretching vibration of adsorbed CHO* becomes visible firstly and then disappears rapidly during the positive potential scanning, indicating the formation of acetaldehyde (CH3CHO) and subsequent deep oxidation to CH3COOH at high potentials. These operando ATR-SEIRAS results exactly confirm that the PA decarboxylation is catalyzed by np-Pt toward C2H4 target along with ethanol, acetaldehyde, and acetate byproducts, in sharp contrast to that in the PA-free electrolyte, where only a band attributed to *OH at 1610 cm−1 can be observed (Fig. 4b and Supplementary Fig. 8b).

Operando ATR-SEIRA spectra of np-Pt in the electrolyte a with and b without 2.0 M PA during positive potential scanning. c Differential charge density distribution of PtO2@Pt(111). Yellow and cyan contours represent charge accumulation and depletion, respectively. d The projected DOS of Pt(111) and PtO2@Pt(111). e Configurations of various reactive species over PtO2@Pt(111) following the non-Kolbe pathway toward C2H4. f Gibbs free energy diagrams of PA decarboxylation to various products over PtO2@Pt(111). g Gibbs free energy diagrams of PA decarboxylation to C2H4 via the non-Kolbe pathway over Pt(111) and PtO2@Pt(111). Source data are provided as a Source data file.
To further elucidate the promoted PA-to-C2H4 activity of np-Pt, we performed DFT calculations33,34. On the basis of the XRD (Fig. 3a) and Raman (Fig. 3f) results, the lowest energy facet of PtO2@Pt(111) was used as a simplified model for np-Pt (Fig. 4c, Supplementary Fig. 9, and Supplementary Data 1), and Pt(111) was also included for comparison (Supplementary Fig. 10). As plotted in Fig. 4c, the differential charge density distributions of PtO2@Pt(111) designate an obvious interfacial electron transfer from Pt(111) to PtO2, with a net 1.71 electrons35,36. The projected density of states (DOS) analysis of the Pt atoms reveals a downshift of the d-band relative to Fermi level (EF) from −1.82 eV for Pt(111) to −2.55 eV for PtO2@Pt(111), as shown in Fig. 4d. These results indicate that the in-situ formation of polarized Pt-PtO2 interface during PA decarboxylation leads to an altered electronic structure and thus intermediate adsorption, which accounts for the improved PA-to-C2H4 activity. In view of the extremely low yield of ethane (C2H6) (Supplementary Table 3), C2H4 is generated mainly via the non-Kolbe rather than the disproportionation route (Fig. 1b), and the corresponding configurations over PtO2@Pt(111) for PA-to-C2H4 conversion through the non-Kolbe pathway are shown in Fig. 4e. To gain insight into the high selectivity of PtO2@Pt(111) toward C2H4, the configuration of PA decarboxylation to various products, including C2H4, C2H6, C4H10, CH3CH2OH, CH3CHO, and CH3COOH over PtO2@Pt(111) were optimized (Supplementary Fig. 11) and the relevant Gibbs free energy diagrams were calculated (Fig. 4f). Initially, the propionate derived from PA in alkaline electrolyte is adsorbed on PtO2@Pt(111) and then decarboxylated to yield *CH2CH3. To proceed, the *CH2CH3 can be deprotonated to *CH2CH2 followed by desorption to generate the C2H4 target product, with the greatest energy uphill of 0.40 eV. Alternatively, the *CH2CH3 is either attacked by hydroxide to yield CH3CH2OH or coupled with another *CH2CH3 to produce C4H10 and C2H4/C2H6 via the Kolbe route and disproportionation, respectively. The generated CH3CH2OH can be adsorbed on PtO2@Pt(111) again and further oxidized to CH3CHO and CH3COOH. Noticeably, the highest energy uphill for C2H4 via non-Kolbe route (0.40 eV) is much lower than those for CH3CH2OH (0.50 eV), and C2H4/C2H6 (via disproportionation pathway), C4H10, and CH3COOH (0.71 eV), suggesting that C2H4 generation on PtO2@Pt(111) via the non-Kolbe route is preferable. To accentuate the critical role of the in situ formed Pt-PtO2 interface, we also calculated the Gibbs free energy diagrams of PA-to-C2H4 via the non-Kolbe route over the Pt(111) facet (Fig. 4g and Supplementary Fig. 12). It is found that the in situ formed Pt-PtO2 interface favors the desorption of *CH2CH2 with a free energy increase of 0.11 eV due to the downshift of the d-band (Fig. 4d), in sharp contrast to that for the Pt(111) facet (0.80 eV). Hence, these DFT calculations collectively validate that the np-Pt featuring unique Pt-PtO2 interface formed in situ facilitates highly selective PA-to-C2H4 conversion, in line with the electrochemical and operando ATR-SEIRAS results.
Bipolar ethylene electrosynthesis
We sought to develop a bipolar electrosynthesis system by pairing anodic PA decarboxylation and cathodic CO2 reduction in a continuous-flow electrolyzer to simultaneously generate C2H4 at both electrodes. To achieve this proof-of-concept, we constructed a flow cell with tungsten doped copper oxides (W-CuOx) and np-Pt as the cathode and anode electrocatalysts, respectively (Fig. 5a). 1.0 M KOH aqueous solution was used as catholyte, and 2.0 M PA (pH = 10.0 ± 0.2) in a mixture of KOH (1.5 M) and K3PO4 (0.5 M) was added to the anode chamber as anolyte. Both cathode and anode chambers were isolated by a piece of anion exchange membrane (AEM). Carbon paper was employed as a gas diffusion layer to support the W-CuOx electrocatalyst.

a Schematic of a bipolar C2H4 electrosynthesis system by pairing CO2 reduction and PA decarboxylation. b FEC2H4 and JC2H4 over W-CuOx for cathodic CO2 reduction under different current densities in a continuous-flow cell. c Stability test of W-CuOx over 100 h in 1.0 M KOH (pH = 14.0 ± 0.2) for CO2 reduction at a current density of −400 mA cm−2. d FEC2H4 over W-CuOx for CO2 reduction and np-Pt for PA decarboxylation under different current densities or voltages with 85% iR correction in a paired continuous-flow cell. e C2H4 production comparison of our paired electrosynthesis system with reported traditional CO2 reduction electrolyzers. f Stability test of the paired electrosynthesis system over 265 h at a current of 400 mA cm−2. g TEA of bipolar C2H4 production in the paired electrosynthesis system at different current densities. Error bars represent the standard deviation of three independent samples. Data are presented as mean values ± standard deviation. Source data are provided as a Source data file.
The cathodic electrocatalyst of W-CuOx was prepared via an in situ electrochemical reconstruction method (see “Methods” for details). A series of physicochemical characterizations, including XRD (Supplementary Fig. 13), operando Raman spectroscopy (Supplementary Fig. 14), XPS (Supplementary Fig. 15), and HR-TEM (Supplementary Fig. 16) corroborate the successful W doping and Cu+ enrichment in W-CuOx due to the electron-attracting property of high valence W cation. The present Cu+ is believed to favor C‒C coupling during CO2 reduction to produce C2+ chemicals including C2H4. Electrochemical measurements reveal that the W-CuOx electrocatalyst achieves a 63.4% FEC2H4 at −800 mA cm−2 (Fig. 5b, Supplementary Fig. 17, Supplementary Fig. 18, and Supplementary Table 4), which is significantly greater than that of the CuOx control (49.4%, Supplementary Fig. 18 and Supplementary Table 5). Additionally, the W-CuOx electrocatalyst can afford a higher JC2H4 of −544.6 mA cm−2 at −0.90 V vs RHE than CuOx control (−143.8 mA cm−2 at −0.93 V vs RHE), as depicted in Supplementary Fig. 18c. These comparisons elucidate the preference of C2H4 generation over W-CuOx, which is further verified by operando ATR-SEIRAS characterizations (Supplementary Fig. 19). The potential-dependent operando ATR-SEIRA spectra demonstrate the more favorable adsorption of CO (*CO), a critical intermediate for C2H4 generation over W-CuOx than CuOx14. In addition to high CO2-to-C2H4 activity, the W-CuOx also exhibits robust durability, as revealed by a stable chronopotentiometric curve at −400 mA cm−2 and a relatively invariant FEC2H4 of 50.7% for 100 h (Fig. 5c).
With the high FEC2H4 for both PA decarboxylation and CO2 reduction in hand, we then paired them together for simultaneous C2H4 generation from PA and CO2 wastes, with an unprecedented faradaic efficiency exceeding 100%. Figure 5d and Supplementary Fig. 20 show that the paired electrosynthesis system can reach a current density of 1000 mA cm−2 at a cell voltage of ~4.9 V, and the FEC2H4 of >107.0% can be achieved at the current density greater than 400 mA cm−2. Notably, a maximum FEC2H4 of 118.7% can be obtained at a current density of 800 mA cm−2 (see Supplementary Table 6 and 7 for details). Specifically, the paired electrosynthesis system can still reach a high FEC2H4 of 109.8% at 1000 mA cm−2, highlighting the great feasibility of converting two wastes of PA and CO2 to valuable C2H4. It is worth noting that such an outstanding performance of our paired electrosynthesis system is far superior to those of the reported C2H4 electrosynthesis systems from merely CO2 based on Cu NN (FEC2H4 of 83% at 212 mA cm−2)37, CuAl (FEC2H4 of 80% at 400 mA cm−2)13, SS-Cu (FEC2H4 of 80% at 710 mA cm−2)38, and others (Fig. 5e, and Supplementary Table 8). In addition, the paired electrosynthesis system also exhibit excellent stability at a high current density of 400 mA cm−2 for 265 h, with unattenuated FEC2H4 over 103.4% (Fig. 5f). We note that the apparent FEC2H4 over 100% arises from the convergent property in our paired electrosynthesis system, wherein the cathode reduces CO2 to C2H4 and the anode simultaneously decarboxylates PA to generate C2H4 as well. It means that the closed-loop flow of electrons from cathode to anode leads to bipolar C2H4 generation with superior electron economy. Finally, a preliminary technoeconomic analysis (TEA, see Supplementary Fig. 21 for details) was conducted to investigate the feasibility of this paired electrosynthesis system39,40,41,42. It suggests that the profitability of the process depends on the cost of renewable electricity, FEC2H4, and operating current density. In this context, a paired electrosynthesis system is economically feasible when the current density is greater than or equal to 200 mA cm−2 (Fig. 5g). The TEA estimates net revenues of ~$244.3 per ton C2H4 synthesized from PA waste and CO2 under commercially relevant current density (400 mA cm−2).
Discussion
In summary, this study realizes the concurrent conversion of both PA and CO2 wastes to value-added C2H4 via an innovative paired electrosynthesis system that seamlessly integrates anodic PA decarboxylation and cathodic CO2 reduction. To accomplish this, nanoporous Pt microparticles and W-doped CuOx were developed as the anode and cathode electrocatalysts, respectively. Combined operando spectroscopy characterizations and DFT calculations reveals that the in situ formed polarized Pt-PtO2 interface optimizes critical intermediates adsorption for efficient and selective PA decarboxylation due to the downshifted d-band, and W doping stabilizes Cu+ to facilitate the CO2-to-C2H4 conversion. As a result, the paired electrosynthesis system achieves a highest FEC2H4 of 118.7% and a large current density of 1000 mA cm−2, and can operate stably for over 265 h at a large current density of 400 mA cm-2, demonstrating the great practicability for C2H4 production. Further improvement could be expected by separately optimizing the areas of two electrodes in the continuous-flow cell. Overall, these findings document important insight into the paired electrosynthesis for carbon neutrality and can be extendable to other electrochemical reactions for efficient generation of valuable chemicals at both electrodes.
Methods
Materials synthesis
Synthesis of anode electrocatalysts for propanoic acid (PA) decarboxylation to C2H4
To deposit Pt on a Ti foil surface, the Ti foil was initially cleaned by ultrasonication in 1.0 M HCl solution for 10 min, and subsequently rinsed with acetone, ethanol, and deionized water. It was then immersed in an electrolyte containing 10 mL of 0.01 M K2PtCl4 (≥99.0%, Aladdin), 10 mL of 0.2 M KCl (≥99.0%, Aladdin), and 0.02 mL of concentrated HCl solution. After bubbling with nitrogen gas (N2) for 10 min, electrodeposition was performed via cyclic voltammetry with a potential range of 0.7 to −0.35 V vs RHE for 50 cycles at a scan rate of 50 mV s−1. The resulting electrode was then washed with deionized water and dried at 80 °C for 2 h. Subsequently, the electrode was calcined in a tubular furnace at 500 °C for 3 h under Ar atmosphere.
Synthesis of cathode electrocatalysts for CO2 reduction to C2H4
First, 50 mg of C4H6CuO4·H2O ( ≥ 99.0%, Aladdin) and 30 mg of Na2WO4·2H2O were dispersed in 5 mL of deionized water to form a mixed solution. The solution was subsequently subjected to ultrasonication for 10 min, and then introduced into 5 mL of 1.0 M NaOH (≥99.0%, Aladdin) solution under stirring for 1 h at ambient temperature. After centrifugation and washing with deionized water, the W-CuO/Cu(OH)2 precursor was obtained. To prepare the electrode for the continuous-flow cell, 5 mg W-CuO/Cu(OH)2 was dispersed in 0.475 mL isopropanol containing 0.025 mL Nafion (~5 wt% in a mixture of lower aliphatic alcohols and water) by ultrasonication for 30 min. The inks were then airbrushed onto a gas diffusion layer (GDL, YSL-30T) as the cathode electrode (3 × 1 cm−2, mass loading: ~1.0 mg cm−2). The synthesis of the W-CuOx electrocatalyst was carried out via in situ electrochemical reconstruction at a current density of −500 mA cm−2 under CO2 reduction conditions for 20 min.
Physicochemical characterizations
Cu K-edge XANES measurements were conducted at the XAFS station of the BL14W1 beam line of the Shanghai Synchrotron Radiation Facility. The morphology and elemental mapping were analyzed by scanning electron microscopy (SEM) (Zeiss SUPRA 55 SAPPHIRE and TESCAN VEGA 3 SBH). 1H nuclear magnetic resonance (1H NMR) spectra were recorded at room temperature (25 °C) on a Bruker AMX-400 to detect the liquid products. High-resolution transmission electron microscopy (HR-TEM) images were obtained on a JEOL JEM-2010 electron microscope with an accelerating voltage of 200 kV. XPS measurements were conducted on an AXISULTRA DLD-600 W XPS from Shimadzu-Kratos Corporation (mono Al Kα 1486.6 eV) and recorded by a hemispherical analyzer with a step size of 0.05 eV. The gas products were self-injected into a gas chromatograph (PANNA A91PLUS) equipped with a TCD and methanizer/FID detectors. X-ray diffraction (XRD) was performed on an X-ray diffractometer (Rigaku, Ultima IV) with Cu Kα (λ = 0.154 nm) radiation at a tube current of 40 mA and a voltage of 40 kV. The active Pt species in both fresh and post np-Pt were scraped from Ti-based substrates for physiochemical characterizations (e.g., XANES, SEM, XPS, TEM, XRD and Raman). Raman measurements were conducted via confocal Raman spectroscopy (LabRAM HR800, Horiba JobinYvon) with a 532 nm laser source.
Electrochemistry measurements
PA decarboxylation to C2H4 in an H-cell
A gas-tight H-cell separated by a Nafion 117 membrane was used. 50 mL of aqueous 2.0 M PA and 0.5 M K3PO4 electrolytes (pH = 10.0 ± 0.2 adjusted by adding KOH) were employed as anolyte, and 50 mL of aqueous 1.0 M KOH electrolytes was employed as catholyte. A Pt foil (Pine Instruments, 1 × 1 cm2) and an Ag/AgCl electrode (Gaoss Union) filled with saturated KCl solution were used as counter electrode and reference electrode, respectively. N2 was injected into the anode at a flow rate of 20 sccm during the test, and the gas products were injected into a gas chromatograph for quantification. The stirring rate of the anolyte (300 rpm) was kept constant during the test. By changing the ratio of K3PO4 to KOH, the pH values of the anolyte can be adjusted. The electrolysis of PA in the H-cell was performed on an electrochemical workstation (CS-310MA, CorrTest).
PA decarboxylation to C2H4 in a flow cell
A commercial flow cell electrolyzer (Gaoss Union, 1 cm2 active area) was used. The np-Pt electrode (1 × 3 cm2) was placed in the anolyte chamber, and the catholyte and anolyte chambers were separated by an anion-exchange membrane (FAA-3-PK-130, Fumapem). An Ag/AgCl (Gaoss Union, filled with saturated KCl solution) electrode and a Pt electrode were employed as reference electrode and counter electrode, respectively. 2.0 M PA and 0.5 M K3PO4 electrolytes with pH = 10.0 ± 0.2 were employed as anolyte, and 1.0 M KOH electrolyte was served as catholyte, respectively. N2 was injected into the gas-tight anolyte with a flow rate of 20 sccm during the test, and the gas products were injected into a gas chromatograph for quantification. The liquid samples were collected after 0.5 h of electrolysis for 1H NMR measurements with dimethyl sulfoxide (DMSO) as an internal standard. The catholyte and anolyte were circulated by a peristaltic pump with a flow rate of 10 mL min−1. Electrolysis in the flow cell was performed on an electrochemical workstation (CS-310MA, CorrTest). The faradaic efficiencies (FEs) were calculated based on the following equation:
where Qx and Qtotal are the charge passed into product x and the total passed charge during the electrooxidation, nx represents the electron transfer number for producing x (0 for C2H6, 2 for C2H4 and CH3CH2OH, 4 for CH3CHO, and 6 for CH3COOH), Nx is the product amount (mol) of x measured by GC or 1H NMR, and F is the Faraday constant (96485 C mol−1).
CO2 reduction in a flow cell
A commercial flow cell electrolyzer (Gaoss Union, 1 cm2 active area) was used. The electrode (1 × 3 cm2) was placed between the gas chamber and catholyte chamber, and the catholyte and anolyte chambers were separated by an anion-exchange membrane (FAA-3-PK-130, Fumapem). An Ag/AgCl (Gaoss Union, filled with saturated KCl solution) electrode and a Pt electrode were employed as reference electrode and counter electrode, respectively. 1.0 M KOH electrolyte was employed as the catholyte and anolyte. The gas flow rate was 20 sccm during CO2 reduction and the gas products were injected into a ten-way valve. The catholyte and anolyte were circulated by a peristaltic pump with a flow rate of 10 mL min−1. The electrolysis in the flow cell was performed on an electrochemical workstation (CS-310MA, CorrTest).
Bipolar C2H4 electrosynthesis in a flow cell
One flow-cell electrolyzer (Gaoss Union, 1 cm2 active area) was used for electrochemical CO2 reduction and PA oxidation. The anolyte is 2.0 M PA in an electrolyte (pH 10.0 ± 0.2) of mixted KOH (1.5 M) and K3PO4 (0.5 M), and the catholyte is 1.0 M KOH (pH = 14.0 ± 0.2), which was separated by an anion-exchange membrane (FAA-3-PK-130, Fumapem). The CO2 flow rate in gas chamber and the N2 flow rate injected into gas-tight anolyte were set to 20 sccm. The catholyte and anolyte were circulated by a peristaltic pump with a flow rate of 15 mL min−1. The electrolysis in the flow cell was performed on a potentiostat (Reference 3000, Gamry) equipped with a current booster.
ECSA measurements
The electrochemical double-layer capacitance of an electrocatalyst was used to evaluate its ECSA. In a typical procedure, the cyclic voltammograms at different scan rates (10, 20, 40, 60, 80, and 100 mV s−1) were collected in a non-faradaic region (0.95–1.05 V vs RHE). The difference between anodic current and cathodic current at 1.0 V under different scan rates was recorded and plotted against the scan rates. The 1/2 of the linear fitting slope for these curves was calculated as the double-layer capacitance (Cdl) for the corresponding electrocatalyst, which is proportional to its ECSAs.
EIS measurements
Electrochemical impedance spectroscopy (EIS) was conducted on a Gamry Interface 5000E electrochemical workstation at frequencies ranging from 0.1 to 100 kHz with a signal amplitude of 5 mV.
Quasi-in situ Raman characterizations
Quasi-in situ Raman measurements were conducted via a confocal Raman spectroscopy (LabRAM HR800, Horiba JobinYvon) by using a 532 nm laser source. After electrolysis at 100 mA cm−2 in a H-cell for 0, 5, 10, 20, 30, and 60 min, the corresponding active Pt species in np-Pt electrodes were scraped from Ti-based substrates and then loaded on glass wafer to obtain time-dependent quasi-in situ Raman spectra. Time-dependent Raman spectra of CuOx and W-CuOx for CO2 reduction were obtained during electrolysis at −0.8 V vs RHE in a flow cell for 0, 600, 1800, and 3600 s, with CuOx and W-CuOx-loaded gas diffusion layer (GDL) as working electrodes, an Ag/AgCl as reference electrode and a Pt-wire as counter electrode.
Operando ATR-SEIRAS characterizations
All spectroscopic measurements were collected with 4 cm−1 resolution and at least 128 coadded scans via a FTIR spectrometer (Nicolet iS50, Thermo Scientific) equipped with a liquid nitrogen-cooled MCT detector. A gold-plated silicon crystal was mounted in a PIKE electrochemical three-electrode cell with an Ag/AgCl reference electrode and a platinum-wire counter electrode. Electrocatalyst ink was prepared by dispersing the corresponding powders (8 mg) in a solution containing isopropanol (920 μL) and 5 wt% Nafion solution (80 μL) followed by ultrasonication for 1 h. A total of 120 μL electrocatalyst ink was dropped onto the Au-plated silicon crystal (see the following details) and left to dry slowly. For PA decarboxylation, tests were conducted in 2.0 M PA in an electrolyte (pH 10.0 ± 0.2) of mixed KOH (1.5 M) and K3PO4 (0.5 M). For CO2 reduction, the electrolyte was 0.5 M KHCO3, which was constantly purged with CO2 during the experiment. A CHI 760E electrochemical workstation (CH Instruments, USA) was used for chronoamperometric tests at potentials ranging from 1.1 V to 2.9 V vs RHE for PA decarboxylation and from −0.1 to −0.7 V vs RHE for CO2 reduction.
Preparation of Au plating solution
In a typical process, 0.29 mM NaAuCl4·2H2O was dissolved in 1.5 mL of deionized water and followed by the addition of 1.53 mM NaOH. The solution turned from a transparent yellowish to a translucent orange. Then, 1.25 mM NH4Cl, 3.76 mM Na2SO3 and 1.25 mM Na2S2O3·5H2O were dissolved in 25 mL of water. The two solution were mixed in a volumetric flask and then 25 mL deionized water was added. Finally, the mixed solution was sonicated for 2 h and incubated overnight for further use.
Au film deposition
The Si prism was first immersed in aqua regia to remove surface film, polished with 0.05 µm Al2O3 slurry, and subsequently sonicated in ethanol and water until the surface was hydrophobic. The cleaned prism was subsequently immersed in 40% NH4F for 2 min to create a hydride-terminated surface. Next, the crystals were immersed in Au seeding solution containing 3.75 mL of Au plating solution and 0.86 mL of 2% HF at 55 °C for 4–5 min. Finally, the crystals were rinsed with deionized water for use.
DFT calculations
Spin-polarized density functional theory (DFT) calculations were carried out using the Vienna ab initio Simulation Package (VASP)43,44. The exchange-correlation effects were described by the Perdew-Burke-Ernzerhof (PBE) functional under the generalized gradient approximation (GGA) framework45,46,47. A plane-wave basis set with a kinetic energy cutoff of 500 eV was employed, and van der Waals interactions were incorporated via Grimme’s DFT-D3 method with empirical dispersion corrections for all simulations48. A 4 × 4 Pt(111) slab model consisting of five atomic layers was constructed to simulate the pristine Pt catalyst. To investigate surface oxidation effects under high-potential conditions, an oxidized counterpart (denoted as PtO2@Pt(111)) was created by replacing the top layer of the Pt(111) slab with an α-PtO2 structural configuration. For both Pt(111) and PtO2@Pt(111) models, the bottom two Pt layers were fixed to maintain bulk characteristics, and upper layers (including surface adsorbates during electrolysis simulations) were fully relaxed. Structural optimizations utilized a Monkhorst-Pack k-point grid of 3 × 3 × 1 for Brillouin zone sampling, whereas a finer 12 × 12 × 1 k-point mesh was adopted for electronic structure analyses, including differential charge density distribution and density of states (DOS) calculations. Self-consistent field (SCF) iterations were considered converged when the total energy and residual forces reached thresholds of 1 × 10−5 eV and 0.05 eV Å−1, respectively. To mitigate spurious interactions between periodic images, a vacuum layer of 20 Å was introduced perpendicular to the slab surface. The Gibbs free energy (ΔG) for each elementary reaction step was evaluated following the computational hydrogen electrode (CHE) model developed by Nørskov et al.49, as expressed by:
Where, ΔE denotes the electronic energy difference directly obtained from DFT calculations. ΔZPE represents the zero-point energy correction, and TΔS corresponds to the entropic contribution at 298.15 K. The pH-dependent term ΔGpH is defined as kBT × ln10 × pH, where kB is the Boltzmann constant. The potential-dependent term ΔGU is expressed as −eU, with e being the elementary charge and U the applied electrode potential. Both ΔZPE and TΔS were derived from vibrational frequency analyses of adsorbed intermediates performed on static catalyst slabs.
Data availability
The data supporting this study are available within the paper and the Supplementary Information. All other relevant source data are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Ethylene Global Market Report 2023: COVID-19 Impact and Recovery to 2030 Research and Markets: (2023).
Rahman, M. Z. The dawn of solar ethylene. Nat. Rev. Chem. 8, 568–568 (2024).
McFarland, E. Unconventional chemistry for unconventional natural gas. Science 338, 340–342 (2012).
Song, R. et al. Ethylene production via photocatalytic dehydrogenation of ethane using LaMn1−xCuxO3. Nat. Energy 9, 750–760 (2024).
Sattler, J. J., Ruiz-Martinez, J., Santillan-Jimenez, E. & Weckhuysen, B. M. Catalytic dehydrogenation of light alkanes on metals and metal oxides. Chem. Rev. 114, 10613–10653 (2014).
Ito, T. & Lunsford, J. H. Synthesis of ethylene and ethane by partial oxidation of methane over lithium-doped magnesium oxide. Nature 314, 721–722 (1985).
Gao, Y. et al. Recent advances in intensified ethylene production-a review. ACS Catal. 9, 8592–8621 (2019).
Dutta, S. Sustainable synthesis of drop-in chemicals from biomass via chemical catalysis: scopes, challenges, and the way forward. Energy Fuels 37, 2648–2666 (2023).
Miller, J. H. et al. Screening and evaluation of biomass upgrading strategies for sustainable transportation fuel production with biomass-derived volatile fatty acids. iScience 25, 105384 (2022).
Li, F. et al. Molecular tuning of CO2-to-ethylene conversion. Nature 577, 509–513 (2020).
Zhang, T. et al. Highly selective and productive reduction of carbon dioxide to multicarbon products via in situ CO management using segmented tandem electrodes. Nat. Catal. 5, 202–211 (2022).
Chen, Y. et al. Efficient multicarbon formation in acidic CO2 reduction via tandem electrocatalysis. Nat. Nanotechnol. 19, 311–318 (2024).
Zhong, M. et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 581, 178–183 (2020).
Ma, W. et al. Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper. Nat. Catal. 3, 478–487 (2020).
Wei, P. et al. Coverage-driven selectivity switch from ethylene to acetate in high-rate CO2/CO electrolysis. Nat. Nanotechnol. 18, 299–306 (2023).
Dinh, C. T. et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 360, 783–787 (2018).
Jiang, N., Zhu, Z., Xue, W., Xia, B. Y. & You, B. Emerging electrocatalysts for water oxidation under near-neutral CO2 reduction conditions. Adv. Mater. 34, 2105852 (2022).
Fang, W. et al. Durable CO2 conversion in the proton-exchange membrane system. Nature 626, 86–91 (2024).
Verma, S., Lu, S. & Kenis, J. A. Co-electrolysis of CO2 and glycerol as a pathway to carbon chemicals with improved technoeconomics due to low electricity consumption. Nat. Energy 4, 466–474 (2019).
Xue, W. et al. Bromine-enhanced generation and epoxidation of ethylene in tandem CO2 electrolysis towards ethylene oxide. Angew. Chem. Int. Ed. 62, e202311570 (2023).
Hioki, Y. et al. Overcoming the limitations of Kolbe coupling with waveform-controlled electrosynthesis. Science 380, 81–87 (2023).
Zhang, B. et al. Ni-electrocatalytic Csp3–Csp3 doubly decarboxylative coupling. Nature 606, 313–318 (2022).
Corma, A., Iborra, S. & Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 107, 2411–2502 (2007).
Leech, M. C. & Lam, K. Electrosynthesis using carboxylic acid derivatives: new tricks for old reactions. Acc. Chem. Res. 53, 121–134 (2020).
Holzhäuser, F. J., Mensah, J. B. & Palkovits, R. (Non-)Kolbe electrolysis in biomass valorization–a discussion of potential applications. Green. Chem. 22, 286–301 (2020).
Mom, R. et al. The oxidation of platinum under wet conditions observed by electrochemical X-ray photoelectron spectroscopy. J. Am. Chem. Soc. 141, 6537–6544 (2019).
Tian, X. et al. Engineering bunched Pt-Ni alloy nanocages for efficient oxygen reduction in practical fuel cells. Science 366, 850–856 (2019).
Huang, Y. F., Kooyman, P. J. & Koper, M. T. Intermediate stages of electrochemical oxidation of single-crystalline platinum revealed by in situ Raman spectroscopy. Nat. Commun. 7, 12440 (2016).
Meng, N. et al. Electrosynthesis of formamide from methanol and ammonia under ambient conditions. Nat. Commun. 13, 5452 (2022).
Xie, Z. et al. Well-defined diatomic catalysis for photosynthesis of C2H4 from CO2. Nat. Commun. 15, 2422 (2024).
Yu, Y., He, Y., Yan, P., Wang, S. & Dong, F. Boosted C–C coupling with Cu–Ag alloy sub-nanoclusters for CO2-to-C2H4 photosynthesis. Proc. Natl. Acad. Sci. USA 120, e2307320120 (2023).
Huang, B. et al. Seeded synthesis of hollow PdSn intermetallic nanomaterials for highly efficient electrocatalytic glycerol oxidation. Adv. Mater. 35, 2302233 (2023).
Liu, S., Govindarajan, N., Prats, H. & Chan, K. Understanding the reaction mechanism of Kolbe electrolysis on Pt anodes. Chem. Catal. 2, 1100–1113 (2022).
Vijh, A. K. & Conway, B. E. Electrode kinetic aspects of the Kolbe reaction. Chem. Rev. 67, 623–664 (1967).
Daelman, N., Capdevila-Cortada, M. & López, N. Dynamic charge and oxidation state of Pt/CeO2 single-atom catalysts. Nat. Mater. 18, 1215–1221 (2019).
Gao, R. et al. Pt/Fe2O3 with Pt–Fe pair sites as a catalyst for oxygen reduction with ultralow Pt loading. Nat. Energy 6, 614–623 (2021).
Wu, H. et al. Selective and energy-efficient electrosynthesis of ethylene from CO2 by tuning the valence of Cu catalysts through aryl diazonium functionalization. Nat. Energy 9, 422–433 (2024).
She, X. et al. Pure-water-fed, electrocatalytic CO2 reduction to ethylene beyond 1,000 h stability at 10 A. Nat. Energy 9, 81–91 (2024).
Lum, Y. et al. Tuning OH binding energy enables selective electrochemical oxidation of ethylene to ethylene glycol. Nat. Catal. 3, 14–22 (2020).
De Luna, P. et al. What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science 364, eaav3506 (2019).
Leow, W. R. et al. Chloride-mediated selective electrosynthesis of ethylene and propylene oxides at high current density. Science 368, 1228–1233 (2020).
Zhou, H. et al. Electrocatalytic upcycling of polyethylene terephthalate to commodity chemicals and H2 fuel. Nat. Commun. 12, 4679 (2021).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Hammer, B., Hansen, L. B. & Nørskov, J. K. Improved adsorption energetics within density functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413–7421 (1999).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Le, J. B., Fan, Q. Y., Li, J. Q. & Cheng, J. Molecular origin of negative component of Helmholtz capacitance at electrified Pt(111)/water interface. Sci. Adv. 6, eabb1219 (2020).
Wei, J. et al. The dynamic nature of CO adlayers on Pt(111) electrodes. Angew. Chem. Int. Ed. 59, 6182–6186 (2020).
Wang, S., Zhu, E., Huang, Y. & Heinz, H. Direct correlation of oxygen adsorption on platinum-electrolyte interfaces with the activity in the oxygen reduction reaction. Sci. Adv. 7, eabb1435 (2021).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFA1600800), the National Natural Science Foundation of China (22479055), the Innovation and Talent Recruitment Base of New Energy Chemistry and Device (B21003), the Natural Science Foundation of Hunan Province (2022JJ20084), and the Science and Technology Innovation Program of Hunan Province (2023RC3034). Also, we are grateful to the High-Performance Computing Centre of Central South University for assistance with the computations.
Author information
Authors and Affiliations
Contributions
B.Y. proposed the idea. B.Y. and B.Y.X. supervised the project. W.X. and H.J. prepared the catalysts and carried out the experiments. J.L. performed the DFT calculations. C.T. analyzed the oxidation mechanism. X.C. conducted the XAS measurements. B.Y., B.Y.X., and W.X. wrote the manuscript. All authors discussed the results. W.X. and H.J. contributed equally.
Corresponding authors
Ethics declarations
Competing interests
B.Y. and W.X. of the Huazhong University of Science and Technology have filed a provisional patent (no. 2023113354766) relative to this manuscript. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yumin Da and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xue, W., Jiang, H., Liu, J. et al. Bipolar ethylene electrosynthesis from CO2 and biowaste acid with total faradaic efficiency over 118%. Nat Commun 16, 9054 (2025). https://doi.org/10.1038/s41467-025-64649-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-64649-8
