
Abstract
Theory predicts that in the absence of selection, a newly formed segmental allopolyploid will become ‘autopolyploidized’ if homoeologous exchanges (HEs) occur freely. Moreover, because selection against meiotic abnormalities is expected to be strong in the initial generations, we anticipate HEs to be uncommon in evolved segmental allopolyploids. Here we analysed the whole-genome composition of 202 phenotypically homogeneous and stable rice tetraploid recombinant inbred lines (TRILs) derived from Oryza sativa subsp. japonica subsp. indica hybridization/whole-genome doubling. We measured functional traits related to growth, development and reproductive fitness, and analysed meiotic chromosomal behaviour of the TRILs. We uncover factors that constrain the genomic composition of the TRILs, including asymmetric parental contribution and exclusive uniparental segment retention. Intriguingly, some TRILs that have high fertility and abiotic stress resilience co-occur with largely stabilized meiosis. Our findings comprise evidence supporting the evolutionary possibility of HE-catalysed ‘allo-to-auto’ polyploidy transitions in nature, with implications for creating new polyploid crops.
Similar content being viewed by others


Main
Polyploidization, or whole-genome doubling (WGD), is a potent evolutionary force driving speciation and creating biodiversity1,2,3,4,5. In plant and yeast, WGD is often coupled with hybridization, leading to the formation of allopolyploids or segmental allopolyploids6,7,8. There are at least two mutually inclusive explanations for a higher tendency of wide hybridization-WGD coupling than hybridization between more similar parents: (1) wide hybrids are more prone to producing unreduced gametes9,10,11,12 and (2) WGD restores normal meiosis by enhancing autosyndetic bivalent pairing, whereas multivalents and allosyndetic bivalent pairing lead to a higher frequency of meiotic abnormalities in polyploids derived from more similar diploid progenitors13.
In addition to overcoming meiotic instabilities14,15, nascent polyploids also face challenges associated with establishment and/or persistence. WGD events may be sporadic over short temporal windows and thus involve few founder individuals, or even one individual. This relative rarity, together with the often-immediate reproductive isolation from parental taxa due to ploidy differences, comprises a strong bottleneck for newly formed polyploids16. This early demographic reality is often magnified by minority cytotype disadvantage17,18, both being population-level processes that might constrain evolvability and hence persistence of new polyploids. Yet, despite these ‘birthing’ challenges, WGD is unmistakably widespread across the tree of life, having occurred repeatedly and commonly in innumerable lineages from across the phylogenetic spectrum of plants19,20,21, demonstrating that the evolvability challenges associated with the nascent stages of polyploidy are frequently surmounted.
It has been posited that a major mechanism mitigating low genetic variation in young polyploids is recurrent formation of the same polyploid species from genetically differentiated parental populations16,22,23,24. This recurrent formation leads to the possibility of gene flow among genetically different polyploids when secondary contacts occur, thereby increasing population-level genetic diversity and novel recombinants due to reshuffled standing variation. This avenue of generating genetic diversity under natural settings is ecologically and biologically idiosyncratic, contingent on evolutionary history, mating systems, phenology and geography, among many other factors.
Another potential source of genetic variation in polyploids is the intrinsic genomic and meiotic instability that characterizes the early stages of polyploid stabilization. It has been widely documented that ‘rapid genomic changes’ often occur immediately following polyploid formation. Moreover, these ‘rapid genomic changes’ are often saltational, and collectively have been termed ‘genome shock’25, an insight made long before most of the responsible mechanisms were understood at the molecular level. Diverse mechanisms are involved in perturbation of meiosis and (epi)genome stability, such as de novo genetic/epigenetic alteration and interactions between diverged parental regulatory networks26,27,28,29,30,31,32,33,34. Notwithstanding a burgeoning literature on the rapid genomic and expression-level changes accompanying polyploidy, the extent to which these mechanisms collectively generate evolutionarily meaningful phenotypic variation in newly formed polyploids remains fragmented, as well as mostly unobserved in natural settings.
Hitherto, the most clearly demonstrated genetic mechanism underlying polyploidy-induced rapid genomic change is homoeologous exchange (HE) due to meiotic homoeologous chromosome recombination25,35. Remarkably, although HE itself is often associated with unbalanced aneuploid gametes (hence reduced fertility), it has occurred ubiquitously in many naturally formed neoallopolyploid plant species7,36,37,38,39,40,41,42,43,44,45. This suggests that not all HEs are necessarily detrimental, and that some may generate adaptive phenotypes35. The occurrence of HE is thought to arise, at least in part, from insufficient genetic differentiation between homoeologous chromosomes and/or the absence of mechanisms that enforce exclusively homologous pairing during meiosis46,47. Accordingly, more HEs are expected to occur in allopolyploids formed between genetically less divergent parental taxa, such as in cases of segmental allopolyploids8,48. It has been posited that HEs themselves lead to HEs by a ‘ratchet-like’ process, in that each additional HE would make homoeologous chromosomes more similar7. As such, in theory, after a sufficient number of generations, all four copies in a still fertile tetraploid of every chromosomal segment would be derived from either, but not both, of the hybridizing parents; that is, each chromosome segment, the sizes of which are determined by recombination breakpoints, will become homozygous (homogeneous) for all four constituent copies. Thus, HE-fuelled intersubgenomic segmental replacement, if it occurs freely and without strong purifying selection, would transform a single starting nascent allopolyploid to an autopolyploid swarm, especially under selfing. This possibility, however, remains to be experimentally tested.
Earlier we reported that synthetic segmental allotetraploids derived by doubling the reciprocal F1 hybrids generated between the rice (Oryza sativa L.) subspecies japonica and indica have a notable level of restored fertility49, consistent with findings in other organisms, for example, yeast6. Unlike interspecific allopolyploids that often become meiotically stable, however, these rice lines were meiotically unstable and manifest rampant HEs in the early generations48. Because each line is expected to be genomically mosaic due to HEs, and meiotically unstable due to high chromosome similarity, an intuitive outcome would be universal deterioration of fertility (fitness) in all lines7,8,50. This theoretical prediction, however, has not been empirically interrogated either.
Here we tested the above theoretical predictions by propagating the segmental allotetraploid rice plants via single-seed-descent selfing for 12 successive generations to generate tetraploid recombinant inbred lines (TRILs). We analysed genome compositions of a total of 202 phenotypically homogeneous and stable TRILs by whole-genome resequencing. We find that these lines have reached 80% homozygosity (for all four copies) on average, and were thus ‘autopolyploidized’, and that each line was, as expected, a genomic mosaic of both parents. Analysis of the TRILs led to the identification of several factors constraining genomic compositions of the mosaics, including genomewide overall asymmetric parental contribution and a single segment that was exclusively uniparental. In addition, HE-enabled segmental repatching of some whole chromosomes to one parent-state, which albeit occurring stochastically, may also cause biased genome composition. Intriguingly, some lines showed high fertility and even gave rise to seed yield surpassing those of the diploid parental cultivars. These high-fertility TRILs also manifested high meiotic stability due to an increase of bivalent formation and the alternate-orientation type of quadrivalent configuration. This suggests that, at odds with theory, genome mosaicism-based autopolyploidization does not necessarily lead to universal exacerbated meiotic instability and associated fertility loss. Moreover, we find that some high-yielding TRILs also showed transgressive resilience to two important abiotic stresses, although in their propagation process no such conditions were applied. Together, our findings unravel an unexpected evolutionary potential of HE-catalysed ‘allo-to-auto’ polyploidy-type transition and point to the feasibility for rapid generation of new polyploid crops by induced WGD of diploid hybrids that are prezygotically compatible but postzygotically isolated (or largely so) at the homoploid level30,31,32,33,34.
Results
Homoeologous exchange-mediated rapid autopolyploidization
Based on whole-genome resequencing, we analysed genome compositions of 202 euploid tetraploid lines (2n = 48) identified by dual-colour oligo-fluorescence in situ hybridization (FISH) (for example, Fig. 1a–c), which had undergone 12 successive selfed generations via single-seed descent post segmental allotetraploidization (Extended Data Fig. 1a) between the japonica and indica subspecies49. These lines included both crossing directions of the diploid F1 hybrids before whole-genome doubling (WGD), with 102 lines of NN99, the japonica cultivar Nipponbare as the maternal parent (named TN9-1 to TN9-102), and 100 lines of 99NN, the indica cultivar 9311 as the maternal parent (named T9N-1 to T9N-100).

a, A typical euploid TRIL karyotype illustrated by dual-colour oligo-FISH. Scale bar, 10 μm. b, Depicted ideogram based on c. c, Caryogram of the 12 rice chromosomes of a typical euploid TRIL. Green and red signals are from FAM-green- and Texas-red-labelled oligo probes, respectively. Scale bar, 10 μm. For a and c, the FISH experiments were repeated independently at least 3 times, and all gave the same results. d, Density plot showing the distribution of genomic homozygosity of the TRILs according to crossing direction; dashed lines represent mean values. n = 102 (NN99) and 100 (99NN). e, Circos plots displaying genomic composition of 8 TRILs, 4 each showing the highest (left) and lowest (right) genomic homozygosities. Different coloured blocks denote the 5 homoeologue ratios. Tracks from outside to inside, left: T9N-9, T9N-10, T9N-99 and TN9-10; right: T9N-19, T9N-32, TN9-26 and TN9-50. f, Violin plots showing genomic homozygosities of the 202 TRILs at the chromosome level for each of the 12 rice chromosomes. Different red letters in the bottom right corner of each panel indicate statistically significant differences in homozygosity among the chromosomes, as determined using two-sided one-way analysis of variance (ANOVA) followed by a post-hoc LSD test (P < 0.05, n = 202). The mean value for each chromosome is depicted by a red dot (±s.d.) and presented as a percentage (black number in the bottom left corner). Dots denote individual data points. The range of genomic homozygosity for each chromosome is provided below each panel in blue.
We found that the vast majority of the tetraploid lines were largely autotetraploidized due to HE-propelled genomewide segmental homogenization, with an average homozygosity (defined as the proportion of chromosomal segments being 4:0 or 0:4 of parental alleles across the entire genome) of 84.1% (from 97.8% to 36.8%), and 81.7% of the lines (165 of 202) reaching 80% all-four-copy segmental homogeneity (Supplementary Table 1). Thus, these lines were quasi-autotetraploids in the form of tetraploid recombinant inbred lines (abbreviated as TRILs). These features did not differ discernibly when the TRILs were stratified into two groups according to crossing directions (P = 0.63 by Mann–Whitney U-test, Fig. 1d and Supplementary Table 1), suggesting that parent of origin and source of cytoplasm imposed little impact on overall progression and extent of segmental homozygosity. However, the degrees of homozygosity differed markedly across the TRILs. Four TRILs showed the highest level of homogenization (96.6–97.8%), while 4 TRILs remained segmentally highly heterozygous, with only 36.8–54.7% of the genomic regions attaining homozygosity (all-four-copy segmental homogeneity) (Fig. 1e). In every TRIL, the 12 chromosomes were highly variable with respect to their homozygosity states. When all TRILs were considered together, chromosomes (Chrs.) 6 and 10 showed the highest and lowest degrees of homozygosity (91.3% and 61.6%, respectively) (Fig. 1f and Supplementary Table 2). In addition, in >20% of the TRILs (41 out of 202), 5 chromosomes (Chrs. 6, 8, 9, 11 and 12) became fully homozygous (Supplementary Table 2). Quasi-autopolyploidy status of the TRILs was also supported by a genomewide k-mer analysis involving comparisons with parental autotetraploids, diploid F1 hybrids and, in particular, the initial segmental allotetraploids (Supplementary Fig. 1 and Supplementary Results).
The rapid autotetraploidization was underpinned by a single powerful mechanism, that is, homoeologous exchanges due to meiotic homoeologous chromosome recombination35. We identified a total of 21,900 HE events in the 202 TRILs, which ranged from 61 to 201 among lines and averaged 108.4 ± 25.3 per line (Fig. 2a and Supplementary Table 1), again without significant difference between the two crossing directions (P = 0.22 by Mann–Whitney U-test, Fig. 2b). The 12 chromosomes manifested marked differences in the numbers of accumulated HEs (Fig. 2c). Taking chromosome length (nt) into consideration, Chrs. 7 and 6 showed the highest (0.35 events per Mb) and lowest (0.21 events per Mb) numbers of HEs, respectively (Fig. 2c). Genomewide, there was no significant difference in the distribution of HEs between the crossing directions (P = 0.23 by one-sample t-test, Fig. 2d,e). Strikingly, 7 chromosomes (Chrs. 4, 6, 7, 8, 9, 10 and 12) showed uniparental composition of all four copies in 47 TRILs (Supplementary Fig. 2). Notably, this exclusive uniparental contribution was unlikely due to the chromosomes in question being inherited intact across all the 12 successive generations (that is, no HE ever occurred); rather, we infer that this had resulted from multiple HEs, whereby both copies of a given parent were replaced by counterparts of the other in all segments along the entire length of these chromosomes. This possibility was further supported by resequencing six still available ancestral S1 individuals of the TRlLs, and 67 independently reconstructed indica–japonica S1 segmental allotetraploids (also between cv. 9311 and Nipponbare); we found that in these 73 S1 individuals, HE already occurred in 759 out of the 876 (73 ×12) chromosomes in total, albeit only one meiosis was involved (Supplementary Fig. 3). This suggests that with an additional 11 generations the TRILs experienced, the probability for any chromosome remaining intact (without HE) is virtually zero. Nonetheless, the fact that not all TRILs contained such exclusive uniparental whole-chromosome restoration, and that the identities of these chromosomes were variable in the TRILs harbouring them (Supplementary Fig. 2) suggest that the phenomenon is probably stochastic.

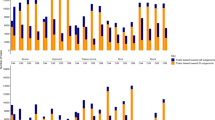
a, Heat maps depicting the genomic landscapes of the 202 whole-genome-resequenced TRILs shown as two groups categorized according to crossing directions. Different colours denote different homoeologue ratios at each locus. Each row represents a TRIL, while the 12 columns represent the 12 rice chromosomes. The position of the centromere in each chromosome is denoted by a red block on each diagrammed chromosome on top of the panel. b, Density and boxplots showing the distributions of HE numbers in the TRILs according to their crossing directions at genomewide scale; pink and blue diagrams represent NN99 and 99NN TRILs, respectively. The P value designates significant difference between the reciprocally derived TRILs (NN99 and 99NN) based on two-sided Mann–Whitney U-test. In boxplots: centre line, median; box limits, lower and upper quartiles; whiskers, minimum and maximum data points. c, The total numbers of HEs (histograms) and the mean HE number (green dots) per Mb on each chromosome of the 202 TRILs. Grey dots in each histogram represent individual data points. Error bars on both histograms and green dots represent s.e.m. Different green letters above the green dots indicate statistically different HE numbers between chromosomes based on two-sided one-way ANOVA followed by a post-hoc LSD test (P < 0.05, n = 202). d, Plot of HE distribution in the TRILs according to their crossing directions at chromosomes scale. The x axis represents the genomic position, while the y axis indicates the number of HEs. The blue line represents the average number of HEs across all samples, the purple area represents the 95% confidence interval (CI), red dots represent TRILs of NN99, and yellow dots represent TRILs of 99NN, the majority of which fall within the confidence interval. Blue dots indicate the positions of centromeres. e, Distribution of HE differences between two crossing directions. P = 0.23 designates no significant difference between the reciprocally derived TRILs (NN99 and 99NN) based on two-sided one-sample t-test. f, Split-half violin plots depict the distribution of parental genomic proportions at the chromosome level and whole-genome scale in all 202 TRILs. Darker and lighter purple colours denote 9311 and Nipponbare (NPB) genomic proportions, respectively. Difference in parental genomic proportions was based on two-sided Mann–Whitney U-test (n = 202). P values are shown on top of each split-half violin plot. For b, d and e: n = 102 (NN99) and 100 (99NN).
The relative contributions of the two hybridizing parents in the TRILs were also different both genomewide and at individual-chromosome levels (Fig. 2f). Specifically, genomewide, the relative contributions of 9311 vs. Nipponbare were 58.3% vs 41.7% when all TRILs were considered together (Fig. 2f). This difference is statistically significant (P < 2.2 × 10−16 by Mann–Whitney U-test), indicating that, overall, 9311 and Nipponbare were the major and minor contributors, respectively, to the genomic compositions of the TRILs, a conclusion that holds irrespective of the crossing direction (P = 0.782, Mann–Whitney U-test; Supplementary Table 1). At the individual-chromosome level, considering all lines together, eight chromosomes (Chrs. 1, 3, 5, 6, 7, 8, 9 and 12) contained significantly more contributions from 9311 than from Nipponbare (P < 0.01, by Mann–Whitney U-test), Chrs. 4, 10 and 11 did not show significant differences between the two parental origins, and only Chr. 2 showed significantly more contribution by Nipponbare than 9311 (Fig. 2f).
Taken together, a general trend of all the TRILs was that they were rapidly autotetraploidized to become quasi-autotetraploids after only 12 generations of successive selfing, despite having been derived from hybridization between two subspecies, and hence, starting as segmental allotetraploids.
Association of HEs and HRs with genomic/epigenomic features
Although diploid F1 hybrids of rice japonica and indica subspecies are often highly sterile, certain genotypic combinations can set some seeds. Thus, F2 segregation populations and RILs can be obtained from sufficiently large number of F1 individuals to study homologous recombination (HR) at the diploid level. Indeed, several previous studies using such intersubspecific rice F2 populations or RILs have investigated the patterns and features of HRs19,51,52. Although some discrepancies were reported by different studies, a consensus conclusion was that there appeared no qualitative differences in HRs between the subspecies and between intraspecific lines with respect to their chromosomal distribution patterns, although the two subspecies were highly different in their respective HR hotspots that were largely (~80%) unique to each subspecies53.
We sought to interrogate whether and to what extent HEs at the tetraploid level would be similar to, or different from, HRs at the diploid level. For this purpose, we used whole-genome resequencing data of a set of 218 diploid RILs (F13) that we developed from the same japonica–indica genotypic combination (Nipponbare and 9311) in both crossing directions. We tabulated the chromosomal distribution and features of HRs in the diploid RILs and compared these to those of HEs from the 202 TRILs. We found that the overall patterns of HRs and HEs were similar across each of the 12 chromosomes, with substantially more events in subtelomeric than in pericentromeric regions and sparse recombination around centromeres; nevertheless, the rates of HRs and HEs differed substantially, as did their patterns at a regional scale and at recombination sites (Extended Data Fig. 2a). We calculated Pearson correlation coefficients between the HEs and HRs at both genomewide and individual-chromosome levels. We found that HRs and HEs showed a significant positive correlation genomewide (Pearson correlation r = 0.61, P < 2.2 × 10−16), and in 11 (except for Chr. 4) of the 12 chromosome pairs (Pearson correlation r = 0.46 to 0.81, P < 0.01) (Fig. 3a).

a, Scatter and line diagrams showing the correlations between regional HE and HR rates at genomewide scale (left) and individual-chromosome level (right). Each dot denotes a 1-Mb window. HE and HR rates are depicted by red and green dots, respectively. A smooth line was fitted using loess function (span = 0.2, degree = 2) for physical positions with HE and HR rates. The x axis denotes the physical positions, and the y axes are HE (left) and HR (right) rates. Correlation coefficients (r) and P values were calculated using two-sided Pearson correlation test and labelled on each diagram accordingly. Shaded areas depict 95% CIs. b,c, Scatterplots displaying the correlations between either HE (b) or HR (c) rate with genomic and epigenomic features, including genomic similarity (defined in Methods), CG DNA methylation (mCG), chromatin accessibility (ATAC-seq), H3K4me3, H3K9ac and Retro-TE content, separately. All genomic and epigenomic features are from cv. Nipponbare, while those concerning cv. 9311 are presented in Supplementary Figs. 4–10. Correlation coefficients (r) and P values, calculated using two-sided Pearson correlation tests, are displayed in each panel. Shaded areas represent 95% CIs. d, Violin plots showing the distribution of HE number per TRIL and HR number per DRIL, with the red dots and red lines representing the mean ± s.d. of the data. The mean, median and s.d. values of HE and HR are shown on the left and right sides, respectively. e, Flag diagram showing the relative proportions of HEs and HRs mapped to genic regions (dark blue) and intergenic regions (green) in TRILs and DRILs. P values indicate significant differences in the relative distributions between HEs and HRs based on two-sided Chi-square test. For a–e, sample sizes are provided in Supplementary Table 25.
We next tested whether parental genomic and epigenomic features, which are known to be determinants or contributors to HRs27,54, were also correlated with HEs. Of the 12 genomic and epigenomic features investigated, 11 were found to be significantly correlated with the rates of both HEs and HRs. Eight of the features (including parental genetic similarity, CHH methylation, miniature inverted repeat transposable elements (MITEs) content, histone modifications by H3K27me3, H3K9ac, H3K4me3 and H3K36me3, and chromatin accessibility) exhibited significant positive correlations with the rates of both HEs and HRs, while three features (CG- and CHG-methylation, and retrotransposon content) showed significant negative correlations with the rates of both HEs and HRs (Pearson correlation P < 0.01, Fig. 3b,c and Supplementary Figs. 4–10). Notably, DNA-transposable element (TE) content showed a weak but significant correlation only with rates of HRs but not HEs (Pearson correlation P = 0.04 and 0.8 for HRs and HEs, respectively, Supplementary Figs. 4–10). These correlations were highly consistent when both parental data were used separately (Supplementary Figs. 4–10). Of note, for all 11 genomic and epigenomic features that were significantly correlated with the rates of both HEs and HRs, values of all the correlation coefficients concerning HE were markedly lower than those for HR (Fig. 3a–c and Supplementary Figs. 4–10), indicating that the correlations between HEs and the genomic/epigenomic features were less tight than they were with HRs. This may suggest a more permissive chromatin environment in the tetraploids than in the diploids for recombination, as supported by our methylome data which showed significantly greater CG and CHG hypomethylation in the S0 tetraploids than in the diploid F1 hybrids (Supplementary Table 3). Our observation of hybridization/polyploidy-induced DNA hypomethylation is consistent with findings in a previous study55.
We further quantified the numbers of accumulated HEs in TRILs and HRs in diploid recombinant inbred lines (DRILs) and found that HEs were twice as numerous as HRs, 108.42 vs 57.40 per line on average (Fig. 3d). We caution that the HR and HE numbers are the cumulative results of 12 consecutive generations, thus the difference is more likely due to a longer temporal window in tetraploids than in diploids before homozygosity was reached, rather than necessarily more recombination events per meiosis in the tetraploids. This scenario is corroborated by our cytogenetically quantified chiasmata data, the cytological manifestation of crossover (CO), which showed that tetraploids had no more than approximately twice the number of chiasmata (including both HRs and HEs) of the diploids (Extended Data Fig. 2b–d). Genomewide, the accumulated HEs are more evenly distributed than HRs, evidenced by significantly lower variance in the number of breakpoints per 500-Kb bin in the TRILs compared with the DRILs when all chromosomes were considered together (81.91 vs 190.50, P < 2.2 × 10−16 by Bartlett’s test) (Extended Data Fig. 2e and Supplementary Table 4). We note, however, that this overall more even distribution of HEs than HRs was contributed by only four chromosomes (Chrs. 4, 7, 11 and 12), whereas Chr. 9 showed the opposite pattern and the remaining seven chromosomes did not show significant differences (Supplementary Table 4). Finally, we analysed genomic features of the breakpoints of a subset of HEs (3,582) and HRs (4,286), which could be reliably delineated within a 3-kb region. We found that the proportions of HE and HR breakpoints that overlapped with gene-body regions (Untranslated regions, exons and introns) were 55.4% and 50.1% (1,986/3,582 for HE and 2,148/4,286 for HR), respectively. Of note, breakpoints of HEs were significantly more enriched in gene-body regions (P < 0.01, Chi-square test) and less enriched in the intergenic regions (other genomic regions except gene-body regions) (P < 0.01, Chi-square test) than were HRs (Fig. 3e and Supplementary Table 5). This genic enrichment of HEs (in TRILs) but not of HRs (in DRILs) coincided with genomewide substantially more hypo-DNA methylation of the regions in the segmental allotetraploids (S0) than in the diploid F1 hybrids relative to the diploid parents (Supplementary Table 3 and Supplementary Results).
Exclusive uniparental contribution of a 1.7-Mb segment
Unexpectedly, based on the whole-genome resequencing data, we found that all 202 TRILs contained a single segment of variable length on Chr. 6, for which all four copies were from 9311. After aligning sequences of the segments, the ‘core segment’ shared by all TRILs was delineated as 1.7-Mb in length and mapped to the position ‘Chr. 6, 7680000–9385000’ (Fig. 4a). We reasoned that this exclusively uniparentally derived segment was unlikely due to chance, given that the TRILs were derived from 202 independent S1 individuals involving reciprocal crosses. Indeed, the probability for all four copies of a given genomic region in all the 202 TRILs to be due to chance was extremely unlikely (P = 5.8 × 10−67, based on a probability density function test).

a, Heat maps depicting the exclusive uniparental (9311) inheritance of a single genomic segment in all TRILs of both crossing directions, its homoeologue ratios in 73 S1 tetraploid individuals and 202 earlier generation (S4) tetraploid individuals. The core segment shared by all 202 TRILs is delimited by two dashed vertical orange lines. b, Heat maps displaying the same chromosomal segment (as in a) in 6 randomly selected TRILs by mapping the resequencing reads against the parental Nipponbare and 9311 reference genomes, respectively, to test for potential influence of different reference genomes, which showed no difference. In a and b, each row represents 1 tetraploid individual. Different colours refer to different homoeologue ratios of Nipponbare and 9311. Information on chromosome and genomic location is given in the graph. c, Gene position and annotation of the 3 functional genes that had transposed from this segment to other genomic loci in the Nipponbare genome. d, Structures of gene OsSOG1 in Nipponbare (top) and 9311 (bottom). Each exon is labelled, along with its corresponding starting position. Vertical lines denote 3 SNPs present in exons, and the corresponding amino acid variants are shown. The annotations of Nipponbare and 9311 OsSOG1 alleles are based on the Nipponbare (NPB) IRGSP-1.0 and 9311 reference genomes, respectively. The gene structure image was created with IBS 2.0.
To further test this conclusion, we resequenced 6 still available ancestral S1 individuals of the TRILs and 67 independently reconstructed S1 euploid individuals, all of which had undergone only one meiosis. We found that 66 of the 73 S1 individuals were heterozygous at the focal region; the remaining 7 individuals were already homozygous at this region, 3 containing all-four-copy 9311 and 4 containing all-four-copy Nipponbare (Fig. 4a). Remarkably, compared with the remaining individuals, which showed an expected fertility range, the 4 plants with all-four-copy Nipponbare at this region manifested severe fertility defects (Supplementary Fig. 11 and Supplementary Results). This suggests that tetraploids with all four-copy Nipponbare at the focal region did occur in earlier generations but would be rapidly purged from progenies due to lower reproductive fitness.
We also reanalysed the whole-genome resequencing data for this focal region of an independent S4 population containing 202 individuals that were available from our previous study48. We found that although experiencing only three rounds of meiosis, 98% (199 out of 202) of the S4 individuals were already homozygous at the focal region with all four copies from 9311 (that is, no allele from Nipponbare), while the remaining 3 individuals were heterozygous at a 1:3 ratio of Nipponbare vs 9311 (Fig. 4a), suggesting strong preferential retention of this segment of 9311 already at the early generations in the tetraploids. We also ruled out a potential bias of reference genomes because using both those of 9311 and Nipponbare produced the same results (Fig. 4b). Together, the exclusive 9311 uniparental inheritance of this 1.7-Mb core segment of Chr. 6 was most probably due to strong positive selection.
Next, we compared the similarity and difference between 9311 and Nipponbare in the 1.7-Mb segment. We found that this region is not more divergent, relative to the rest of the genome, between the two rice subspecies, both overall for the entire segment and for each of the orthologous gene pairs (shared genes) located in this segment (Supplementary Tables 6 and 7). Furthermore, based on OrthoFinder56, we identified 148 and 65 annotated genes (of the total 248 and 162 annotated to the two subspecies genomes, respectively) that are uniquely present within this 1.7-Mb segment of the Nipponbare and 9311 genomes (Extended Data Fig. 3a and Supplementary Table 8). Again, we found that the proportions of Nipponbare- and 9311-uniquely annotated genes within the 1.7-Mb segment are not significantly different from those of randomly sampled same-sized segments of Chr. 6 (excluding the focal segment), or genomewide (P > 0.05, Chi-square test) (Supplementary Tables 6 and 8). Together, our results suggest that the exclusive uniparental retention of this 1.7-Mb segment of 9311 might be due to strong negative selection against the Nipponbare counterpart, because the latter’s complete retention causes defects in genomic and/or organismal functionality (for example, fertility) of the TRILs, as further detailed below.
We inquired whether the phenomenon might be linked to a few ‘driver’ genes (parental-specific or common but differentiated) contained within this 1.7-Mb segment. Blast P and Swiss-Prot predictions indicated that 21 of the 9311-specific genes (65) are involved in cysteine-type peptidase, esterase, ADP-binding and DNA-binding activities, nucleic-acid-binding, zinc-ion-binding, metal-ion-binding, signal-recognition-particle and transferase activity, while the remaining 44 genes are functionally unknown. Importantly, among the 9311-specific genes, 3 were found to have transposed from the original 1.7-Mb focal region to other genomic loci (rather than physical loss) in the Nipponbare genome (Fig. 4c and Supplementary Table 8). Two of the genes (LOC_Os07g37340 and LOC_Os12g33944) encode a putative histone H2A and an ELF (EARLY FLOWERING) protein, respectively, while the third gene (LOC_Os09g24390) codes for zinc knuckle family protein (Fig. 4c). The Ka/Ks (nonsynonymous-to-synonymous substitution) values of all three transposed genes are significantly lesser than 1, suggesting that they are strongly non-neutral (Supplementary Table 9 and Supplementary Results). By contrast, although transposition of 30 genes from the 1.7-Mb focal region to other loci was also identified in the 9311 genome, none has known or predicted function (Supplementary Table 8 and Supplementary Results), consistent with >50% (87 of the 148) Nipponbare-specific genes being transposable elements (TEs), with the others being unknown-function genes (Supplementary Table 8).
Of note, of the 97 common genes in the segment, 3 with potentially differentiated functions (harbouring missense variants between Nipponbare and 9311 alleles) are involved in responses to DNA damage or DNA repair (Supplementary Table 8). These 3 genes are OsSOG1 (known to function in DNA damage response; LOC_Os06g15410 is its human homologue), EEPD1 (known to be involved in DNA damage repair via homologous recombination) and LOC_Os06g15360 (encoding RAD23, a component of the DNA damage repair complex) (Supplementary Table 8). Incidentally, the 9311 haplotypes of these 3 genes were found to prevail among the 3,023 rice cultivars (https://iric.irri.org/projects/3000-rice-genomes-project) (Supplementary Table 10 and Supplementary Results). Moreover, based on the SIFT (sorting intolerant from tolerant) scores for mutation types, the functions of at least OsSOG1 seem to differ between the two rice subspecies, with the indica OsSOG1 allele potentially exhibiting greater functionality than the japonica allele (SIFT value: 0.03), with an A→G transition resulting in a lysine-to-arginine amino acid substitution (Fig. 4d, Extended Data Fig. 3b,c and Supplementary Results). Together, our results suggest that the exclusive uniparental contribution of the 1.7-Mb segment from the 9311 parent to all the TRILs is probably obligatory, possibly driven by multifaceted functional constraints.
Besides this 100% uniparental inheritance, we identified 12 additional segments by relaxing the criterion stringency to 75–85%. These 12 segments ranged from 0.38- to 13.8-Mb in size and mapped to different chromosomes, with six and six being biased towards 9311 and Nipponbare, respectively (P < 2.2 × 10−16 by one-sample proportions test, Extended Data Fig. 4).
We also identified two segments located on Chrs.1 and 6 (Extended Data Fig. 4) that were biasedly selected probably due to cytonuclear interactions at earlier generations, because at least one maternal copy was present in 90% (180 out of 202) of the lines, significantly higher than expected (P < 0.01, Chi-square test)48. We noted, however, that the 10% (22 out of 202) lines that did not have this homo-cytonuclear genomic make-up showed lower fitness (less seed set) than the other 90%, although these differences were not statistically significant (Supplementary Table 11). This is unexpected given the tight correlation of cytonuclear genomic composition and fertility in earlier generations of the tetraploids48. Perhaps in this case, cytonuclear incompatibility is genetic context dependent, and this phenomenon might be transitory in the earliest evolutionary stages of allopolyploids.
Meiotic stability was variable and evolvable in the TRILs
In principle, HE-mediated rapid homogenization of two divergent genomes would transform an allotetraploid or segmental allopolyploid into an autotetraploid containing a single genome with four identical copies of every chromosome, but each being a mosaic of the two parental genomes7,8. This chromosomal homogeneity would most probably lead to multivalent formation and unbalanced gametes, the result being lowered fertility and fitness. To test this prediction, we compared meiotic chromosome behaviour at metaphase of meiosis I of two TRILs that showed the highest fertilities (TN9-81 and T9N-14) with two TRILs that showed the lowest fertilities (TN9-16 and TN9-17) among the 202 lines (Supplementary Table 12). As controls, we also included the S1 segmental allotetraploids (NN99-S1 and 99NN-S1) and two sensu stricto autotetraploids (Nipponbare-4X and 9311-4X) from newly doubled diploid parental cultivars, Nipponbare and 9311. As expected, we found that NN99-S1 and 99NN-S1 manifested the highest frequencies of bivalents and lowest frequencies of quadrivalents among the tetraploids, while Nipponbare-4X and 9311-4X showed the opposite (Fig. 5a–c). Of note, of the two high-fertility TRILs, one (T9N-14) did not show reduced bivalent frequency and increased total quadrivalent frequency than the most stable S1 segmental allotetraploids (NN99-S1 and 99NN-S1), while the other (TN9-81) showed only marginal differences; in contrast, both low-fertility TRILs (TN9-16 and TN9-17) showed substantially greater differences from the S1 segmental allotetraploids in both bivalent and quadrivalent pairing frequencies (Fig. 5c). More strikingly, of the quadrivalents that could be discernibly classified as chained, alternate and adjacent configurations, the alternate type, which is known as most likely to proceed to a 2:2 balanced chromosome segregation during anaphase13, showed similar frequencies between the two high-fertility TRILs and the S1 segmental allotetraploids, while both low-fertility TRILs and the sensu stricto autotetraploids (Nipponbare-4X and 9311-4X) showed significantly lower frequencies of this quadrivalent configuration (Fig. 5c). Of note, based on further evaluation of the meiotic chromosome behaviour of additional TRILs with varying levels of genomic homozygosity, we did not find a general correlation between meiotic stability and genomic homozygosity (P > 0.8 for both bivalents and quadrivalents, Supplementary Fig. 12).

The autotetraploids are NPB-4X and 9311-4X, and the reciprocal S1 segmental allotetraploids are NN99 and 99NN. a,b, Representative images of chromosome pairing behaviour at metaphase/diakinesis of meiosis I (MI) in PMCs from 5 independent inspections, n = 3 for each genotype. Different chromosomal pairing configurations (adjacent quadrivalent, alternate quadrivalent, bivalent, chain and ambiguous) are indicated. Scale bars, 10 μm. c, Tabulated number (mean ± s.d.) and percent of the various types of pairing configuration at metaphase/diakinesis are from PMCs of at least 3 euploid plants for each of the 8 kinds of tetraploids. Different red letters represent statistical differences in multiple comparisons across the 8 tetraploid types by two-sided one-way ANOVA followed by a post-hoc LSD test (P < 0.05, in total quadrivalents and bivalents columns) and two-sided Chi-square test (FDR-adjusted P < 0.05, in chain, adjacent, alternate and ambiguous columns). d–o, Microscopy images of iodine-potassium-iodide-stained pollens from the diploid parents (Nipponbare and 9311), the reciprocal diploid hybrids (N9-F1 and 9N-F1), the parental autotetraploids (NPB-4X and 9311-4X), the first-generation reciprocal segmental allotetraploids (NN99-S1 and 99NN-S1), two low-fertility TRILs (TN9-16 and TN9-17), and two high-fertility TRILs (TN9-81 and T9N-14), respectively. Scale bars, 100 μm. The snapshot images are representative of each genotype based on 5 independent inspections, n = 3. p, Quantified proportions of fertile pollens across the 12 genotypes. Different red letters indicate statistically significant differences among the 12 genotypes based on two-sided one-way ANOVA followed by a post-hoc LSD test (P < 0.05), and sample sizes are provided in Supplementary Table 25. In boxplots: centre line, median; box limits, lower and upper quartiles; whiskers, minimum and maximum data points. q,r, DAPI-stained images of mature pollens from diploid F1 hybrid (N9-F1) (q) and TRIL (TN9-16) (r). Scale bars, 10 μm. Representative figure from 5 independent inspections, n = 3 for each genotype. s, A combined peak diagram of the DNA content of pollen nuclei from both diploid (red) and tetraploid (blue) rice genotypes (n = 3) based on flow cytometry data. Representative figure from 3 independent experiments, n = 3 for each genotype.
Taken together, our data indicate that (1) meiotic instability is closely associated with fertility, as expected; (2) not all autotetraploidized TRILs necessarily show increased meiotic instability; rather, some can maintain, if not enhance, the level of meiotic stability of their segmental allotetraploid progenitors due to stabilized frequencies of both bivalent formation and the alternate type of quadrivalent configuration, which however is genomic context dependent.
We also assessed pollen fertility of these plants and found that although the trait varied significantly among the tetraploid lines, it did not show a correlation with seed fertility (Fig. 5d–p). Moreover, we did not find evidence for unreduced gametes in the tetraploid lines. Together, neither reduced pollen fertility (to the extent observed) nor unreduced gametes contribute to the striking difference in organismal reproductive fitness between the high- and low-fertility TRILs (Fig. 5q–s).
Phenotypic diversity and agronomic performance of TRILs
To evaluate the phenotypic consequences of variable genomic compositions of the TRILs, we measured 20 morphological and 3 physiological traits in the 202 TRILs along with their diploid parental cultivars (Nipponbare and 9311), autotetraploids of both parents (Nipponbare-4X and 9311-4X), reciprocal diploid F1 hybrids (N9-F1 and 9N-F1) and reciprocal S1 segmental allotetraploids (NN99-F1 and 99NN-F1) in 2 consecutive years (2021–2022) under normal paddy-field conditions, except for germination rate, which was assayed under controlled laboratory conditions (Fig. 6a–d, and Supplementary Tables 13 and 14). Results for the 19 morphological and 3 physiological traits fully concurred between the 2 years (Extended Data Fig. 5), with the key observation being a high level of diversity for all 23 measured traits (Extended Data Fig. 6). Transgressive phenotypes were observed for 20 traits in at least 1 TRIL, and for 8 traits in >50% (129 out of 202) of the TRILs (Extended Data Fig. 6). Overall, all types of tetraploids showed reduced fertility and hence lower seed yield relative to their diploid parental cultivars, as expected; nonetheless, 2 TRILs (TN9-81 and T9N-14) showed significantly higher seed yields in both years under normal paddy-field conditions (Fig. 6g and Supplementary Fig. 13). The high-yield performance of TN9-81 and T9N-14 can be largely attributed to 6 component traits that showed transgressivity, that is, plant height, panicle length, spikelet number per panicle, grain number per panicle, fertility rate and thousand-kernel weight, all of which exceeded the 75th percentile at population level (Supplementary Table 15).

Diploid parents are Nipponbare and 9311, reciprocal diploid F1 hybrids are N9 and 9N, autotetraploids of diploid parents are NPB-4X and 9311-4X, and reciprocal S1 segmental allotetraploids are NN99-S1 and 99NN-S1. a, Overall plant architecture showing plant height, tiller number and tiller angle. b, Illustration and quantification of primary (N) and secondary (n) branch numbers per panicle. c, Fertility per panicle, or the relative amount of filled (top) vs empty (bottom) kernels of a given panicle taken from each of the studied rice plant groups. d, Grain length and grain width. e,f, Illustration of traits reflecting tolerance to two abiotic stresses, 150 mM NaCl (e) and chlorate (2 mM KClO3) (f) by the various rice plant groups. g–i, Quantification of yield per plant (g) and survival rate after salt stress (h) or chlorate treatment (i). Data are means ± s.e.m. Black dots represent individual data points. Different red letters above each histogram indicate statistically significant differences among the various genotypes based on two-sided one-way ANOVA followed by a post-hoc LSD test (P < 0.05), and sample sizes are provided in Supplementary Table 25.
We noted that when comparing the TRILs as two groups according to their crossing directions, 9 traits (flag-leaf length, primary-branch number, thousand-kernel weight, flag-leaf width, secondary-branch number, grain length, panicle length, spikelet number and tiller number) showed significant differences (Extended Data Fig. 7; P < 0.01 by Mann–Whitney U-test). This suggests that although selection for more compatible cytonuclear genomic composition appeared to have largely eroded compared with the earlier generation48, the cytoplasmic origin did affect phenotypes of the TRILs, consistent with previous reports in several organisms at the diploid level57,58. Interestingly, all crossing direction-impacted traits in the TRILs showed superior performance in the direction where the maternal parent performed better than the paternal parent (Extended Data Fig. 7), suggesting a maternal effect in growth and development of the tetraploids.
We also tested possible correlations between genome homozygosity and phenotypic performances in the TRILs. We find that genome homozygosity exhibited a significant negative correlation with 6 traits: plant height, secondary-branch number, spikelet number, grain density, germination rate and yield, and a significant positive correlation with seed length (all Pearson correlation P < 0.05), but no significant correlation with the remaining 17 traits including meiotic stability (Supplementary Fig. 12). These results suggest that there might be no generic correlations between the degrees of genomic homozygosity and overall organismal performance in the TRILs. Instead, genomic context (that is, the variably reshuffled genomic compositions due to HE-mediated mosaicism) is probably the major determinant.
We next assessed performance of the TRILs along with their two diploid parental cultivars, the diploid F1 hybrids (9N-F1 and N9-F1), the S1 segmental allotetraploids (NN99-S1 and 99NN-S1) and autotetraploids of both parents under two abiotic stress conditions, NaCl (salinity) and chlorate (KClO3) (sensitivity to this chemical is a reliable indicator of tolerance to nitrogen (N) deficiency in rice)59,60. In this case, the two diploid parental cultivars are known to show different tolerances to these two stress conditions, with cv. Nipponbare being more tolerant to salinity and less sensitive to chlorate (that is, less tolerant to N deficiency) than cv. 9311 (refs. 61,62). Results indicated that under NaCl (150 mM for 7 days) treatment, 10 TRILs exhibited greater tolerance than the more tolerant parent cv. Nipponbare, while under KClO3 treatment (2 mM for 4 days), 13 TRILs showed significantly higher sensitivity to KClO3 (denoting higher tolerance to N deficiency) than the more sensitive parent cv. 9311, with 3 TRILs showing better performance to both stresses, that is, transgressive phenotypes (Fig. 6e,f,h,i and Supplementary Fig. 14). Consistent results were obtained in 3 independent experiments. Given that only a small proportion of the TRILs showed enhanced tolerance compared with both parents under either or both stress conditions and the S1 segmental allotetraploids showed tolerance levels intermediate of the diploid parents (Fig. 6e,f,h,i), our results indicate that neither WGD per se nor its coupling with a merger of intact parental subgenomes (hybridization) was sufficient to confer the stress-tolerant phenotypes; instead, the traits are apparently conditioned by specific genomic compositions enabled by the accumulated HEs at the tetraploid level. The emergence of the stress-tolerant traits in the TRILs is remarkable given the finite population size of the TRILs and the absence of purposeful selection in any of their preceding generations when potentiating genetic variations were apparently more attainable, that is, the HEs were segregating.
To gain some mechanistic insights on the evolved stress-tolerance traits in the TRILs, we performed integrated transcriptome and methylome analyses on root tissue of salt-stressed and mock-grown seedlings. We made several interesting observations: (1) under salt stress, global gene expression profiles of the two salt-tolerant TRILs were more similar to their own mock plants than to other samples, while profiles of the salt-sensitive TRILs showed similar disparities from all remaining samples, consistent with significantly more differentially expressed genes (DEGs) in the salt-sensitive than in the tolerant TRILs (Fig. 7a and Extended Data Fig. 8a); (2) the 133 ‘salt-stress vs mock’ upregulated DEGs specific to both salt-tolerant TRILs are overrepresented by genes involved in transmembrane transport, tetrapyrrole binding and respiratory burst, whereas the 505 ‘salt-stress vs mock’ downregulated DEGs unique to both salt-sensitive TRILs are enriched in biological pathways involved in response to oxidative stress, transmembrane transport and response to stress (Fig. 7b–d); (3) although salt stress induced substantial CHH hypermethylation in all four studied TRILs, and all with a notable enrichment of differentially methylated regions (DMRs) in the 3-Kb upstream region of gene transcription start sites (TSS) (Extended Data Fig. 8b and Supplementary Fig. 15), we identified 27 genes that exhibited concomitant CHH hypermethylation and upregulated expression in the two salt-tolerant TRILs only. These genes are significantly enriched in stress-related GO terms, including electron transfer, oxidoreductase activity and membrane function, and contain key regulators of osmotic stress tolerance, such as AP2/EREBP transcription factors and NADPH oxidases (Fig. 7e–g).

a, Principal component analysis (PCA) showing cluster stratification based on genomewide gene expression profiles of two salt-tolerant TRILs (ST-TRIL1 and ST-TRIL-2) and two salt-sensitive TRILs (SS-TRIL1 and SS-TRIL2) under both mock control and salt-stress conditions. PC1 and PC2 represent the first and second principal components which respectively account for 32.8% and 13.3% of the variability. The results indicate that global gene expression profiles of the two salt-tolerant TRILs are more similar to their own mock counterparts than to the other samples, while profiles of the two salt-sensitive TRILs are similarly departed from all remaining samples including their own mock counterparts. This suggests that the salt-tolerant and salt-sensitive TRILs are poles apart in their robustness to the stress. b, Venn diagrams showing the relationships of salt stress vs mock DEGs among the 4 TRILs. Numbers indicate the counts of DEGs for various comparisons. Groups I and II (top) highlight the salt stress-induced DEGs that are uniquely upregulated in each of the two salt-tolerant TRILs, while Groups III and IV (bottom) show the salt stress-induced DEGs that are uniquely downregulated in each of the two salt-sensitive TRILs. c, Expression patterns of genes of the 4 groups (I–IV) across the 4 TRILs (depicted by a heat map) and their significantly enriched gene ontology (GO) terms. d, Heat map depicting expression patterns of the 9 identified genes associated with stress-induced biotic respiratory burst in the 4 TRILs, highlighting their significant overexpression in the two salt-tolerant TRILs in response to the stress compared with the two salt-sensitive TRILs. e, Venn diagram showing an intersection analysis reflecting correlation of the Group I genes in c with CHH hypermethylation in the two salt-tolerant TRILs. f, GO enrichment analysis was conducted on 27 genes—those at the centre of the overlap across the 3 gene sets in e—that exhibited both CHH hypermethylation and upregulated expression only in the two salt-tolerant TRILs. g, Heat map depicting the comparison of CHH methylation (blue) and expression levels (red) under the salt stress vs mock conditions for genes that exhibited concomitant CHH hypermethylation and upregulated expression only in the two salt-tolerant TRILs. For c, d and g, the colour scale indicates the gene expression/methylation level normalized by Z-score.
Discussion
Homoeologous exchange catalyses biodiversity of polyploidy
We have reported that rampant homoeologous exchanges (HEs) occurred in the earlier selfed progenies (S4) of the synthetic segmental allotetraploids parented by the japonica and indica rice subspecies48. However, whether HEs could become fixed to enable persistence of the homoeologue-recombinant genotypes that are self-sustainable remained unknown. Here we find that each of the 202 TRILs had a unique genome composition and a distinct composite organismal phenotype (including fitness) due to differences in the number, location and length of fixed HEs. Given that in theory, many different TRILs can be generated from HEs even starting from a single S0 segmental allotetraploid individual, and that each TRIL can become genetically homogeneous after relatively few generations of selfing, our results suggest that HE is a powerful mechanism whereby genetic and phenotypic diversity can be rapidly generated in newly formed allopolyploids.
It can be imagined that, due to decoupling in the newly formed TRILs of co-evolved genes, pathways and networks within each of the hybridizing parental subspecies, the bulk of TRILs is probably maladaptive and inferior in competition with parental taxa, especially under parental environments. However, it is possible that some rare recombinant genotypes might show enhanced fitness in survivorship or reproduction under a specific disturbed environment or novel ecological niche63, which may facilitate their demographic establishment. Thus, our results have revealed a new facet of allopolyploidy-mediated evolution, that is, HE-fuelled genetic differentiation comprising an ‘allo-to-auto’ ploidy-type transition, and which in the process generates novel and transgressive phenotypes. Our empirical data have showcased that this transition process can be highly creative and therefore might be evolutionarily relevant in any taxa for which the polyploidy process is set in motion by hybridization between parents bearing different, but not too different, genomes. Here, the two rice subspecies diverged ~0.44 million years ago64,65 with nucleotide identities being 76.3 ± 26.8% genomewide, and 97.1 ± 5.3% in protein-coding genes based on our analyses (Extended Data Fig. 9 and Supplementary Results). Thus, in principle, our results can be recapitulated in any diploid (sub)species pairs with similar genetic divergences. However, it should be cautioned that one additional critical condition might also be required, that is, the absence of strong homoeologous pairing suppressor (HPSs) genes such as Ph1 in wheat66. Thus, under circumstances where these two essential conditions (that is, moderate parental genetic divergence and absence of HPS) are met, HE by itself appears to be a potent driver of evolutionarily relevant diversification under polyploidy. We should acknowledge, however, that our current data cannot rule out the possibility of the presence of HPSs in the rice tetraploids because it is also possible that the intersubspecific subgenome divergence is insufficient to be discriminated by HPSs during meiotic pairing. This possibility is raised because HPSs usually evolve from pre-existing meiosis genes, such as the Ph1 candidate gene TaZIP4-B2 in polyploid wheat67.
An additional consideration is the relevance of this study to the sorting-out of phylogenetics and systematics of polyploid complexes, which often are challenging. The potential of multiple origins combined with HE-generated diversification comprise conditions that might generate a phenotypically bewildering array of natural derivatives, which of course continue to evolve and diversify and themselves interbreed to various extents68,69. Although a similar awareness of this issue has been raised recently on the basis of a study of synthetic allopolyploids in Brassica, it is from a different aspect, that is, it documents spontaneous reconstitution of a novel subgenome due to merging of whole chromosomes from two parental subgenomes after artificial allopolyploidization70. In contrast, our findings explicitly suggest that some of today’s extant autopolyploid species may have evolved from archaic allopolyploids through HEs, underscoring the need to redirect efforts towards looking for two or more, rather than one, of their possible diploid progenitors, followed by comparative genomic analyses.
The ‘allo-to-auto’ transition is under genetic constraint
Although meiotic HEs occurred rampantly during the selfing process of the indica–japonica tetraploid rice, these are not without intrinsic genetic constraints. As such, although highly variable, the tetraploid recombinant genotypes may not have unlimited genotypic space to explore. Indeed, we document at least two aspects of strong genetic constraints that limit the genomic composition of the TRILs.
One constraint is that all 202 TRILs showed asymmetric contributions by the hybridizing diploid parents, with cv. 9311 contributing more than cv. Nipponbare. Our analyses of transcriptome and methylome of the diploid F1 hybrids and the immediate-generation allotetraploids suggest that the asymmetrical parental contribution to the TRILs was not contributed by overall functional subgenome dominance (Supplementary Fig. 16), as proposed for many other systems71. We posit that the phenomenon in our case might be underpinned by selection for better genome function (for example, genome stability) coupled with higher organismal fitness (for example, germination and fertility) in the process of evolving into the TRILs.
The second constraint is that all TRILs contained a common Chr. 6 segment that was derived exclusively from the cv. 9311 parent. Our results have parallels with findings from the homoploid hybrid Italian sparrow (Passer italiae) in which all four island populations were found to contain a genomic region invariably inherited from the same maternal parent species72. Further analyses in that study showed that this uniparental-derived region was enriched for genes involved in DNA repair, and authors postulated that uniparental inheritance of multigenic DNA repair pathways may need parent-specific inheritance for proper functionality72. However, because no population of the species was derived from the reciprocal cross, and mitonuclear genes were also enriched in the uniparental region, it remains possible that the selective force in that study was cytonuclear compatibility, whereas DNA repair genes were mere hitchhikers.
In our study, we find that all studied TRILs of both reciprocal crosses contain the same Chr. 6 segment of the cv. 9311 parent in all four copies (irrespective of it being the female or male parent), and thus we can unequivocally rule out the possibility of selection for cytonuclear compatibility.
In fact, in our earlier work with an independent cohort of S4 segmental allotetraploid populations, we also observed an overrepresentation of the homo-9311 (Nipponbare:9311 = 0:4) segments on Chrs. 6 and 8, and we posited then that the biased parental legacy might result from selection for early flowering, because the ancestral S1 individuals were grown in northeastern China48. Here, given that all generations of the independently constructed TRILs were grown in a phenologically non-restrictive locality (Tianjin, China), no selection for earlier flowering is expected. We thus suspect that our earlier proposed scenario, which invokes demographic contingency, cannot fully explain the phenomenon. Instead, we find that three DNA repair-related genes, including OsSOG1, an important player in DNA damage responses in rice73, are probably functionally differentiated between the parental alleles due to missense mutations in critical domains. This lends support to the previous proposition that DNA repair genes need uniparental origin for proper function in admixed hybrid genomes72. In addition, we also identified three functionally important genes, which had transposed to other genomic loci in the Nipponbare genome. Assuming these genes are essential to genome function and/or organismal fitness of the TRILs, the selective retention of the single 1.7-Mb segment of 9311 is apparently more attainable than the otherwise co-retention of this segment of Nipponbare and additional segments harbouring these transposed genes. Nevertheless, we acknowledge that the mechanisms underlying the exclusive retention of this 1.7-Mb segment of 9311 remains speculative and requires further experimental validation.
Restabilized meiosis and high fertility of some TRILs
According to traditional genetic tenets, autopolyploids should be meiotically more unstable than allopolyploids, and accordingly, the former are often less fertile than the latter. Thus, it is expected that due to HE-catalysed homogenization, the quasi-autotetraploids (TRILs) we developed would exhibit exacerbated meiotic instability and reduced fertility. Indeed, we find that majority of the TRILs manifested this predicted outcome due to significantly decreased proportions of bivalent and increased proportions of quadrivalent pairing compared with the immediately formed segmental allotetraploids. However, this general trend did not apply to all of the TRILs. Specifically, at least two (TN9-81 and T9N-14) of the 202 arbitrarily developed TRILs (without selection for any organismal traits including fertility at any of the 12 generations) that manifested the highest fertilities actually showed a meiosis that is as stable as or even more stable than that of the S1 segmental allotetraploids (NN99-S1 and 99NN-S1) containing largely intact subgenomes (undergoing only one meiosis). We further show that the restabilized meiosis of these two high-fertility TRILs is mainly due to concurrent higher levels of bivalent and the alternate-type quadrivalent pairing configurations compared with the low-fertility lines.
Of note, our results agree with a previous meta-analysis, which concluded that with respect to neopolyploidy, there is no evidence to support the traditional view that autopolyploids possess lower fertility than allopolyploids13. Because meiosis stability is often causally linked to fertility in neopolyploidy, it may be suggested that autopolyploidy might rapidly evolve stable meiosis. Indeed, this has been observed in synthetic polyploids of several other plant taxa after multigenerational selection for higher fertility13,74. Together, it may be suggested that rapid evolution of meiotic stability in newly formed plant polyploids is probably a common biological property, consistent with the often recurrent and ubiquitous occurrence of historical WGDs in the entire plant kingdom1,5,23,75. However, the genetic and/or epigenetic basis underlying rapid evolution of meiotic stability in nascent plant polyploids remains largely mysterious, although some intriguing insights have emerged recently, such as the rapid evolution of crossover interference and reduced crossover rate28,76.
Some TRILs have agronomic potential
Compared with their wild progenitors, domesticated crops possess a suite of distinct traits collectively known as ‘domestication syndromes’. Relative to natural selection, conscious human selection is more intense and thus more effective, enabling rapid fixation of domestication traits, as recognized by Darwin37. Recent studies have highlighted this, showing that domestication typically is a protracted process in which repeated hybridizations between the focal domesticating crops with their wild progenitors or relatives may have been essential to generate their diversification and broad adaptation to different agro-ecological environments77. A related issue is that the utilization of wild germplasm in crop breeding programmes entails extensive backcrossing to purge wild alleles of domestication syndrome-determining genes. In contrast, the approach we report here does not involve wild taxa, and as such, the stabilized TRILs used a ploidy difference from their hybridizing parental cultivars, which themselves are already highly adapted. Thus, the TRILs may more directly become novel crops if showing robust superior agronomic performance under a given agricultural setting. A classic example of allopolyploidization-enabled one-step creation of a ploidy-altered novel crop is the octaploid Triticale (BBAADDRR), which was achieved by hybridizing bread wheat (Triticum aestivum, BBAADD) with rye (Secale cereale, RR), both being domesticated crops. We note that in contrast to this classical approach in which each allopolyploidization event generates only one homogeneous ploidy-increased progeny line, our ‘allo-to-auto’ ploidy-type transition approach produces many different TRILs, each of which has a unique suite of composite phenotypes for breeders to evaluate. We show here that these phenotypes include multiple transgressive agronomically important features, underscoring the potential agricultural application of HE-driven rapid crop germplasm innovation.
Advantages of the TRILs compared with diploid heterosis
We consider that our japonica–indica intersubspecific tetraploid rice hybrid system has at least two salient advantages over diploid rice F1 hybrids, that is, fertility restoration and potential intersubspecific heterosis exploitation. Diploid japonica–indica F1 hybrids are notoriously known as having low fertility, posing a major hindrance to agricultural use of its otherwise large heterosis. In comparison, the immediately genome-doubled S0 segmental allotetraploids show significantly enhanced fertility but are meiotically unstable. This suggests that, if stable and divergent TRILs can be developed, they may serve as crossing parents for ‘tetraploid hybrid rice’ breeding (heterosis utilization). Alternatively, if the developed TRILs have agronomic performances that are already similar to, or even better than, the diploid F1 hybrids (for example, as shown in Arabidopsis intraspecific hybrid progenies)31,78, they may directly become tetraploid rice cultivars. Our study has clearly shown, as a proof-of-concept, concrete feasibility of the second possibility, while the first possibility remains to be tested by crossing the selected stable TRILs.
Methods
Plant materials and phenotyping
A total of 202 rice quasi-tetraploid recombinant inbred lines were obtained through 12 generations of selfing by single-seed descent from reciprocal segmental allotetraploid rice, generated via intersubspecific hybridization (japonica cv. Nipponbare × indica cv. 9311) followed by colchicine-induced whole-genome doubling (Extended Data Fig. 1a). All TRILs, along with the control genotypes, were grown in an experimental paddy field in Tianjin, China. Phenotyping was conducted for 20 morphological and 3 physiological traits related to vegetative and reproductive growth, including fertility, over 2 consecutive years (2021–2022), except for germination rate, which was measured under controlled laboratory conditions. Each line was evaluated using at least 30 (10 in year 2021, and 20 in year 2022) independent individuals. In parallel, a diploid reciprocal recombinant inbred line (DRIL) population comprising 218 lines derived from the same parents as the TRILs was included (Extended Data Fig. 1a). In addition, 6 available ancestral S1 individuals of the TRILs and 67 independently reconstructed S1 segmental allotetraploids were used in this study. Details of the construction and pedigrees for TRILs and DRILs, morphological data comparisons and statistical analyses are provided in Supplementary Methods.
Dual-colour Oligo-FISH
Dual-colour Oligo-FISH analysis was performed essentially as reported79. Two sets of rice oligo libraries were designed using Chorus2 software (v.2.0)80 (https://github.com/zhangtaolab/Chorus2) and labelled with FAM-green and Texas red using a direct labelling protocol79. FISH was performed as reported81. Slides were examined under an Olympus fluorescence microscope and digitally photographed.
Nucleic acid extraction, sequencing and data analyses
Genomic DNA for whole-genome resequencing was extracted from young leaves of one euploid individual for each TRIL, one randomly selected individual of each DRIL, and 73 S1 segmental allotetraploids. For RNA-seq and whole-genome bisulfite sequencing, RNA and DNA were respectively extracted from immature panicles (<2 cm) and roots of key representative genotypes (detailed in Extended Data Fig. 1b). Library construction and sequencing were performed following standard protocols. Detailed procedures for nucleic acid extraction, sequencing and preliminary data analysis are provided in Supplementary Methods.
Homozygosity, HE loci and breakpoints in the TRILs
Genomic composition and HEs were determined essentially as reported48. Genomic homozygosity is defined as the proportion of homozygous chromosomal segments (parental allele composition 4:0 or 0:4) across the entire genome. For each TRIL, homozygosity was calculated as the total length of homozygous segments divided by the total genome size (Extended Data Fig. 10a). For fine mapping of HE breakpoints within a 5-Kb genomic bin, homoeologous ratios between Nipponbare and 9311 based on each single-nucleotide polymorphism (SNP) were calculated to locate an HE breakpoint into two adjacent SNPs. Only the length of determined HE breakpoint region <3 Kb were kept for further analyses. Detailed procedures are provided in Supplementary Methods.
Estimation of recombination rates using co-parental DRILs
A high-density linkage map of rice was constructed using our DRIL population that contains 218 lines. In total, 2,383 recombination bins showing variable genotypes across all DRILs were used in construction of the high-density linkage map. Local recombination rate was estimated using the loess method in MareyMapOnline (https://lbbe-shiny.univ-lyon1.fr/MareyMap-Online/)82. HR breakpoints in DRILs were identified using the same approach as that used for HE breakpoints in TRILs. Details are provided in Supplementary Methods.
Defining genomic similarity of parental genomes
Comparative genomic analysis of the two diploid parental genomes was conducted following a published pipeline83, with the rice japonica cv. Nipponbare (MSU7.0) and indica cv. 9311 reference genomes (http://ricerc.sicau.edu.cn/RiceRC). Genomic similarity between the two diploid parental genomes was defined as the proportion of genomic segments with identical nucleotide sequences within each 1-Mb window, as illustrated in the schematic diagram in Extended Data Fig. 10b. Detailed procedures and the calculation formula are provided in Supplementary Methods.
Cytogenetic analysis of meiosis, pollen fertility and ploidy
Cytogenetic analysis of meiosis was performed as previously described84 using young panicles from all tetraploid control genotypes and eleven representative TRILs, including two with the highest and two with the lowest fertilities. In parallel, pollen fertility was assessed using the iodine-potassium iodide staining method85, and pollen ploidy was determined by flow cytometry following established protocols86. These analyses were conducted for all control genotypes (both diploid and tetraploid) and four TRILs (the same two with the highest and two with the lowest fertilities).
Bioinformatic Analyses
This study encompassed a range of bioinformatics analyses, including whole-genome resequencing, transcriptome, methylome, ATAC-seq and ChIP-seq analyses, as well as TE content analysis in the parental genomes. The relationships between the analyses, and the plant materials used are shown in Extended Data Fig. 1b. Additional analyses included predicting functional differentiation between parental alleles of the 9311 Chr. 6 segment, examining parental genetic differences in the three DNA repair-related genes, and conducting haplotype analysis of these three genes in the 3K Rice Genome (RG) Panel. Detailed methodologies for each analysis are provided in Supplementary Methods.
Assessing salt tolerance and nitrogen-use efficiency
All 202 TRILs, together their diploid parental cultivars (Nipponbare and 9311), were assessed for salt tolerance and nitrogen-use efficiency. Seeds were germinated at 28 °C in the dark for 3 days and then transferred to seed trays floating on a culture solution with slow-rotation aeration under a 16/8 h light/dark photoperiod in a greenhouse. The salt and chlorate treatments were applied when the seedlings reached the 4-leaf and 2-leaf stages, respectively. For salt treatment, plants were treated with culture solution containing 150 mM NaCl for 7 days and allowed to recover for another 7 days. For chlorate treatment, the plants were treated with modified culture solution (using 2 mM KClO3 to replace 1.44 mM NH4NO3) for 4 days, followed by 7 days recovey. Three independent experiments were conducted for each of the treatments, and mock controls were included in all experiments (Supplementary Fig. 14). Survival rates were tabulated. In addition, an independent salt-treatment experiment was performed using two salt-tolerant TRILs and two salt-sensitive TRILs. Seedlings (4-leaf stage) were treated with 125 mM NaCl for 24 h to collect root tissues from both mock and salt-stressed groups for RNA-seq analysis.
Statistics
All statistical tests in this study were performed using basic packages in R (v.4.2.0, https://www.r-project.org).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The clean data for genome resequencing of the 202 tetraploid inbred recombinant lines (TRILs), 73 S1 tetraploid individuals and 218 diploid recombinant inbred lines (DRILs), as well as the transcriptome and methylome data of diploid parents, F1 hybrid, S0 allotetraploid, and representative salt-tolerant and -sensitive TRILs generated in this study have been deposited in the Sequence Read Archive (SRA) under the accession code PRJNA1224316 (https://www.ncbi.nlm.nih.gov/sra/). ChIP-seq data for H3K27me3, H3K9ac, H3K4me3 and H3K36me3 (note that H3K36me3 data are only available for Nipponbare), as well as ATAC-seq data from both Nipponbare and 9311, were retrieved from publicly available databases87,88,89. These datasets are also available in the SRA under the accession numbers SRR034623, SRR034636, SRR034649, SRR094791 and SRR10991557. Source data are provided with this paper.
Code availability
The custom Perl and R scripts used in this study are from our previously published work48 and are publicly available via GitHub at http://github.com/wuying003/HE-identification-for-allotetraploid-rice.
References
Wendel, J. F. Genome evolution in polyploids. Plant Mol. Biol. 42, 225–249 (2000).
Otto, S. P. & Whitton, J. Polyploid incidence and evolution. Annu. Rev. Genet. 34, 401–437 (2000).
Soltis, P. S., Marchant, D. B., Van de Peer, Y. & Soltis, D. E. Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev. 35, 119–125 (2015).
Wendel, J. F. The wondrous cycles of polyploidy in plants. Am. J. Bot. 102, 1753–1756 (2015).
Van de Peer, Y., Mizrachi, E. & Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424 (2017).
Charron, G., Marsit, S., Hénault, M., Martin, H. & Landry, C. R. Spontaneous whole-genome duplication restores fertility in interspecific hybrids. Nat. Commun. 10, 4126 (2019).
Gaeta, R. T. & Chris Pires, J. Homoeologous recombination in allopolyploids: the polyploid ratchet. New Phytol. 186, 18–28 (2010).
Mason, A. S. & Wendel, J. F. Homoeologous exchanges, segmental allopolyploidy, and polyploid genome evolution. Front. Genet. 11, 1014 (2020).
Buggs, R. J. A., Soltis, P. S. & Soltis, D. E. Does hybridization between divergent progenitors drive whole-genome duplication? Mol. Ecol. 18, 3334–3339 (2009).
Buggs, R. J. A. et al. Transcriptomic shock generates evolutionary novelty in a newly formed, natural allopolyploid plant. Curr. Biol. 21, 551–556 (2011).
Barker, M. S., Arrigo, N., Baniaga, A. E., Li, Z. & Levin, D. A. On the relative abundance of autopolyploids and allopolyploids. New Phytol. 210, 391–398 (2016).
Spoelhof, J. P., Soltis, P. S. & Soltis, D. E. Pure polyploidy: closing the gaps in autopolyploid research. J. Syst. Evol. 55, 340–352 (2017).
Ramsey, J. & Schemske, D. W. Neopolyploidy in flowering plants. Annu. Rev. Ecol. Evol. Syst. 33, 589–639 (2002).
Comai, L. The advantages and disadvantages of being polyploid. Nat. Rev. Genet. 6, 836–846 (2005).
Otto, S. P. The evolutionary consequences of polyploidy. Cell 131, 452–462 (2007).
Soltis, P. S. & Soltis, D. E. The role of hybridization in plant speciation. Annu. Rev. Plant Biol. 60, 561–588 (2009).
Levin, D. A. Minority cytotype exclusion in local plant populations. Taxon 24, 35–43 (1975).
Bohutínská, M. et al. Polyploids broadly generate novel haplotypes from trans-specific variation in Arabidopsis arenosa and Arabidopsis lyrata. PLoS Genet. 20, e1011521 (2024).
Marand, A. P. et al. Meiotic crossovers are associated with open chromatin and enriched with Stowaway transposons in potato. Genome Biol. 18, 203 (2017).
Leebens-Mack, J. H. et al. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685 (2019).
Rice, A. et al. The global biogeography of polyploid plants. Nat. Ecol. Evol. 3, 265–273 (2019).
Doyle, J. J., Doyle, J. L., Brown, A. H. & Palmer, R. G. Genomes, multiple origins, and lineage recombination in the Glycine tomentella (Leguminosae) polyploid complex: histone H3-D gene sequences. Evolution 56, 1388–1402 (2002).
Soltis, P. S., Soltis, D. E. & Chase, M. W. Angiosperm phylogeny inferred from multiple genes as a tool for comparative biology. Nature 402, 402–404 (1999).
Tate, J. A., Soltis, D. E. & Soltis, P. S. in The Evolution of the Genome (ed. Gregory, T. R.) 371–426 (Elsevier, 2005).
McClintock, B. The significance of responses of the genome to challenge. Science 226, 792–801 (1984).
Adams, K. L. & Wendel, J. F. Polyploidy and genome evolution in plants. Curr. Opin. Plant Biol. 8, 135–141 (2005).
Choi, K. & Henderson, I. R. Meiotic recombination hotspots – a comparative view. Plant J. 83, 52–61 (2015).
Gonzalo, A. All ways lead to Rome—meiotic stabilization can take many routes in nascent polyploid plants. Genes 13, 147 (2022).
Shimizu, K. K. Robustness and the generalist niche of polyploid species: genome shock or gradual evolution? Curr. Opin. Plant Biol. 69, 102292 (2022).
Fujimoto, R., Taylor, J. M., Shirasawa, S., Peacock, W. J. & Dennis, E. S. Heterosis of Arabidopsis hybrids between C24 and Col is associated with increased photosynthesis capacity. Proc. Natl Acad. Sci. USA 109, 7109–7114 (2012).
Greaves, I. K., Groszmann, M., Wang, A., Peacock, W. J. & Dennis, E. S. Inheritance of trans chromosomal methylation patterns from Arabidopsis F1 hybrids. Proc. Natl Acad. Sci. USA 111, 2017–2022 (2014).
Greaves, I. K. et al. Twenty-four–nucleotide siRNAs produce heritable trans-chromosomal methylation in F1 Arabidopsis hybrids. Proc. Natl Acad. Sci. USA 113, E6895–E6902 (2016).
Greaves, I. K. et al. Trans chromosomal methylation in Arabidopsis hybrids. Proc. Natl Acad. Sci. USA 109, 3570–3575 (2012).
Chodavarapu, R. K. et al. Transcriptome and methylome interactions in rice hybrids. Proc. Natl Acad. Sci. USA 109, 12040–12045 (2012).
Deb, S. K., Edger, P. P., Pires, J. C. & McKain, M. R. Patterns, mechanisms, and consequences of homoeologous exchange in allopolyploid angiosperms: a genomic and epigenomic perspective. New Phytol. 238, 2284–2304 (2023).
Bertioli, D. J. et al. The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat. Genet. 51, 877–884 (2019).
Darwin, C. On the origin of species by means of natural selection, or the preservation of favoured races in the struggle for life. Br. Foreign Med. Chir. Rev. 25, 367–404 (1860).
Chester, M. et al. Extensive chromosomal variation in a recently formed natural allopolyploid species, Tragopogon miscellus (Asteraceae). Proc. Natl Acad. Sci. USA 109, 1176–1181 (2012).
Chu, Y. et al. Homoeologous recombination is recurrent in the nascent synthetic allotetraploid Arachis ipaënsis × Arachis correntina4x and its derivatives. G3 11, jkab066 (2021).
Flagel, L. E., Wendel, J. F. & Udall, J. A. Duplicate gene evolution, homoeologous recombination, and transcriptome characterization in allopolyploid cotton. BMC Genomics 13, 302 (2012).
He, Z. et al. Extensive homoeologous genome exchanges in allopolyploid crops revealed by mRNAseq-based visualization. Plant Biotechnol. J. 15, 594–604 (2017).
Hurgobin, B. et al. Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 16, 1265–1274 (2018).
Lashermes, P., Hueber, Y., Combes, M. C., Severac, D. & Dereeper, A. Inter-genomic DNA exchanges and homeologous gene silencing shaped the nascent allopolyploid coffee genome (Coffea arabica L.). G3 6, 2937–2948 (2016).
Zhang, Z. et al. Homoeologous exchanges occur through intragenic recombination generating novel transcripts and proteins in wheat and other polyploids. Proc. Natl Acad. Sci. USA 117, 14561–14571 (2020).
Edger, P. P. et al. Origin and evolution of the octoploid strawberry genome. Nat. Genet. 51, 541–547 (2019).
Bomblies, K. & Madlung, A. Polyploidy in the Arabidopsis genus. Chromosome Res. 22, 117–134 (2014).
Addo Nyarko, C. & Mason, A. S. Non-homologous chromosome pairing: sequence similarity or genetic control? Trends Genet. 38, 419–421 (2022).
Wu, Y. et al. Genomic mosaicism due to homoeologous exchange generates extensive phenotypic diversity in nascent allopolyploids. Natl Sci. Rev. 8, nwaa277 (2021).
Xu, C. et al. Genome-wide disruption of gene expression in allopolyploids but not hybrids of rice subspecies. Mol. Biol. Evol. 31, 1066–1076 (2014).
Sybenga, J. Chromosome pairing affinity and quadrivalent formation in polyploids: do segmental allopolyploids exist? Genome 39, 1176–1184 (1996).
Epstein, R. et al. Exploring impact of recombination landscapes on breeding outcomes. Proc. Natl Acad. Sci. USA 120, e2205785119 (2023).
Tian, Z. et al. Do genetic recombination and gene density shape the pattern of DNA elimination in rice long terminal repeat retrotransposons? Genome Res. 19, 2221–2230 (2009).
Marand, A. P. et al. Historical meiotic crossover hotspots fueled patterns of evolutionary divergence in rice. Plant Cell 31, 645–662 (2019).
Underwood, C. et al. Epigenetic activation of meiotic recombination near Arabidopsis thaliana centromeres via loss of H3K9me2 and non-CG DNA methylation. Genome Res. 28, 519–531 (2018).
Wang, L. et al. DNA hypomethylation in tetraploid rice potentiates stress-responsive gene expression for salt tolerance. Proc. Natl Acad. Sci. USA 118, e2023981118 (2021).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Barton, N. H. The role of hybridization in evolution. Mol. Ecol. 10, 551–568 (2001).
Roux, F. et al. Cytonuclear interactions affect adaptive traits of the annual plant Arabidopsis thaliana in the field. Proc. Natl Acad. Sci. USA 113, 3687–3692 (2016).
Teng, S. et al. QTLs and candidate genes for chlorate resistance in rice (Oryza sativa L). Euphytica 152, 141–148 (2006).
Zhang, Z. S. et al. Rice NIN-LIKE PROTEIN 3 modulates nitrogen use efficiency and grain yield under nitrate-sufficient conditions. Plant Cell Environ. 45, 1520–1536 (2022).
Gao, Z. et al. The indica nitrate reductase gene OsNR2 allele enhances rice yield potential and nitrogen use efficiency. Nat. Commun. 10, 5207 (2019).
Hu, B. et al. Variation in NRT1.1B contributes to nitrate-use divergence between rice subspecies. Nat. Genet. 47, 834–838 (2015).
Rieseberg, L. H. et al. Major ecological transitions in wild sunflowers facilitated by hybridization. Science 301, 1211–1216 (2003).
Ma, J. & Bennetzen, J. L. Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl Acad. Sci. USA 101, 12404–12410 (2004).
Zhu, Q. & Ge, S. Phylogenetic relationships among A-genome species of the genus Oryza revealed by intron sequences of four nuclear genes. New Phytol. 167, 249–265 (2005).
Griffiths, S. et al. Molecular characterization of Ph1 as a major chromosome pairing locus in polyploid wheat. Nature 439, 749–752 (2006).
Rey, M.-D. et al. Exploiting the ZIP4 homologue within the wheat Ph1 locus has identified two lines exhibiting homoeologous crossover in wheat-wild relative hybrids. Mol. Breed. 37, 95 (2017).
Doyle, J. J. & Egan, A. N. Dating the origins of polyploidy events. New Phytol. 186, 73–85 (2010).
Grant, V. Plant Speciation (Columbia Univ. Press, 1981).
Katche, E. et al. Stable, fertile lines produced by hybridization between allotetraploids Brassica juncea (AABB) and Brassica carinata (BBCC) have merged the A and C genomes. New Phytol. 230, 1242–1257 (2021).
Cheng, F. et al. Gene retention, fractionation and subgenome differences in polyploid plants. Nat. Plants 4, 258–268 (2018).
Runemark, A. et al. Variation and constraints in hybrid genome formation. Nat. Ecol. Evol. 2, 549–556 (2018).
Nishizawa-Yokoi, A. et al. SUPPRESSOR OF GAMMA RESPONSE 1 plays rice-specific roles in DNA damage response and repair. Plant Physiol. 191, 1288–1304 (2023).
Li, L., Liu, K. H. & Sheen, J. Dynamic nutrient signaling networks in plants. Annu. Rev. Cell Dev. Biol. 37, 341–367 (2021).
Jiao, Y. et al. Ancestral polyploidy in seed plants and angiosperms. Nature 473, 97–100 (2011).
Morgan, C. et al. Evolution of crossover interference enables stable autopolyploidy by ensuring pairwise partner connections in Arabidopsis arenosa. Curr. Biol. 31, 4713–4726.e4 (2021).
Purugganan, M. D. Evolutionary insights into the nature of plant domestication. Curr. Biol. 29, R705–R714 (2019).
Wang, L. et al. Hybrid mimics and hybrid vigor in Arabidopsis. Proc. Natl Acad. Sci. USA 112, E4959–E4967 (2015).
Liu, X. et al. Dual-color oligo-FISH can reveal chromosomal variations and evolution in Oryza species. Plant J. 101, 112–121 (2020).
Zhang, T. et al. Chorus2: design of genome‐scale oligonucleotide‐based probes for fluorescence in situ hybridization. Plant Biotechnol. J. 19, 1967–1978 (2021).
Zhang, H. et al. Persistent whole-chromosome aneuploidy is generally associated with nascent allohexaploid wheat. Proc. Natl Acad. Sci. USA 110, 3447–3452 (2013).
Siberchicot, A., Bessy, A., Guéguen, L. & Marais, G. A. B. MareyMap online: a user-friendly web application and database service for estimating recombination rates using physical and genetic maps. Genome Biol. Evol. 9, 2506–2509 (2017).
Li, G. et al. Patterns of chromosomal variation, homoeologous exchange, and their relationship with genomic features in early generations of a synthetic rice segmental allotetraploid. Int. J. Mol. Sci. 24, 6065 (2023).
Cheng, Z. Analyzing meiotic chromosomes in rice. Methods Mol. Biol. 990, 125–134 (2013).
Mu, N. et al. MUS81 is required for atypical recombination intermediate resolution but not crossover designation in rice. New Phytol. 237, 2422–2434 (2023).
Storme, N. & Geelen, D. The Arabidopsis mutant jason produces unreduced first division restitution male gametes through a parallel/fused spindle mechanism in meiosis II. Plant Physiol. 155, 1403–1415 (2011).
He, G. et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell 22, 17–33 (2010).
Liang, Z. et al. Reorganization of the 3D chromatin architecture of rice genomes during heat stress. BMC Biology 19, 53 (2021).
Edgar, R., Domrachev, M. & Lash, A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002).
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFF1003303), the National Natural Science Foundation of China (31991211), the Key Research and Development Program of Jilin Province (20210202003NC) and by the US NSF plant genome programme.
Author information
Authors and Affiliations
Contributions
Y.W., B.L., L.G. and J.F.W. conceived and designed the study. X.H., J.L., G.L., Z.Z., T. Lian, C.X. and L.G. conducted all the analyses. X.H., B.Z., T. Luo, R.L. and X.C. performed the experiments. X.H., J.L., G.L. and X.L. did the field phenotyping. X.H., Y.W., L.G., B.L. and J.F.W. wrote the manuscript with input from all authors. All authors contributed substantially to this study, read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Plants thanks Feng Cheng, Leandro Quadrana and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Construction of designed diploid and tetraploid rice systems used for the various study contents.
a, The construction processes and pedigrees of tetraploid recombinant inbred lines (TRILs) and diploid recombinant inbred lines (DRILs). Both TRILs and DRILs were developed after 12 consecutive generations of selfing via single-seed descent (SSD). The arrows indicate key events, including hybridization, colchicine-induced whole-genome duplication (WGD), and selfing. b, Specification of the study contents (green boxes) and the corresponding plant materials used (pink boxes). WGS and WGBS are abbreviations for whole-genome sequencing and whole-genome bisulfite sequencing, respectively. In total, eight genotypes served as parental lines for, or comparison controls with, the TRILs and DRILs: diploid parents (cv. Nipponbare and 9311), autotetraploids (Nipponbare-4X and 9311-4X), diploid F1 hybrids (N9-F1 and 9N-F1), and segmental allotetraploids (NN99-S0/S1 and 99NN-S0/S1).
Extended Data Fig. 2 Chromosomal distribution of homoeologous exchanges (HEs) in the TRILs, homologous recombinations (HRs) in the DRILs, and cytological visualization of crossovers (COs) in all genotypes.
a, Chromosomal distribution of breakpoints of HEs in 202 TRILs and HRs in 218 DRILs, based on whole-genome resequencing data. Each vertical bar represents a breakpoint, with purple indicating HEs and green indicating HRs. Chromosomes are denoted by thick grey lines, and centromeres are marked by red dots. b-d, Cytological visualization and quantification of chiasmata (the cytological manifestation of CO events) during meiosis I (MI) in various genotypes, including diploid parents (Nipponbare and 9311), diploid F1 hybrids (N9-F1 and 9N-F1), autotetraploids (NPB-4X and 9311-4X), segmental allotetraploids (NN99-S1 and 99NN-S1), two TRILs with low-fertility (TN9-16 and TN9-17) and two TRILs with high-fertility (TN9-81 and T9N-14). b-c, representative images of chiasmata in a diploid F1 hybrid (N9-F1) and a TRIL (TN9-16), respectively. The experiment was repeated independently for five times, and all gave the same results. Bars = 10 μm. d, Quantification of the number of chiasmata during MI for the 12 genotypes, with red letters indicating statistically significant differences among the 12 genotypes based on two-sided one-way ANOVA followed by a post-hoc LSD test (P < 0.05). Box plot elements: centre line, median; box limits, lower and upper quartiles; whiskers, minimum and maximum data points. e, A line chart depicting the distribution of breakpoints of HEs (upper) and HRs (lower) aggregated from the 202 TRILs and 218 DRILs, respectively. A 500-Kb sliding window was used for tabulation, and centromere positions are indicated on each chromosome with black dots. NPB is short for Nipponbare. Sample sizes (n) in d are provided in Supplementary Table 25.
Extended Data Fig. 3 Nucleotide sequence comparison of the ‘core’ 1.7-Mb segment on Chromosome 6 between the diploid parents, experimental validation of single nucleotide polymorphisms (SNPs) in OsSOG1, and predicted functional differentiation in OsSOG1 between the rice subspecies (indica vs. japonica).
a, Diagrammatic depiction of the quantitative (copy number) or qualitative (presence vs. absence) correspondence of the annotated genes between Nipponbare and 9311 within the ‘core’ 1.7-Mb segment on chromosome 6, which fall into five groups. Blue and yellow filled boxes represent genes located within the focal region, while unfilled boxes indicate genes outside the region. b, IGV (Integrative Genomics Viewer) screenshots showing four SNPs between the OsSOG1 alleles of Nipponbare (NPB) and 9311, based on reference genomes. Nature of the SNPs (sense, missense or intronic) are indicted. c, Alignment of the full-length coding sequences of the two parental alleles of OsSOG1 based on cDNA cloning and Sanger sequencing, which validated the SNPs identified in b. The potential functional consequences of the two missense SNPs located in the exons are predicted based on the SIFT scores accessed from the Ensembl Plants database (https://plants.ensembl.org/).
Extended Data Fig. 4 Chromosomal segments showing distorted segregation in the TRILs, identified by whole-genome resequencing.
In total, 14 segments were identified. Of these, 12 segments showed distorted segregation but no evidence for parent-of-origin effect. These include six segments (Group 1) biased towards the 9311 parent (at distortion threshold 85%) and six segments (Group 2) biased towards Nipponbare (at distortion threshold 75%). Two additional segments (Group 3) showed clear parent-of-origin effect, that is, biased towards the maternal parent (at distortion threshold 90%), suggesting they were likely under cytonuclear selection. Chromosomal positions and sizes of all three groups of distorted segregation segments are indicated. Each row in the heatmap denotes a single TRIL, with the five possible homoeologous ratios of Nipponbare vs. 9311 alleles labelled with different colors. Crossing directions of the TRILs are labeled on the left side of the heatmap.
Extended Data Fig. 5 Correlations between two independent sets of phenotypic data for 22 traits.
The data were based on field testing in two consecutive years (2021 - 2022) of the S12 and S13 TRILs at the same locality (Tianjin, China, 39.1252° N, 117.0153° E). The x-axis and y-axis are the data of the S12 and S13 TRILs, respectively. For each trait, correlation coefficient (r) and P-values were calculated by two-sided Pearson correlation test and labeled on each diagram. Shaded areas depict 95% confidence intervals. The units for the data presented in each scatter plot are labeled in the lower-right corner of the corresponding plot. Sample size (n) = 202 for each trait.
Extended Data Fig. 6 Frequency distribution of 23 traits across the various genotypes.
The vertical yellow and green bars in the histograms denote the NN99 and 99NN TRILs, respectively, alongside the diploid parents, Nipponbare (dashed blue line) and 9311 (dashed red line), the diploid F1 hybrids (light green and dark blue dashed lines), and two high-yielding TRILs (dashed purple and brown lines). The numbers in red and blue at the top corner of each histogram denote the proportions (%) of TRILs exhibiting over-transgressivity (higher than the higher diploid parent) and under-transgressivity (lower than the lower diploid parent), respectively, for each trait. NPB stands for Nipponbare. Sample sizes (n) = 102 and 100 for NN99 and 99NN TRILs, respectively.
Extended Data Fig. 7 Differences in the distribution of nine morphological traits between the TRILs due to parent-of-origin effects.
The gray and black columns represent the NN99 and 99NN TRILs, respectively, while the dashed vertical lines denote the mean values of Nipponbare (red) and 9311 (blue). Of note, all these nine traits also showed distinct differences between the diploid parents, Nipponbare and 9311; moreover, in each trait, the maternal parent is superior to the paternal parent, indicating maternal dominance in the parent-of-origin effects on these traits. The P labeled in each panel indicates statistically different distribution of the traits in each comparison based on two-sided Mann-Whitney U test. Sample sizes (n) = 102 and 100 for NN99 and 99NN TRILs, respectively.
Extended Data Fig. 8 Analyses of genome-wide alteration of gene expression and DNA methylation in salt-tolerant and -sensitive TRILs in response to the stress.
a, Volcano plots showing differentially expressed genes (DEGs) in the two salt-tolerant TRILs (ST-TRIL1 and ST-TRIL2) and two salt-sensitive TRILs (SS-TRIL1 and SS-TRIL2) vs. their respective mock controls. The dashed line on the x-axis indicates a significance threshold of P = 0.05, while the dashed lines on the y-axis represent log2 (FoldChange) values of ± 1. The number of DEGs is indicated above each panel, with blue and red representing down- and up-regulated genes, respectively, while black representing genes with no significant difference in expression. The x-axis represents log2 (FoldChange), and the y-axis represents the -log10 (FDR-adjusted P). b, DNA methylation patterns in all three cytosine contexts (CG, CHG, and CHH) within gene bodies, DNA transposable elements (DNA-TE), and retrotransposons (Retro-TE), and their 3-Kb upstream and downstream regions in the two salt-tolerant TRILs (ST-TRIL1 and ST-TRIL2) and two salt-sensitive TRILs (SS-TRIL1 and SS-TRIL2). The green lines represent methylation levels in the mock control, while the red lines represent methylation levels under salt treatment.
Extended Data Fig. 9 Comparative genomic analyses between the parental genomes.
a, Synteny and rearrangement (SyRI) plot depicting comparisons between Nipponbare and 9311 for each of the 12 chromosomes. Alignments were constructed using MUMmer. SyRI was used to identify variations. Syntenic regions, inversions, translocations, and duplications are highlighted by different colors. b, Sequence difference between the Nipponbare and 9311 genomes across the 12 chromosomes. The numbers on the left panel represent the average values and standard deviations (SDs) of genomic differences for each chromosome, while the right panel shows genomic difference per 1-Mb window across the genome by a line chart, with red dots indicating the centromere positions. c, Conserved synteny between Nipponbare and 9311 in each chromosome. Syntenic blocks are linked by shaded bands, and non-syntenic (non-matching) regions are shown as white gaps between Nipponbare and 9311. d, A violin plot showing the identity value of syntenic genes shared between the Nipponbare and 9311 genomes.
Extended Data Fig. 10 Definition and calculation of genomic homozygosity and similarity.
a, Diagrammatic illustration of the strategy used in calculating ‘genomic homozygosity’, which is defined as the proportion of homozygous segments across the entire genome. The genome is divided into discrete 5-Kb bins, and the proportion of homozygous segments is determined based on diagnostic SNP-stratified genotyping data. For example, within a 1-Mb region comprising 200 bins, the genomic composition of each bin is calculated based on genotyping data (see Supplementary Methods in Supplementary Information for details). The homozygosity of this 1-Mb region was calculated to be 50% if it contains 100 homozygous bins (homoeolog ratios of 4:0 or 0:4) and 100 heterozygous bins (homoeolog ratios of 3:1, 1:3 or 2:2). b, Schematic depiction of the calculation of genome similarity using a 1-Mb segment of chromosome 1. The gray areas represent syntenic regions, which are conserved genomic segments shared between the two genomes. The different colored blocks within these syntenic regions indicate the various kinds of sequence variations, such as insertions, deletions, or SNPs. Genome similarity is quantified by comparing the extent of sequence conservation and variation within these syntenic regions. Panel b was generated using BioRender with permission.
Supplementary information
Supplementary Information
Supplementary Methods, Results and Figs. 1–19.
Supplementary Tables
Supplementary Table 1–25.
Source data
Source Data Figs. 1–7 and Extended Data Figs. 2, 5–9
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Han, X., Li, J., Li, G. et al. Rapid formation of stable autotetraploid rice from genome-doubled F1 hybrids of japonica–indica subspecies. Nat. Plants 11, 743–760 (2025). https://doi.org/10.1038/s41477-025-01966-2
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41477-025-01966-2
