
Abstract
Pseudomonas aeruginosa is a widespread nosocomial pathogen with a significant to cause both severe planktonic acute and biofilm-related chronic infections. Small RNAs (sRNAs) are noncoding regulatory molecules that are stabilized by the RNA chaperone Hfq to trigger various virulence-related signaling pathways. Here, we identified an Hfq-binding sRNA in P. aeruginosa PAO1, PqsS, which promotes bacterial pathogenicity and pseudomonas quinolone signal quorum sensing (pqs QS) system. Specifically, PqsS enhanced acute bacterial infections by inducing host cell death and promoting rhamnolipid-regulated swarming motility. Meanwhile, PqsS reduced chronic infection traits including biofilm formation and antibiotic resistance. Moreover, PqsS repressed pqsL transcript, increasing PQS levels for pqs QS. A PQS-rich environment promoted PqsS expression, thus forming a positive feedback loop. Furthermore, we demonstrated that the PqsS interacts and destabilizes the pqsL mRNA by recruiting RNase E to drive degradation. These findings provide insights for future research on P. aeruginosa pathogenesis and targeted treatment.
Similar content being viewed by others


Introduction
Pseudomonas aeruginosa is an opportunistic pathogen that is prevalent in hospital environments1. This bacterium is one of the most commonly isolated pathogens and is responsible for high rates of morbidity and mortality in patients worldwide2. Indeed, P. aeruginosa infections are responsible for a large number of hospital-acquired cases of chronic infections (such as implants, intubation, and cystic fibrosis) and acute infections (such as pneumonia and bacteremia)3. P. aeruginosa chronic colonization is associated with high-density biofilm formation with matrix-enclosed aggregates that are difficult to eliminate4. Under specific environmental conditions, P. aeruginosa disperses from the initial infection site to other parts of the body, leading to acute systemic dissemination. A series of virulence factors are released during P. aeruginosa infections, such as LasB elastase, and rhamnolipids (RLs)5. Among them, RLs are involved in mediating the swarming motility6. The expression of these virulence factor is controlled by quorum sensing (QS) systems, two-component systems, and cyclic diguanosine monophosphate (c-di-GMP) signaling7. Four interconnected QS systems, las, rhl, pqs, and iqs, control collective behaviors in a bacterial population and signal molecules in a density-dependent manner to regulate bacterial pathogenicity during infections8.
Pseudomonas quinolone signal, 2-heptyl-3-hydroxy-4 (1H)-quinolone (PQS), is the signaling molecule of the pqs QS system, specifically secreted by Pseudomonas species9. The pqs QS system is subject to positive autoinduction because the LysR-type transcriptional regulator PqsR (MvfR) binds to the promoter region of pqsABCDE (PpqsA), triggering transcription once activated by PQS. The precursor 2-aminobenzoylacetate (2-ABA) is first converted into 2-heptyl-4-quinolone (HHQ) and then transformed into PQS. In addition, 2-ABA is also converted into 2-n-heptyl-4-hydroxyquinoline-N-oxide (HQNO) by the mono-oxygenase PqsL10. PQS and HQNO are typically produced at similar levels in P. aeruginosa PAO1, but deletion of pqsL can lead to the overproduction of PQS11. At high cell densities, large amounts of PQS signaling molecules can be detected in the bacterial culture supernatant12 and play multiple roles in P. aeruginosa virulence and biofilm formation13. For example, PQS causes the death of the host Caenorhabditis elegans and disrupts the intestine epitheliumin in mice14. In addition, the cell division factor, ZapE, regulates biofilm formation by influencing PQS productions15. Bacterial membrane vesicles (MVs) are nanoscale vesicle structures that carry various hydrophobic molecules, proteins, and nucleic acids16, and have been identified as essential components of the extracellular matrix in biofilms17,18. PQS also contributes to P. aeruginosa MV formation and is frequently detected in MVs19,20.
Bacterial small RNAs (sRNAs), particularly trans-encoded sRNAs, are non-coding regulatory molecules that are typically 50–500 nucleotides long and participate in various biological processes in bacteria21. sRNAs regulate their targets through multiple mechanisms, such as transcriptional or translational termination, in response to various environmental signals, including phosphate and ammonium22,23. The major bacterial RNA-binding protein host factor required for bacteriophage Qβ RNA replication (Hfq), a member of the ubiquitous Lsm/Sm protein family, binds to various sRNAs for stabilization and recognition of target mRNAs24,25. The roles of various important P. aeruginosa sRNAs participating in infection mechanisms, such as QS, type three secretory system (T3SS), and biofilm formation, have been thoroughly investigated26,27. For example, the sRNA PhrS activates quinolone synthesis through PqsR and enhances the pqs QS system under low-oxygen conditions28. Another P. aeruginosa sRNA, PrrF, was required for PQS production and bacterial virulence in response to low-iron conditions in a murine model of acute lung infection29.
In this study, we aimed to investigate the Hfq-binding sRNA, P. aeruginosa pqs QS regulating sRNA (PqsS), in P. aeruginosa PAO1 pathogenesis and to elucidate the molecular mechanisms underlying its regulatory pathways. We showed that PqsS promoted bacterial acute infections, while repressed biofilm-related chronic infection traits. PqsS repressed PqsL to reduce HQNO production and facilitate PQS production for pqs QS system and MVs formation, while PQS-rich environment further promotes PqsS expression, thus forming a positive feedback loop. Specifically, PqsS interacted with and destabilized the pqsL mRNA by recruiting RNase E to drive degradation.
Methods
Bacterial growth and cell culture
P. aeruginosa strains, plasmids, and primers used in this study are provided in Supplementary Table 1. Bacteria were cultured in tryptic soytone broth (TSB) (1.7% tryptone, 0.3% soytone, 0.25% glucose, 0.5% NaCl, 0.25% K2HPO4), Luria–Bertani (LB) broth (1% tryptone, 0.5% yeast extract, 0.5% NaCl), or ABTGC B-medium (0.1% MgCl2, 0.1% CaCl2, 0.1% FeCl3) supplemented with 10% A10, 0.2% glucose, and 0.2% casamino acids. The media was solidified with 1.5% Bacto Agar (Difco, USA). The overnight-grown subcultures were transferred into 96-well plates, and their growth curves were measured for cell density analysis (OD600) for 24 h using a Tecan Spark plate reader (Tecan Group Ltd., Switzerland). To assess whether different PQS concentrations affected PqsS expression, we generated different PQS concentrations TSB medium (1, 10, and 100 μM, respectively) by supplementing PQS-free TSB with PQS. After overnight culture, PAO1 WT bacteria was collected and washed existing PQS by PBS, then add new medium with different PQS concentrations for to treating 1 h. For cell culture, the murine RAW 264.7 (ATCC®TIB-71™) and human A549 (ATCC®CCL-185) cell lines were grown in DMEM medium supplemented with 10% fetal bovine serum (Gibco, USA), and incubated at 37 °C in a 5% CO2, humidified incubator.
P. aeruginosa mutant, knockdown (KD), and complement strains construction
P. aeruginosa PAO1 deletion mutant strains were generated by homologous recombination. Briefly, the mutants were constructed by overlapping PCR to contain a gentamicin-resistance cassette. The mutant fragments were inserted into pK18, a suicide vector, to produce gene knockout plasmids. Each knockout plasmid was transformed into E. coli strain RK600 and conjugally transferred from RK600 to PAO1. The resultant integrants were selected on agar containing 60 µg/mL gentamicin (Gm60). To resolve merodiploids, the Gm-resistant colonies were streaked to produce single colonies on LB+Gm60 plates containing 10% sucrose. Potential mutants without gentamicin-resistance cassettes were screened by PCR using corresponding flanking primers and were confirmed by Sanger sequencing. P. aeruginosa PA14 pqsS gene knockdown (KD) strains were generated by Clustered Regularly Interspaced Short Palindromic Repeats. The IPTG-inducible system driving dCas9 in pUC18-mini-Tn7T-Plac-dCas9 was replaced with a tetracycline-inducible system for better performance in P. aeruginosa, resulting in plasmid pSZU368. pSZU368 was constructed with an sgRNA targeting pqsS. Transformation of pSZU368-sgRNApqsSintoP. aeruginosa PA14 was achieved by tri-parental mating. Complement strains were constructed as follows: ΔpqsS + : ΔpqsS mutant expressing pqsS from a rescue construct using the pMiniCTX1-pqsS plasmid ‘s own promoter; ΔpqsS + pJV300-pqsS: a promoter-free pJV300 plasmid using the pqsS promoter.
RNA isolation and RNA-seq
Bacterial strains were grown in TSB/ LB until the exponential phase. Then, total RNA was isolated using an EASYspin Plus bacterial microRNA kit (Aidlab, China) and purified using a Ribo-Zero rRNA Removal Kit (Epicentre Biotechnologies, USA). The RNA integrity was determined by TapeStation RNA ScreenTape with RNA size distributions. For Nanopore RNA-seq, total RNA was tailed with poly(A) using E. coli Poly(A) Polymerase (NEB, UK). Then, nanopore RNA-seq was performed using a Ligation Sequencing Kit (SQK-LSK109) with an ONT Barcoding Expansion Kit (EXP-NBD114). Artemis was used to show the mapping profiles of our RNA-seq results30. For Illumina RNA-seq, The libraries of RNA samples were generated using NEBNext R Ultra™ Directional RNA Library Prep Kit for Illumina R (NEB, USA). The libraries were sequenced using an Illumina Hiseq platform to generate paired-end reads. P. aeruginosa RNA-seq data analysis was performed as previously reported31.
Northern blotting
Northern blotting was performed using the NorthernMax Kit (Invitrogen, USA). After RNA extraction, 5 μg of total RNA was separated on a 1.2% agarose gel containing formaldehyde. The gels were then electro-blotted onto Brightstar Plus nylon membranes (Applied Biosystems, USA) and immobilized at 85 °C for 30 min. The specific biotin-labeled single-stranded DNA probes (Supplementary Table 1) with optimized concentrations were added to fresh hybridization buffer, and the blots were incubated with a hybridization buffer overnight. 5S rRNA was used as an internal control. After washing, the blots were further washed with Wash and Block Buffer, and then the signal was detected using the biotin chemiluminescence detection kit (Beyotime, China). Hybridization signals were visualized on an Amersham Imager 680. FIJI/ImageJ software was used to measure band intensities.
Fluorescent primer extension
A 6-carboxyfluorescein (FAM)-labeled oligonucleotide complementary to the sequence 99 to 135 bp downstream pqsS transcription start site (TSS) was used for primer extension reactions. Total RNA (2 μg) and 1 μM FAM-labeled primer were incubated at 70 °C for 5 min, before reverse transcription at 42 °C for 60 min. The samples were then incubated with 20 μg RNase A at 37 °C for 30 min, after which cDNA was ethanol precipitated and dissolved in 15 μL nuclease-free water. Finally, the products were analyzed on an ABI 3730 genetic analyzer.
RNA In vitro transcription (IVT), protein purification and protein-RNA EMSA
DNA templates were transcribed into PqsS RNA using a T7 High-Efficiency Transcription Kit (TransGen, China). The purified RNA was checked on an 8% Tris-Acr-Urea gel. P. aeruginosa Hfq protein was purified as described previously24. The PqsS sRNA was in vitro transcribed. For electrophoretic mobility shift assay (EMSAs), a 4 μM stock of Hfq was prepared in binding buffer (20 mM Tris-HCl pH 8.0, 40 mM NaCl, 10 mM KCl, 1 mM MgCl2), and 20 μM stocks of the RNAs were prepared in DEPC water (RNase free). The RNAs were incubated at 50 °C for 3 min before the proteins were added. Native PAGE (8%) was used to study complex formation. Hfq was titrated into PqsS RNAs at different ratios. The RNA concentration was kept constant at 2 μM. After 15 min of incubation at 37 °C, the samples were mixed with 5×EMSA loading buffer (50% v/v glycerol, 50% v/v binding buffer, 5 mM DTT), before loading onto the gel. The gels were run at 4 °C in 1×TBE running buffer and stained with SYBR gold.
Molecular docking
The Hfq PDB structure file was extracted from 7OGM (RCSB PDB ID) by ChimeraX (UCSF)32. PqsS sRNA was modified and docked with Hfq by Hdock server33. Pymol 1.4 (Schrodinger, LLC) was used to map the interactions between protein and potential ligand34. The distance of hydrogen bonds between molecules less than 4.0 Å was recognized as interactive molecules.
Cell death and adhesion assay
Mid-log phase bacteria (OD600 = 0.6) grown in LB broth were centrifuged and resuspended in PBS to infect RAW264.7 murine macrophages or A549 human lung epithelial cells grown in DMEM medium supplemented with 10% FBS at an MOI of 100. After centrifugation of 24-well culture plates, bacterial phagocytosis by macrophage was allowed to proceed for 30 min before washing three times with sterile PBS and adding fresh DMEM media supplemented with 400 μg/mL gentamicin. For cell death assay, after 2 h infection, the culture supernatants were collected for detection of LDH activities. The LDH activities were detected by using a commercial LDH cytotoxicity kit (Yeasen Bio, China) according to standard procedures. For cell adhesion assay of bacteria on A549 human lung epithelial cells, after 2 h infection, unadhered bacteria were washed by PBS and cells were lysis with 1% Triton. Adhered bacteria were plated on LB agar plates. The percent adherence of PAO1 WT, ΔpqsS, and ΔpqsS+ strains was compared to inocula by plate counts. All the assays were performed in triplicate. Mid-log phase bacteria (OD600 = 0.6) grown in the LB broth were centrifuged and resuspended in phosphate-buffered saline (PBS) to infect RAW264.7 murine macrophages or A549 human lung epithelial cells grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS at an multiplicity of infection of 100. After centrifugation of 24-well culture plates, bacterial phagocytosis by macrophage was allowed to proceed for 30 min before washing thrice with sterile PBS and adding fresh DMEM media supplemented with 400 μg/mL gentamicin. For the cell death assay, culture supernatants were collected 2 h after infection of LDH activity. LDH activity was detected using a commercial LDH cytotoxicity kit (Yeasen Bio, China), according to standard procedures. For the cell adhesion assay of bacteria on A549 human lung epithelial cells, after 2 h of infection, unadhered bacteria were washed with PBS, and cells were lysed with 1% Triton. Adherent bacteria were plated onto Luria Broth (LB) agar plates. The percent adherence of PAO1 WT, ΔpqsS, and ΔpqsS+ strains was compared to inocula by plate counts. All assays were performed in triplicate.
Galleria mellonella larva infection model
Fifth-stage G. mellonella (wax moth) larvae (Huiyude Bio, China) that were bright and white without gray-black spots with a limited mass ranging from 250 to 350 mg were randomly placed in groups of 20 and starved for 12 h before injection. P. aeruginosa PAO1 WT, ΔpqsS, and ΔpqsS+ strains were grown overnight in the LB liquid media at 37 °C and 220 rpm. Overnight cultures were diluted 1:100 in fresh LB broth and grown to an OD600 of 0.8. Cultures were centrifuged, washed in sterile PBS, and resuspended at an OD600 of 0.1. Serial dilutions were prepared in PBS to a final concentration of 5000 CFU/ml. Each larva was swabbed with 70% ethanol to prevent contamination of the injection site and injected with 10 μL of bacterial dilution using a 50 μL Hamilton syringe. The negative control group was injected with 10 μL of sterile PBS. Then, every five injected larvae were incubated in 10 cm plates at 37 °C, and the number of dead larvae was monitored for the next 48 h. Larvae were scored as dead if they failed to respond to mechanical stimulation of the head. Statistical analysis was performed using the Mantel-Cox test, and p values were corrected using the Benjamini–Hochberg method.
Rhamnolipids quantification
Rhamnolipids were extracted from supernatants (2 mL) using ethyl acetate. The extraction was performed twice, and the collected organic fractions were left to evaporate overnight. The resulting white solid precipitates were dissolved in water (100 μL) and mixed with a 0.19% (wt/vol) orcinol solution (900 μL) freshly prepared in 50% H2SO4. The samples were heated at 80 °C for 10 min and cooled to room temperature for 10 min before measuring absorbance at 421 nm. Three biological and three technical replicates were performed to ensure experimental reproducibility.
Swarming motility
The swarming motilities of PAO1 WT, ΔpqsS, and ΔpqsS+ strains were examined by using 0.5% (w/v) agar and 8 g/L nutrient broth supplemented with 0.5% (w/v) glucose. Bacterial culture (1 μL; OD600 = 1) was inoculated at the center of the Petri dishes and sat for 30 min, then incubated at 30 °C for 24 h for visualizations. Bacteria spreading from the inoculation spot was measured with a sliding caliper. Three biological and three technical replicates were performed to ensure experimental reproducibility.
Biofilm formation
Biofilm formation was quantified by crystal violet staining. Overnight LB-grown bacterial cultures were diluted in fresh medium (1:100) and incubated in 2 mL of LB in 96-well culture plates at 37 °C for 24 h with 50 rpm allowing stable biofilm formation. After removing the spent media, we removed the loosely associated bacteria by washing them with water two times, and the remaining bacteria were stained with 0.1% crystal violet for 20 min. Then, the crystal violet stain was discarded while the stained biofilms were washed twice thoroughly with ddH2O and air-dried. The stained biofilm was then dissolved into 200 µL of 95% ethanol and quantified at 630 nm using a Tecan Infinite 200 Pro plate reader.
Confocal microscopy imaging of biofilm
Confocal microscopy imaging of biofilm was adapted from the previous protocol35. To visualize bacterial biofilm formation, we cultured overnight grown bacterial cultures in 20 mm glass-bottom culture dishes (Biosharp, China). After 24 h of incubation at 37 °C with 50 rpm allowing stable biofilm formation, the supernatant was removed, and the well surface was rinsed briefly with 0.9% NaCl. One milliliter of 0.9% NaCl containing 1 μM of SYTO 9 (Invitrogen, USA) was added to the well, and the plate was incubated for 15 min in the dark. Fluorescent images were acquired with a confocal microscope (Zeiss LSM 900, German) for monitoring fluorescence and analyzed using the Comstat software. Experiments were performed in triplicate, and representative images are shown. To evaluate the colistin resistance, after 12 h biofilm formations, samples were challenged with 10 μg/mL colistin overnight. Planktonic cultures were then discarded, and the biofilms grown in the plates were washed with sterile PBS once before being stained with the LIVE/DEAD BacLight kit (Invitrogen, USA) for 20 min.Cells stained green with SYTO-9 indicated live cells, while cells stained red with propidium iodide (PI) indicated dead cells.
Macrophage phagocytosis and bacteria survival assay
The method for macrophage intracellular survival assays was adapted from the previous protocol36. Mid-log phase bacteria (OD600 = 0.6) grown in LB broth were centrifuged and resuspended in PBS to infect RAW264.7 macrophages grown in DMEM medium supplemented with 10% FBS at an MOI of 100. After centrifugation of 24-well culture plates, bacterial phagocytosis was allowed to proceed for 30 min before washing three times with sterile PBS and adding fresh DMEM media supplemented with 400 μg/mL gentamicin. After 2 h infection, macrophages were lysed by using 1% Triton X-100 and the number of viable bacteria was determined by subsequent plating onto LB agar plates. The percentage of survival represents the ratio between the number of bacteria at time 2 h and the number of bacteria internalized after phagocytosis.
Cellular ROS production measurement by DCFH-DA staining in flow cytometer
ROS production level in RAW264.7 cells cultured for 2 h after phagocytosis was detected by Reactive Oxygen Species Assay Kit (Beyotime, China). Briefy, RAW264.7 cells were infected with the isolates respectively at a Multiplicity of Infection (MOI) of 100 at 37 °C in 5% CO2 incubator. After 2 h infection, cells were washed with PBS and incubated with the 2’-7’dichlorofluorescin diacetate (DCFH-DA) (final concentration 10 μM) reagent at 37 °C for 20 min in DMEM medium without FBS. The cells were washed with PBS and harvested. The stained cells were analyzed by using a Cytofex S flow cytometer (Beckman, USA). All samples were assayed with lasers emitting at 488 nm, and the fluorescence was collected by 530/30 nm bandpass filter. The flow cytometric data were analyzed using FlowJo software.
Real-time (RT)-quantitative (q) PCR
Bacterial strains were grown in the LB medium until they reached the mid-exponential phase (OD600 ~ 0.6). Total bacterial RNA was extracted using an EASYspin Plus bacterial microRNA kit (Aidlab, China) and treated with RNase-free DNase I to eliminate genomic DNA contamination. The RNA concentration was measured using a Nanodrop 2000 spectrophotometer (Thermo Fisher). One microgram of the diluted extracted RNA was converted to cDNA using the PrimeScript RT Reagent Kit with gDNA Eraser (Perfect Real Time) (TaKaRa, Japan). Validated primers (Supplementary Table 1) and ChamQ Universal SYBR qPCR Master Mix (Vazyme, China) were added to the cDNA. The mixture was amplified using QuantStudio 1 (Applied Biosystems, USA), and three replicates were obtained. The data were collected using QuantStudio Real-Time PCR software v1.3, normalized to endogenous rpsL levels, and analysed using the comparative critical threshold (CT) method. All reactions were conducted in triplicate. Furthermore, to test the effect of the PqsS sRNA on target gene pqsL mRNA stability, the corresponding bacteria were grown to reach an OD600 of 1.0, followed by treatment with 100 µg/mL rifampicin. At each indicated time point after rifampicin treatment, the same volume of bacteria was harvested, and the corresponding RNA was extracted immediately for RT-qPCR.
Green fluorescence protein (GFP) reporter gene assay
For the pqsA promoter-gfp and pqsS promoter-gfp reporter fusion strains, the corresponding plasmid was introduced into corresponding strains by electroporation. Transformants were selected on LB agar plates containing 100 μg/ml carbenicillin. Overnight cultures (grown in LB medium at 37 °C, shaking at 200 rpm) were diluted in ABTGC medium to an OD600nm of 0.02 (~2.5 × 108 CFU/mL). A blank control (ABTGC medium) was used. The microtiter plate was incubated at 37 °C in a Tecan Infinite 200 Pro plate reader (Tecan Group Ltd., Mannedorf, Switzerland) to measure the cell density (OD600nm) and GFP fluorescence (excitation at 485 nm, emission at 535 nm) at 1 h intervals for 24 h.
LC‐MS/MS analysis of PQS, HQNO, and HHQ
1 mL overnight bacteria culture fluid (OD 1.0) was centrifuged at 10,000 × g for 10 min, and the supernatant was filtered through a 0.2 μm Millex Syring Filter. Then, the bacterial supernatants were resuspended in an equal volume of methanol and stored at −20 °C. The extract was subjected to LC‐MS/MS with a C18 reverse-phase column, as previously reported37. Briefly, a C18 reverse-phase column connected to a mass spectrometer was used to separate the hydroxyl-alkyl-quinolones using a water/acetonitrile gradient. Positive electrospray in the MRM mode with 261023 mTorr argon and 30 V as the collision gas were employed to quantify hydroxyl-alkyl-quinolones using the ion transitions HHQ 244.159, HQNO 260.159, PQS 260.175. All MS experiments in this study were performed on a Q Exactive Orbitrap Mass Spectrometers (Thermo Scientific, USA). Analyst software was applied for data acquisition and processing.
Western blotting
To analyze the PqsL protein levels, total proteins were separated by 12% sodium dodecyl sulfate-polyacrylamide and transferred onto polyvinylidene difluoride membranes. The membranes were treated with 5% non-fat milk for 1 h and incubated with the primary antibodies against the FLAG-tag, RNA Polymerase α (α-RNAP), Hfq, or RNase E at 4 °C overnight, followed by washing in tris-buffered saline (TBST). The blots were further incubated with a secondary goat anti-mouse-HRP antibody (Abcam, UK) at room temperature for 1 h. The blots were washed in TBST before detection with enhanced chemiluminescence reagent and an ECL Western Blotting Substrate Kit (Abcam, UK). FIJI/ImageJ software was used to measure band intensities.
RNA-RNA electrophoretic mobility shift assays (REMSA)
DNA templates were transcribed into PqsS (+, negative-strand), PqsS (-, positive-strand, positive control), pqsL mRNA, using a T7 High-Efficiency Transcription Kit (TransGen, China). The purified RNA was checked on an 8% Tris-Acr-Urea gel. Then, REMSA was performed with PqsS (+, 0.5, 1, 2, and 4 μM), PqsS (-, positive control, 1 μM), pqsL mRNA (1 μM), in 10× REMSA binding buffer. The reaction mixtures were incubated for 2 min at 85 °C and then at 37 °C for 30 min. RNA was separated on an 8% native polyacrylamide gel electrophoresis (PAGE) using a Native-PAGE Preparation kit (Sangon, China), stained with SYBR™ Gold Nucleic Acid Gel Stain (Invitrogen, USA) for 10 min, and visualized under a UV light.
RNase E cleavage assay
N-terminal RNase E (1-529 aa) was purified as described previously38. In vitro transcribed pqsS pqsL, and rpsL DNA templates were generated using a T7 High-Efficiency Transcription kit (TransGen, China). Transcripts of the indicated concentrations were incubated in REMSA binding buffer for 2 min at 85 °C, followed by incubation at 37 °C for 30 min. Then, RNase E was added to a final concentration of 7.5 μM, and the samples were incubated for 30 min at 37 °C. Then, the samples were subjected to electrophoresis on a 1.2% agarose gel containing formaldehyde. After electrophoretic transfer to a Brightstar Plus nylon membrane, the membranes were crosslinked by 85 °C. The specific biotin-labeled primer at an optimized concentration, was added to fresh hybridization buffer, and the blots were incubated in buffer overnight. Then, the signal was detected using the biotin chemiluminescence detection kit (Beyotime, China). Hybridization signals were visualized on an Amersham Imager 680.
Extraction and purification of P. aeruginosa membrane vesicles (MVs)
Bacterial strains were grown in TSB until the exponential phase. Bacterial cells were harvested by centrifugation at 8000 × g, 4 °C for 15 min. The supernatant was passed through a 500 mL Vacuum bottle with a 0.45 µm filter (Biofil, China) and then concentrated in Masterflex Easy-Load with 100 KD Vivaflow membranes (Sartorius AG, German). The concentrated supernatants were incubated with 2 μg/mL RNase A/T1 (Thermo Scientific) for 1 h at 37 °C before centrifugation at 50,000 × g for 2 h at 4 °C to pellet the crude MVs. The MVs were purified by density gradient centrifugation (ranging from 15 to 60%) at 100,000 × g with Opti-Prep Gradient Medium (Sigma Aldrich, USA) for 16 h at 4 °C. The 35–45% layer containing purified MVs were washed in PBS for 1.5 h at 4 °C. Final purified MVs pellets were resuspended in PBS.
Characterization of MVs by Transmission electron microscopy (TEM), and Nanoparticle tracking analysis (NTA)
Purified MVs were visualized by negative staining TEM. In brief, 5 μL of the MVs were incubated for 1 min on a glow-discharged 200-mesh carbon grid (Zhongjingkeyi Technology, China) and contrasted with a phosphotungstic acid solution. Subsequently, the grids were viewed on an HT7700 transmission electron microscope (Hitachi, Japan) operating at 100 kV and fitted with a high-sensitivity real-time charge-coupled device camera. For NTA, the quantification and size characterizations of the MVs were performed on a NanoSight NS300 (Malvern Instruments Ltd, UK). For the size and concentration measurements, a 448 nm laser in scatter mode was used. Briefly, MV samples were diluted (500×) in PBS to obtain a concentration within the recommended measurement range (1–10 × 108 particles/mL). Using a 1 mL syringe, the sample was injected into the instrument and videos were captured in triplicate for 30 s. The mean values for size and concentration were analyzed using the NanoSight (NTA software, version 3.0). For northern blotting, we performed a RNase digestion step to remove RNAs outside MVs, samples were incubated with 10 ng/μL RNase A/T1 (Thermo Scientific, USA) for 30 min at 37 °C, diluted 1/100 with water and pelleted at 50,000 × g for 1 h to remove the external RNase.
Phylogenetic analysis of pqsS and bioinformatics analyses
Public complete genomes from 83 P. aeruginosa strains marked as human isolates were downloaded from the Pseudomonas genome database (access date 12 August 2021)39, which were then used to build a nucleotide database with makeblastdb40. BLASTn was then used to search the pqsS gene against this database. Nucleotide sequences used to construct the phylogenetic tree were aligned using MAFFT, and a maximum-likelihood tree was constructed using PhyML of Unipro Ugene and FigTreev1.4.3 based on the General Time Reversible (GTR) model41 of nucleotide substitution with c-distributed rates among sites. IntaRNA was used to predict the interaction region between RNAs. Softberry Boot Programmable Read-Only Memory (BPROM) was used to predict bacterial promoters and potential upstream proteins based on transcription factor-binding sites.
Statistical analysis
All statistical analyses were performed using GraphPad Prism software; the data were analyzed by a two-sided Mann–Whitney U test and Benjamini-Hochberg-corrected P values. The data represent the means ± SD of three independent experiments unless otherwise indicated. P < 0.05 were considered statistically significant. All false discovery rate controls were performed with the Benjamini-Hochberg procedure, and a false discovery rate of 10% (q < 0.1) was selected as the significant threshold. Statistical details for all tests performed can be found in the Fig. legends.
Results
The PqsS is an Hfq chaperone-binding sRNA in P. aeruginosa PAO1
By analysing publicly available High-throughput Global sRNA target identification by Ligation and sequencing (Hi-GRIL-seq) data of sRNAs from P. aeruginosa strain PAO142, we screened them by removing known sRNAs and retaining unknown sRNAs with lengths between 200 and 500 nt. The remaining 12 sRNAs are listed in Supplementary Table 2. Among them, a potential sRNA IG_minus_901047_901519 was renamed to PqsS, which is encoded by the intergenic region between the genes PA0826 and PA0827 (Fig. 1a). To confirm that PqsS is an sRNA that is transcribed into PAO1, northern blotting was performed using a PqsS-specific probe. The results showed that pqsS was transcribed in PAO1 wild-type (WT) and the complementary ΔpqsS mutant of this strain (ΔpqsS+), but not in the ΔpqsS mutant (Fig. 1b). We then introduced a specific promoter-less plasmid (pJV300) containing pqsS with its predicted promoter region into the ΔpqsS mutant. Northern blotting showed that this plasmid also led to normal expression of PqsS sRNA compared to that observed in PAO1 WT and normal complement strains. These results revealed that PqsS is an sRNA that is independently transcribed from the reverse strand of the genome, with a size of approximately 230–250 nucleotides in PAO1 WT (Fig. 1b). Furthermore, 5’ monophosphate-dependent terminator exonuclease (TEX) degrades processed sRNAs while sparing mature sRNAs43. The published high-throughput sequencing data for TEX-treated P. aeruginosa RNAs44 also included PqsS, confirming that PqsS is an independently transcribed mature sRNA (Supplementary Fig. 1a).

a Upper panel: the position of the pqsS in the PAO1 genome between PA0826 and PA0827, yellow box; PA0826, red box; pqsS, blue box; ssrA, green box; PA0827. Downward panel: pqsS sequences: −35 and −10 boxes of the putative promoter region, green; transcription start site, blue; pqsL mRNA binding site, red; Hfq protein binding site, highlighted; and termination site, purple. b Northern blotting was performed with a specific probe directed against PqsS sRNA in RNA preparations from PAO1 WT, ΔpqsS, and ΔpqsS mutant expressing pqsS from a rescue construct using the pMiniCTX1-pqsS plasmid ‘s own promoter (ΔpqsS+) or a pJV300 plasmid using the pqsS promoter (ΔpqsS + p) (n = 3). 5S rRNA was used as the loading control. Densitometric analysis of the bands was performed with ImageJ. c Northern blotting was performed with a specific probe directed against PqsS sRNA in RNA preparations from PAO1 WT, Δhfq, and Δhfq+ strains (n = 3). 5S rRNA was used as a loading control. Densitometric analysis of the bands was performed using the ImageJ software. d Electrophoretic mobility shift assays (EMSA) with PqsS sRNA in the presence of different concentrations of Hfq protein. e Docking analysis of the exact interaction sites in the Hfq hexamer-PqsS sRNA complex. The seven key interaction bases (A40, C42, C113, C116, G134, G149, and G190) of PqsS sRNA are shown in red.
BPROM online tool analysis suggested a putative unique promoter region for pqsS upstream of its TSS. Next, we determined the TSS of pqsS by fluorescent primer extension and found that it was identical to that predicted by BPROM analysis (Supplementary Fig. 1b). We then mapped the TSS (+1) of the pqsS coding sequence to positions A5,362,926 (adenine at 901,479 bp of the forward strand) in the PAO1 genome (6,264,404 bp total)45 (Fig. 1a). The total size of PqsS was determined to be 239 nt by direct nanopore RNA-seq of P. aeruginosa PAO1, which generated long reads for full-length RNAs in an unbiased manner (Supplementary Fig. 1c). The termination site of the pqsS coding sequence was mapped to T5,363,165 (thymine at 901,240 bp on the forward strand) of the genome (Fig. 1a). Moreover, RT-qPCR results showed that the mRNA levels of the adjacent genes PA0826, PA0827, and ssrA did not change after pqsS mutation (Supplementary Fig. 1d), indicating that PqsS functions independently of the adjacent genes without inducing a polar effect. Using the online tool RNAfold, we predicted that the secondary structure of PqsS comprised 5 hairpin loops, 11 internal loops, and 5 bulges (Supplementary Fig. 2a).
The Hfq protein is a major sRNA chaperone that binds to sRNAs and protects them from degradation by cellular nucleosidases26. Therefore, we examined the effect of hfq deletion on PqsS abundance using northern blotting. The presence of PqsS RNA in WT and Δhfq+ strains, which was absent from the Δhfq strain, indicated that Hfq promoted PqsS abundance (Fig. 1c). We then verified the interaction between PqsS and Hfq using a protein-RNA EMSA with in vitro transcribed PqsS sRNA and purified Hfq protein. EMSA reactions were performed using different concentrations of Hfq protein but a fixed amount of PqsS sRNA. We successfully observed Hfq-PqsS complexes and found that the band intensity increased with increasing Hfq concentration (Fig. 1d), confirming a direct interaction between the Hfq protein and PqsS sRNA in vitro.
We performed a P. aeruginosa Hfq hexamer-PqsS sRNA docking analysis to predict the exact interaction sites. Seven PqsS sRNA bases, A40 (adenine, position 40 from the TSS), C42 (cytosine), C113 (cytosine), C116 (cytosine), G134 (guanine), G149 (guanine), and G190 (guanine), appeared to bind to Hfq (Fig. 1a, e). These sRNA bases interacted with four Hfq amino acid residues: Y25 (tyrosine, position 25 from the start codon), G29 (glycine), K31 (lysine), and T49 (threonine) (Supplementary Fig. 2b). Finally, to confirm that the seven nucleotides (A40, C42, C113, C116, G13, G149, and G190) constituted the key sequence of PqsS required for binding to Hfq, we repeated the EMSA using a mutated PqsS (PqsS*-Hfq) sRNA (A40, C42, C113, C116, G13, G149, and G190 were complementary to U40, G42, G113, G116, C13, C149, and C190) generated by in vitro transcription. No interactions were observed between PqsS*-Hfq sRNA and Hfq proteins (Supplementary Fig. 2c). These results showed that PqsS is an sRNA in P. aeruginosa PAO1 that binds to the Hfq chaperone via seven key nucleotides (A40, C42, C113, C116, G13, G149, and G190).
PqsS promotes PAO1-mediated acute infections including host cell death and RL-regulated swarming motility
Growth curves showed that PAO1 WT, ΔpqsS, and ΔpqsS+ strains grew at similar rates in vitro (Fig. 2a). Therefore, we tested the influence of PqsS on bacterial virulence in vitro and in vivo using macrophage killing and Galleria mellonella infection assays, respectively. We leveraged RAW264.7 murine macrophages and A549 human lung epithelial cells as representative cell models of P. aeruginosa infection. We infected both cell types with PAO1 WT, ΔpqsS, and ΔpqsS+ complementary strains and then determined cytosolic lactate dehydrogenase (LDH) release as an indicator of cytotoxicity. We found that RAW264.7 macrophages infected with ΔpqsS released 2.13-fold and 1.99-fold less LDH compared to those infected with PAO1 and ΔpqsS+ strains, respectively (Fig. 2b). For A549 epithelial cells, we found that the cells infected with ΔpqsS released 3.24-fold and 3.09-fold less LDH compared to those infected with PAO1 and ΔpqsS+ strains, respectively (Supplementary Fig. 3a). Besides, we performed bacterial adherence assay of PAO1 WT, ΔpqsS, and ΔpqsS+ strains comparing inocula by plate counts. The results showed that PqsS promoted P. aeruginosa adherence to A549 human lung epithelial cells (Supplementary Fig. 3b). In the in vivo Galleria mellonella infection model, we found that the ΔpqsS strain showed 2.03-fold and 1.97-fold lower mortality compared to PAO1 WT and ΔpqsS+ strains at 24 h post-infection. The untreated and PBS control samples showed very low mortality rates when used as negative controls (Fig. 2c).

a Growth curves of PAO1 WT, ΔpqsS, and ΔpqsS+ strains. Data represent the means ± standard deviations (SD) (n = 6). b LDH assay for cell death in RAW 264.7 murine macrophages infected with PAO1 WT, ΔpqsS, and ΔpqsS+ strains. Data represent the means ± SD (n = 3, **P ≤ 0.01, Mann–Whitney U test). c In vivo measurement of virulence of the PAO1 WT, ΔpqsS, and ΔpqsS+ strains in a Galleria mellonella infection model (n = 10, Mantel-Cox test for statistics, **P ≤ 0.01). Each treated group was injected with a 10 μL bacteria dilution, the negative control group was injected with 10 μL sterile PBS, whereas the untreated group was not injected. d The production of rhamnolipids by PAO1 WT, ΔpqsS, and ΔpqsS+ strains (n = 3, *P ≤ 0.05, Mann–Whitney U test). e Swarming motility of PAO1 WT, ΔpqsS, and ΔpqsS+ strains. All strains were observed after 24 h of growth in 0.5% semisolid medium (8 g/L Nutrition broth, 5 g/L glucose) at 30 °C (n = 3).
RLs are key virulence factors produced by P. aeruginosa that damage the host cells46. As expected, RL production by the ΔpqsS strain was decreased 1.92-fold and 1.85-fold compared to that of PAO1 and the ΔpqsS+ strains, respectively (Fig. 2d). Bacterial swarming motility is directly related to RL-mediated acute infection phenotypes. We thus performed a swarming motility assay and found that the growth radii decreased by 1.89-fold and 1.85-fold in the ΔpqsS mutant (38 mm) compared to the PAO1 WT (72 mm) and the corresponding complement (71 mm) strains, respectively (Fig. 2e). Taken together, these results suggest that PqsS promotes RL production in P. aeruginosa, swarming motility, and macrophage death under in vitro conditions along with the killing of G. mellonella under in vivo conditions to enhance acute infections.
PqsS reduces PAO1-mediated chronic infections including biofilm formation, antibiotic resistance, macrophage survival, and reactive oxygen species (ROS) productions
Biofilm formation by P. aeruginosa is a well-known cause of chronic infections47. The effect of PqsS on biofilm formation was determined using crystal violet staining. The biofilm formation of ΔpqsS mutant was increased by 1.59-fold and 1.64-fold, which was comparable to that of the PAO1 WT and ΔpqsS+ strains, respectively (Supplementary Fig. 3c). Confocal laser scanning microscopy was used to observe the surface-attached biofilms. Consistent with the results from CV staining, the ΔpqsS mutant showed a 1.87-fold and 1.96-fold increase in biomass attached to the surface compared to the WT and ΔpqsS+ strains, respectively (Fig. 3a, b). P. aeruginosa biofilm formation directly contributes to bacterial antibiotic resistance. Hence, we evaluated how PqsS affected the resistance against colistin, which is a “last-resort” polymyxin antibiotic against multidrug-resistant Gram-negative pathogens48. Confocal imaging results showed that the ΔpqsS mutant developed 2.37-fold and 2.26-fold increased antibiotic-tolerant subpopulations in response to colistin treatment than the PAO1 WT and ΔpqsS+ strains, respectively (Fig. 3c, d).

a Confocal laser scanning microscopy (CLSM) of biofilms from PAO1 WT, ΔpqsS, and ΔpqsS+ strains in glass-bottom culture dishes (n = 3), scale bar = 20 μm. b Biomass were quantitatively analysed using the Comstat software and are expressed as μm3/μm2. Data represent the means ± SD (n = 3, *P ≤ 0.05, Mann–Whitney U test). c CLSM of biofilms after treatment of PAO1 WT, ΔpqsS, and ΔpqsS+ strains treatment with a medium containing 10 μg/mL colistin (n = 3), scale bar = 20 μm. d The Live/dead ratio of biofilms after treatment of PAO1 WT, ΔpqsS, and ΔpqsS+ strains treatment with a medium containing 10 μg/mL colistin. Data represent the means ± SD (n = 3, **P ≤ 0.01, Mann–Whitney U test). e Survival of PAO1 WT, ΔpqsS, and ΔpqsS+ strains upon phagocytosis by RAW264.7 cells. The results are expressed as the percentage of surviving bacteria compared to the number of bacteria internalized after 2 h after phagocytosis. Data represent the means ± SD (n = 3, **P ≤ 0.01, Mann–Whitney U test). f Flow cytometry for measurement of intracellular reactive oxygen species (ROS) levels in RAW264.7 cells at 2 h after phagocytosis of PAO1 WT, ΔpqsS, and ΔpqsS+ strains. Data are presented as the means ± SD (n = 3). g Quantification of intracellular ROS in RAW 264.7 cells treated in (f). Data are presented as the means ± SD (n = 3, *P ≤ 0.05, Mann–Whitney U test).
Chronic infection of P. aeruginosa is associated with its ability to resist immune clearance and survive in phagocytic cells49. Therefore, we tested whether PqsS affected the phagocytosis rate and intracellular survival of PAO1 in RAW264.7 macrophage infection assay. The pqsS mutant behaved similarly to the WT strain with phagocytosis by macrophages (Supplementary Fig. 3d); however, the pqsS mutant was less sensitive to macrophage killing than the WT or complemented strains, with a 3.01-fold and 2.95-fold increase in the bacterial survival rate, respectively (Fig. 3e).
Macrophages produce reactive oxygen species, leading to the destruction and clearance of pathogens50. Next, we measured the intracellular ROS production in macrophages after infection with PAO1 WT, ΔpqsS, and ΔpqsS+ strains. We found that the ROS levels within macrophages infected with ΔpqsS were 1.91-fold and 1.87-fold lower than those in the PAO1 WT and ΔpqsS+ strains, respectively (Fig. 3f, g). These results indicate that PqsS represses P. aeruginosa PAO1 chronic infection phenotypes, including biofilm formation, antibiotic resistance, macrophage survival, and ROS production.
PqsS inhibits pqsL to increase PQS levels, thereby promoting pqs QS system in PAO1
To identify target genes and signaling pathways associated with PqsS, we extracted total RNA (Supplementary Fig. 4a) and performed RNA-seq to detect global gene expression profile differences between the PAO1 WT and ΔpqsS strain. In total, 292 genes (130 repressed and 162 activated) were differentially transcribed (>2-fold) between the two strains (Fig. 4a, Supplementary Table 3). Among these, pqsL, which encodes a monooxygenase in P. aeruginosa pqs QS system, was also predicted by TargetRNA2 as a putative PqsS target (Supplementary Table 4). The results of RT-qPCR analysis were consistent with RNA-seq data, which showed that the pqsL mRNA levels increased by 2.1-fold and 2.0-fold in ΔpqsS compared to those in PAO1 and ΔpqsS+ strains, respectively (Fig. 4b).

a A volcano plot of the differentially expressed genes (DEGs) determined for PAO1 WT and ΔpqsS strains by RNA-seq. FC, fold change; padj, adjusted p value. b Relative quantification of pqsL, pqsA, pqsB, pqsC, pqsD, pqsE, and pqsH expression in PAO1 WT, ΔpqsS, and ΔpqsS+ strains determined by RT-qPCR. The data are presented as the relative transcript abundance of the indicated genes compared to the internal control transcript for rpsL using the comparative Ct method (2−∆∆CT). Data represent the means ± SD (n = 3, *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001, Mann–Whitney U test). c Expression of PpqsA−gfp reporter fusions in PAO1 WT, ΔpqsS, ΔpqsS + , ΔpqsL, ΔpqsLΔpqsS, and ΔpqsR strains. The relative fluorescence intensity (reflected as GFP/OD600) was measured in representative strains containing PpqsA−gfp (n = 3). d Qualification of PQS concentrations measured in the supernatants of PAO1 WT, ΔpqsS, ΔpqsS + , ΔpqsL, ΔpqsLΔpqsS, and ΔpqsR cultures. Data represent the means ± SD (n = 3, *P ≤ 0.05, Mann–Whitney U test). e Qualification of HQNO concentrations measured in the supernatants of PAO1 WT, ΔpqsS, ΔpqsS + , ΔpqsL, ΔpqsLΔpqsS, and ΔpqsR cultures. Data represent the means ± SD (n = 3, *P ≤ 0.05, Mann–Whitney U test). f Northern blotting analysis of PqsS in PAO1 WT after treatment with the medium containing exogenous 1, 10, or 100 μM PQS for 1 h. 5S rRNA was used as the loading control. Densitometric analysis of the bands was performed using the ImageJ software.
P. aeruginosa pqs QS system utilizes the pqsABCDE operon and PqsH to synthesise PQS molecules. Specifically, the PQS precursor anthranilic acid was converted by PqsA to produce anthraniloyl-CoA51, which then formed 2-aminobenzoylacetyl-CoA (2-ABA-CoA) catalyzed by PqsD52. This was then converted into 2-ABA by PqsE, formed HHQ by PqsBC53 and finally converted into PQS by PqsH54. Thus, to examine how PqsS sRNAs influence pqs QS system, the transcription of pqsA, pqsB, pqsC, pqsD, pqsE, and pqsH was examined via RT-qPCR in PAO1 WT, ΔpqsS, and ΔpqsS+ strains. The results of RT-qPCR results showed the genes were repressed in ΔpqsS strain, which is consistent with our RNA sequencing results (Fig. 4b, Supplementary Table 3). PQS is sensed by its specific receptor PqsR, also known as multiple virulence factor regulator (MvfR), in P. aeruginosa55. Hence, we knocked out pqsR in the negative control. We transformed PAO1 WT, ΔpqsS, ΔpqsS + , ΔpqsL, ΔpqsLΔpqsS, and ΔpqsR strains with the PpqsA−gfp reporter plasmid14 to construct PAO1 pqs QS system bioreporter strains. Here, the pqsA gene expression was monitored to determine the activity of the PAO1 pqs QS system56. Growth curves showed that PAO1-pqsA-gfp, ΔpqsS-pqsA-gfp, ΔpqsS + -pqsA-gfp, ΔpqsL-pqsA-gfp, ΔpqsLΔpqsS-pqsA-gfp, and ΔpqsR-pqsA-gfp strains grew at similar rates in vitro (Supplementary Fig. 4b). Using PAO1 PpqsA−gfp and ΔpqsR PpqsA−gfp as positive and negative controls, respectively, we found that the expression of pqsA-gfp was decreased in ΔpqsS PpqsA−gfp compared to that in PAO1 PpqsA−gfp and ΔpqsS+ PpqsA−gfp strains. Interestingly, pqsA-gfp expression in the ΔpqsLΔpqsS PpqsA−gfp strain was similar to that in ΔpqsL PpqsA−gfp strain, which itself was higher than that in ΔpqsS PpqsA−gfp strain (Fig. 4c). These results suggested that PqsS activated the P. aeruginosa PAO1 pqs QS system by repressing pqsL.
Growth curves showed that PAO1 WT, ΔpqsL, ΔpqsLΔpqsS, and ΔpqsR strains grew at similar rates in vitro (Supplementary Fig. 4c). Monooxygenase PqsL is responsible for converting precursor 2-ABA into HQNO via a PQS-branched pathway10. Hence, we performed LC-MS/MS to measure the production of PQS, the PQS derivative HQNO, and the PQS precursor HHQ. As expected, the ΔpqsS mutant produced 1.9 μg/mL of PQS, which was lower compared to 3.8 μg/mL and 3.6 μg/mL observed in PAO1 WT and ΔpqsS+ strains, respectively, while ΔpqsL (5.5 μg/mL) and ΔpqsLΔPqsS (5.3 μg/mL) strains produced more PQS to similar degrees. The ΔpqsR strain produced virtually no PQS, as expected in negative control (Fig. 4d). For HQNO, ΔpqsS produced more HQNO of 5.7 μg/mL compared to 3.6 μg/mL and 3.5 μg/mL in PAO1 WT and ΔpqsS+ strains, respectively. However, ΔpqsL, ΔpqsLΔPqsS, and ΔpqsR strains produced no HQNO (Fig. 4e). HHQ exhibited the same trend as PQS (Supplementary Fig. 4d). Thus, PqsS increases PQS and HHQ production, while decreasing HQNO levels, primarily by inhibiting pqsL.
The PQS concentration of normal P. aeruginosa secreted in liquid cultures fluctuated from approximately 1 to 60 μM12,57,58. To assess whether different PQS concentrations affected PqsS expression, after stimulation with exogenous PQS, northern blotting showed that the abundance of PAO1 WT PqsS sRNA was increased by 1.82-fold and 2.53-fold in 10 μM and 100 μM PQS TSB than that in 1 μM PQS TSB, respectively (Fig. 4f). These results confirmed that PqsS represses pqsL to stimulate pqs QS systems and increase PQS production in P. aeruginosa PAO1, whereas high concentrations of PQS further promote PqsS expression, thus forming a positive feedback loop.
PqsS binds to the pqsL mRNA to decrease its stability with RNase E cleavage
The antibiotic rifampicin targets bacterial RNA polymerase and is frequently used to inhibit bacterial RNA synthesis and suspend transcription59. Researchers have used rifampicin to evaluate the inhibitory effect of sRNA on target mRNA stability at the post-transcriptional level, termed the rifampicin transcription inhibition assay60. To determine the exact molecular mechanism by which PqsS represses pqsL mRNA, we tested mRNA stability using a rifampicin transcription inhibition assay. Here, pqsL mRNA was degraded much more slowly in ΔpqsS (half-life: 9.1 min) than in WT (3.2 min) and ΔpqsS + (3.3 min) strains, respectively (Fig. 5a). The mRNA stability assay revealed that PqsS sRNA decreased pqsL mRNA stability at the post-transcriptional level, further reducing pqsL mRNA levels. We then used western blotting to measure the amounts of FLAG-tagged PqsL (PqsL-FLAG) in PAO1, ΔpqsS, and ΔpqsS+ strains. The pqsS deletion increased PqsL-FLAG production by 2.15-fold, and complementation with pqsS in ΔpqsS restored PqsL-FLAG production (Fig. 5b). Thus, PqsS appears to reduce PqsL production by decreasing the stability of pqsL mRNA.

a Relative quantification of pqsL mRNA by RT-qPCR in rifampicin-treated PAO1 WT, ΔpqsS, and ΔpqsS + strains for 0–15 min. Data represent the percentage of pqsL transcripts relative to time point zero. Data represent the means ± SD (n = 3). b Representative western blot analysis of PqsL in protein preparations from PAO1 WT, ΔpqsS, and ΔpqsS + strains. RNA Polymerase α (α-RNAP) served as a loading control (n = 3). Protein size (kDa) is indicated on the right side. Densitometric analyses of the bands were performed using the ImageJ software. c Predicted base pairing between PqsS sRNA and pqsL mRNA. Red letters indicate the predicted binding motif with perfect base pairing. In the in vitro transcribed PqsS*-pqsL sRNA, the CGGCUC motif was mutated to GCCGAG. d REMSA with a fixed amount of pqsL mRNA and an increasing amount of PqsS sRNA. e In vitro cleavage of pqsL mRNA by RNase E in the absence or presence of PqsS sRNA (n = 3). Densitometric analyses of the bands were performed using the ImageJ software. f Proposed molecular mechanism model of PqsS sRNA inducing pqsL mRNA decay by recruiting RNase E.
Next, we predicted the complementary 12-nucleotide regions that could mediate the interaction between PqsS sRNA (UGCCGGCUCGCC) and pqsL mRNA (UGGGAGCCGACG) using IntaRNA with a six-nucleotide binding motif (CCGCUC) input for PqsS sRNA (Figs. 1a, 5c). The pqsL mRNA-binding region was observed in the predicted PqsS sRNA secondary structure (Supplementary Fig. 2a). To test for a direct interaction between PqsS and pqsL mRNA in vitro, we performed RNA-RNA EMSA (REMSA) using in vitro transcribed PqsS and pqsL mRNA. REMSA reactions were performed using different concentrations of PqsS and a fixed amount of pqsL mRNA. We observed the PqsS-pqsL mRNA complex with the band intensity appearing enhanced with increasing PqsS concentrations. These findings confirm the direct interaction between PqsS sRNA and pqsL mRNA in vitro (Fig. 5d). To confirm that the 6-nucleotide motif (CCGCUC) was the PqsS sequence required for pqsL mRNA binding, we performed REMSA using a mutated PqsS (PqsS*-pqsL) sRNA (the motif CCGCUC was mutated to GCCGAG) generated by in vitro transcription. No interaction was observed between PqsS*-pqsL sRNA and pqsL mRNA (Supplementary Fig. 4e), indicating that the PqsS CCGCUC motif was critical for PqsS binding to pqsL mRNA.
After discovering that PqsS sRNA decreased pqsL mRNA stability, we aimed to explore the underlying mechanism. The best previous example of one bacterial sRNA that binds to target mRNA coding sequence (CDS) and inhibits its mRNA stability is MicC61. Specifically, MicC inhibits ompD mRNA by binding to its CDS and recruiting RNase E for degradation. RNase E is the most important bacterial RNase for degrading mRNAs62; thus, we hypothesized that PqsS reduces the stability of pqsL mRNA CDS by facilitating RNase E-mediated cleavage. We performed in vitro RNase E cleavage assays and found that incubation with purified RNase E led to the degradation of the pqsL mRNA CDS. Moreover, the addition of PqsS sRNA increased the degradation of pqsL mRNA CDS by RNase E (Fig. 5e). However, we did not observe any additional degradation of pqsL mRNA CDS upon the addition of PqsS*-pqsL sRNA. PqsS sRNA did not exhibit any additional degradation effect on the rpsL mRNA internal control upon the addition of RNase E (Supplementary Fig. 4f). Thus, we demonstrated the molecular mechanism underlying the interactions of PqsS sRNA-Hfq protein-pqsL mRNA-RNase E in which the Hfq-PqsS complex decreased pqsL mRNA stability by recruiting RNase E to drive degradation (Fig. 5f).
PqsS promotes PAO1 MVs productions and abundantly detected in MVs
Given that PQS contributes significantly to the majority of planktonic MV formation in P. aeruginosa19, we next investigated whether PqsS sRNA also contributes to P. aeruginosa MV production. We performed an optimized P. aeruginosa PAO1 MV purification procedure to obtain pure MV without flagella, pili, or Pf4 phage contamination (Supplementary Fig. 5). After MV purification, protein quantification by BCA demonstrated that the total protein amount was reduced by 39% in MVs from the ΔpqsS mutant (111.3 ng/μL) compared to those from the PAO1 WT (182.5 ng/μL). ΔpqsR mutant (10.9 ng/μL) was used as the negative control (Fig. 6a). The negative staining transmission electron microscopy results clearly showed that the numbers of MVs decreased in ΔpqsS mutant compared to those in the PAO1 WT strain. They were nearly 100–200 nm in size and were spherical structures harboring membrane bilayers (Supplementary Fig. 6a). The nanoparticle tracking analyses (NTA) were used to further quantify the number of MVs, which decreased from 7.8 × 1011 particles/mL in PAO1 WT to 4.2 × 1011 particles/mL in ΔpqsS mutant. In contrast, ΔpqsR mutant (5.6 × 1010 particles/mL) was used as the negative control with nearly unchanged size distributions (Fig. 6c, Supplementary Fig. 6b).

a Identification of protein content of MVs in PAO1 WT, ΔpqsS, and ΔpqsR strains via BCA. Data represent the means ± SD (n = 3, *P ≤ 0.05, Mann–Whitney U test). b Negative staining transmission electron microscopy (TEM) identification of MVs in PAO1 WT and ΔpqsS strains (n = 3). Scale bar = 200/100 nm. c NTA of MV concentrations in PAO1 WT, ΔpqsS, and ΔpqsR strains. Data represent the means ± SD (n = 3, *P ≤ 0.05, Mann–Whitney U test). d Northern blotting analysis of PqsS from PAO1 WT and its purified MVs samples (n = 3). 5S rRNA was used as the loading control. Densitometric analysis of the bands was performed using the ImageJ software. e Western blot analysis of Hfq in protein preparations from PAO1 WT and MVs samples. Total protein served as a loading control (n = 3). Densitometric analyses of the bands were performed using the ImageJ software. f Western blot analysis of RNase E in protein preparations of PAO1 WT and MVs samples. Total protein served as a loading control (n = 3). Densitometric analyses of the bands were performed using the ImageJ software.
We also performed northern blotting to determine whether PqsS could be detected in the PAO1 MVs. PqsS abundance in MVs was 2.89-fold higher than that in PAO1 WT within the same amount of total RNA extract (Fig. 6d). In addition, the key bacterial sRNA-binding protein Hfq could help sRNAs pack into MVs63. Specifically, the C-terminal regions (CTR) of Hfq form amyloid-like structures with fibrils that disrupt bacterial membranes, allowing Hfq to be translocated into the periplasm and exported into MVs64. Western blotting was performed to measure the amount of Hfq protein in PAO1 cells and MVs. Hfq protein abundance increased by 4.36-fold in MVs compared to that in PAO1 WT within the same amount of total protein extract. This result indicates that Hfq might ensure sRNA stability in MVs (Fig. 6e). A recently published proteome also showed that RNase E protein levels were decreased by 7.9-fold in MVs compared to those in their parent bacteria65, which implied that sRNAs are less likely to be degraded by RNase E in MVs. Next, western blotting was used to measure the amount of RNase E in PAO1 cells and MVs. The abundance of RNase E protein decreased by 4.17-fold in MVs compared to that in PAO1 WT within the same amount of total protein extract (Fig. 6f). These results revealed that PqsS sRNA and Hfq protein were abundant in the MVs. Moreover, MVs may protect sRNAs from RNase E.
In addition, we performed comparative genomic analysis to investigate whether pqsS is present in other pathogenic P. aeruginosa strains. Genome alignment analysis of 83 representative P. aeruginosa strains revealed that pqsS was highly conserved and widely distributed in 59 strains (59/83) (Supplementary Fig. 7a, b and Supplementary Table 5); however, pqsS was not present in other non-P. aeruginosa isolates from the Pseudomonas genus (0/117). To investigate whether homologous PqsS in other P. aeruginosa strains also suppressed pqsL mRNA levels, we knocked down the pqsS homolog in P. aeruginosa PA14 and quantified the pqsL mRNA levels by RT-qPCR. The mRNA level in the mutant was increased by 1.7-fold compared to that in the corresponding WT strain (Supplementary Fig. 7c), implying that PqsS-mediated pqsL regulation is conserved in other P. aeruginosa strains. Moreover, analysis of publicly available clinical RNA-seq data from P. aeruginosa samples showed that the transcription of pqsS was increased in human sputum samples66,67 and SARS-CoV2 infected samples (patients investigated were diagnosed with a secondary P. aeruginosa infection)68 compared to the bacterial liquid culture control (Supplementary Table 6). These results reveal that pqsS is conserved in pathogenic P. aeruginosa strains and is clinically relevant.
Discussion
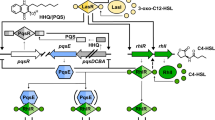
P. aeruginosa employs various QS systems to coordinate group behavior and optimize the expression of various regulatory proteins and Hfq-binding sRNAs69. In the present study, we explored the potential relationship between Hfq-binding sRNAs and pqs QS systems in P. aeruginosa. We found that a novel Hfq-binding sRNA-mediated mechanism enabled P. aeruginosa to promote pqs QS and mediate acute infections (Fig. 7). We first identified PqsS as an Hfq-binding sRNA in PAO1 (Fig. 1). PqsS promoted P. aeruginosa acute infections in a macrophage model in vitro and G. mellonella in vivo and enhanced RL-mediated swarming motility (Fig. 2). PqsS decreased P. aeruginosa biofilm-related chronic infection traits including macrophage survival and ROS production (Fig. 3). Furthermore, we identified pqsL as a target gene of PqsS and found that PqsS increased PQS production in an enhanced pqs QS system. PQS further promotes PqsS expression, thus forming a positive PqsS-PQS feedback loop (Fig. 4). We also showed that the PqsS-Hfq complex inhibited pqsL mRNA by binding to its CDS and recruiting RNase E for degradation (Fig. 5). Finally, PqsS promoted PAO1 MV production and was abundantly detected in the MVs (Fig. 6). These findings highlight an extra sRNA-centered QS signal amplification strategy in P. aeruginosa communities, in addition to traditional bacterial QS communication. PqsS enhances P. aeruginosa infection. Hence, it may be a potential drug target for the development of anti-P. aeruginosa therapy.

A novel sRNA named PqsS promotes pqs quorum sensing (QS) with the assistance of the chaperone protein Hfq. Specifically, high levels of PQS promotes PqsS expression and forms a PqsS-Hfq complex. Then, this complex decreases pqsL mRNA stability by recruiting RNase E to drive degradation, thereby enhancing PQS productions for pqs QS and MVs formation. PQS further promotes PqsS expression, thus forming a positive PqsS-PQS feedback loop. Therefore, PqsS promotes acute bacterial infections including host cell death, rhamnolipids production, and swarming motility, while reducing chronic infections, such as biofilm formation, cellular survival, and host cell ROS production.
P. aeruginosa expresses various sRNAs under specific microenvironmental conditions, such as different ion, oxygen, and metabolite concentrations, to regulate acute or chronic infection mechanisms. Acute infection mechanisms of P. aeruginosa include host cell death, virulence factor synthesis, flagella or pili-mediated motility, and T3SS, whereas chronic infection mechanisms include biofilm formation, antibiotic tolerance, and reduced sensitivity to phagocytes70. The classification of P. aeruginosa sRNAs based on acute and chronic infection regulation is not currently clear; thus, we summarized the previously studied sRNAs into three groups: 1. sRNA can simultaneously promote or inhibit acute and chronic infections simultaneously (eg, ErsA increases motility-related acute infections and biofilm formation-related chronic infections under low oxygen levels71); 2. sRNA promote acute and reduced chronic infections (e.g., PrrF1/2 promotes acute bacterial lung infection while reducing biofilm-related chronic infections in response to low-iron conditions29,72); 3. sRNA promote chronic infections and reduce acute infections (e.g., RsmY/Z promotes biofilm formation to allow bacteria to adopt a sessile lifestyle, mediating chronic infection under conditions involving high c-di-GMP levels73,74 while reducing the expression of type IV pili and flagella to downregulate acute infection70,75). In this study, we report a novel sRNA, PqsS, which enabled P. aeruginosa to promote pqs QS, favor acute infections, and repress chronic infections by sensing high PQS concentrations (Fig. 7). Based on our classification method, PqsS belongs to the type two sRNA, which promotes acute infections and reduces chronic infections, similar to PrrF1/229,72. Hence, it may serve as the biomarker for P. aeruginosa acute infections. Our novel findings and those of previous sRNA-related studies highlight that bacteria employ multiple sRNAs to control their acute or chronic infections in response to different microenvironmental cues.
To date, multiple molecular mechanisms have been discovered by which bacterial sRNAs downregulate target mRNAs76,77,78, including the following: 1. sRNAs pair with the ribosome binding site, a Shine-Dalgarno (SD) sequence, within the 5’ untranslated region (UTR) of the target gene to prevent the translation initiation of its protein (eg, PrrF1/2 repressed antR mRNA translation by binding to its 5’ UTR region79); 2. sRNAs pair with the CDS region of the target mRNA, resulting in the degradation of the target mRNA by recruiting RNases and shortening the half-life of the target mRNA to induce mRNA decay (e.g., MicC inhibits ompD mRNA by binding to its CDS and recruiting RNase E for degradation61); 3. sRNAs cannot change the mRNA quantity but modify the conformation of the mRNA of the target gene such that the gene cannot be expressed normally (e.g., SR1 inhibits ahrC translation by inducing structural changes downstream of the RBS site80). In our study, we first predicted the interaction region between PqsS sRNA and pqsL mRNA within the CDS region (484-473) of pqsL mRNA; thus, the inhibitory mechanism cannot be type 1. We tested the influence of PqsS sRNA on pqsL mRNA levels using qRT-PCR (Fig. 4b). The results showed that sRNA PqsS decreased the expression level of pqsL mRNA; thus, we did not consider type 3. Therefore, we examined the type 2 mechanism by measuring the influence of PqsS sRNA on pqsL mRNA stability to determine whether PqsS sRNA mediated transcript decay. Our mRNA stability results revealed that PqsS sRNA decreased pqsL mRNA stability at the post-transcriptional level, further reducing pqsL mRNA levels (Fig. 5a). We also found that PqsS sRNA increased pqsL mRNA CDS degradation by RNase E (Fig. 5e). Thus, we elucidated the molecular mechanism underlying the induction of pqsL mRNA decay via the recruitment of RNase E by PqsS sRNA (Fig. 5f).
Many P. aeruginosa sRNAs have been studied for their roles in biofilm formation. However, most previous studies have focused on the regulatory effects of sRNAs inside bacteria. These regulatory effects on biofilm formations are indirect. For example, the expression of SicX small interfering RNA (sRNA) is induced under low oxygen levels to promote chronic biofilm formation81. Recently, extracellular RNAs (eRNAs), whose spatial position is closer to the actual biofilm structure than that of endobacterial RNAs, has garnered significant interest. Substantial amounts of eRNAs such as ssrA and crcZ can be detected within biofilms, contributing to their structural integrity and viscosity within biofilms82. However, a stable eRNA source in the biofilm matrix remains elusive due to the natural instability of RNA molecules. MVs are essential components of P. aeruginosa biofilms18,83,84. Another recent study showed that the total RNAs of MVs contained 60% short RNA fragments85. We hypothesized that small regulatory eRNAs may also contribute directly to biofilm matrix formation (i.e. as biofilm matrix components). Thus, MVs may be a valuable source for identifying novel bacterial sRNAs and revealing their functions in biofilm regulation. Our study revealed that a specific sRNA, PqsS, was abundantly detected in MVs and decreased biofilm formation (Figs. 3a, b, and 6d). However, the total eRNA promotes biofilm formation82. Hence, we propose that this is due to a dynamic homeostasis in biofilm formation, indicating that not all sRNAs facilitate biofilm formation. Under a specific microenvironment, bacteria express chronic infection-regulated sRNA, such as SicX, to prefer a biofilm lifestyle, whereas when the condition changes, P. aeruginosa may utilize sRNAs, such as acute infection-regulated PqsS, to switch to a planktonic lifestyle.
Bacterial MVs can fuse with nearby membrane-structured organisms to exchange cargo, thereby playing an important role in inter-cellular communication83. MVs can carry various hydrophobic molecules, proteins, and nucleic acids that travel long distances and target other bacteria for nutrient acquisition. For example, TseF, a P. aeruginosa T6SS effector incorporated into MVs, directs MVs to their parent bacteria by binding to the surface receptors, FptA and OprF, to acquire MV-associated iron86. In addition, MVs may interfere with other bacteria in nearby ecological niches, thus helping bacterial cells compete for nutrients in a limited environment87. For instance, P. aeruginosa MVs contain a high level of hydrophobic PQS signaling molecules88, which can travel a long distance (cm scale, much longer than the normal QS diffusion distance) at the solid surface to eradicate S. aureus colonies89. Despite multiple studies investigating the virulence factors encapsulated in P. aeruginosa MVs, the potential intercellular regulatory roles of MV-carried RNA components remain largely unknown and warrant further investigation.
Comparative genomics analysis showed that PqsS also exists in various clinical P. aeruginosa-infected samples (Supplementary Table 6). Thus, determining the exact host cell targets and the regulatory network of PqsS during infection requires further investigation. Bacterial MV-enriched sRNAs have been reported to modulate host cell functions. In P. aeruginosa, MV-enriched sRNA-sRNA52320 (a 23-nt tRNA fragment) attenuated interleukin-8 secretion in human airway epithelial cells90. Other bacteria also use MV-enriched sRNAs to modulate host-microbe interactions. For example, E. coli sRNA Ile-tRF-5X is released via MVs and promotes human HCT116 cell MAP3K4 expression91. In another study, Legionella pneumophila MVs translocated the sRNA RsmY into host cells and mimicked eukaryotic microRNAs to reduce host defense signaling pathways92. The results of our study, together with previous findings, enrich our understanding of the fascinating world of sRNAs and their intricate roles in bacterial pathogenesis.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
RNA-seq data have been deposited into the Sequence Read Archive database (https://www.ncbi.nlm.nih.gov/sra) with the bioProject accession number: PRJNA982285 (Nanopore PAO1 RNA-seq), PRJNA916110 (ΔpqsS RNA-seq).
References
Curran, C. S., Bolig, T. & Torabi-Parizi, P. Mechanisms and targeted therapies for pseudomonas aeruginosa lung infection. Am. J. Respir. Crit. Care Med. 197, 708–727 (2018).
Rice, L. B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 197, 1079–1081 (2008).
Ciofu, O., Moser, C., Jensen, P. & Høiby, N. Tolerance and resistance of microbial biofilms. Nat. Rev. Microbiol. 20, 621–635 (2022).
Lichtenberg, M. et al. The structure-function relationship of Pseudomonas aeruginosa in infections and its influence on the microenvironment. FEMS Microbiol. Rev. 46. https://doi.org/10.1093/femsre/fuac018 (2022).
Balasubramanian, D., Schneper, L., Kumari, H. & Mathee, K. A dynamic and intricate regulatory network determines Pseudomonas aeruginosa virulence. Nucleic Acids Res. 41, 1–20 (2013).
Caiazza, N. C., Shanks, R. M. & O’Toole, G. A. Rhamnolipids modulate swarming motility patterns of Pseudomonas aeruginosa. J. Bacteriol. 187, 7351–7361 (2005).
Ahator, S. D. & Zhang, L. Small is mighty-chemical communication systems in Pseudomonas aeruginosa. Annu. Rev. Microbiol. 73, 559–578 (2019).
Papenfort, K. & Bassler, B. L. Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 14, 576–588 (2016).
Lin, J., Cheng, J., Wang, Y. & Shen, X. The Pseudomonas Quinolone Signal (PQS): not just for quorum sensing anymore. Front. Cell Infect. Microbiol. 8, 230 (2018).
Soh, E. Y. et al. Disruption of the Pseudomonas aeruginosa Tat system perturbs PQS-dependent quorum sensing and biofilm maturation through lack of the Rieske cytochrome bc1 sub-unit. PLoS Pathog. 17, e1009425 (2021).
Rampioni, G. et al. Unravelling the genome-wide contributions of specific 2-Alkyl-4-Quinolones and PqsE to quorum sensing in Pseudomonas aeruginosa. PLoS Pathog. 12, e1006029 (2016).
Florez, C., Raab, J. E., Cooke, A. C. & Schertzer, J. W. Membrane distribution of the pseudomonas quinolone signal modulates outer membrane vesicle production in Pseudomonas aeruginosa. mBio 8. https://doi.org/10.1128/mBio.01034-17 (2017).
Yang, L., Nilsson, M., Gjermansen, M., Givskov, M. & Tolker-Nielsen, T. Pyoverdine and PQS mediated subpopulation interactions involved in Pseudomonas aeruginosa biofilm formation. Mol. Microbiol. 74, 1380–1392 (2009).
Yang, L. et al. Effects of iron on DNA release and biofilm development by Pseudomonas aeruginosa. Microbiology 153, 1318–1328 (2007).
Liu, X. et al. Cell division factor ZapE regulates Pseudomonas aeruginosa biofilm formation by impacting the pqs quorum sensing system. mLife 2, 28–42 (2023).
Toyofuku, M., Schild, S., Kaparakis-Liaskos, M. & Eberl, L. Composition and functions of bacterial membrane vesicles. Nat. Rev. Microbiol. 21, 415–430 (2023).
Colombo, M., Raposo, G. & Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 30, 255–289 (2014).
Toyofuku, M., Nomura, N. & Eberl, L. Types and origins of bacterial membrane vesicles. Nat. Rev. Microbiol. 17, 13–24 (2019).
Cooke, A. C., Nello, A. V., Ernst, R. K. & Schertzer, J. W. Analysis of Pseudomonas aeruginosa biofilm membrane vesicles supports multiple mechanisms of biogenesis. PLoS ONE 14, e0212275 (2019).
Schwechheimer, C. & Kuehn, M. J. Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat. Rev. Microbiol. 13, 605–619 (2015).
Hör, J., Gorski, S. A. & Vogel, J. Bacterial RNA biology on a genome scale. Mol. Cell 70, 785–799 (2018).
Jia, T. et al. A novel small RNA promotes motility and virulence of enterohemorrhagic Escherichia coli O157:H7 in response to ammonium. mBio. 12. https://doi.org/10.1128/mBio.03605-20 (2021).
Jia, T. et al. The phosphate-induced small RNA EsrL promotes E. coli virulence, biofilm formation, and intestinal colonization. Sci. Signal. 16, eabm0488 (2023).
Dendooven, T., Sonnleitner, E., Bläsi, U. & Luisi, B. F. Translational regulation by Hfq-Crc assemblies emerges from polymorphic ribonucleoprotein folding. Embo J. 42, e111129 (2023).
Gebhardt, M. J. et al. Hfq-licensed RNA-RNA interactome in Pseudomonas aeruginosa reveals a keystone sRNA. Proc. Natl Acad. Sci. USA 120, e2218407120 (2023).
Wang, C., Ye, F., Kumar, V., Gao, Y. G. & Zhang, L. H. BswR controls bacterial motility and biofilm formation in Pseudomonas aeruginosa through modulation of the small RNA rsmZ. Nucleic Acids Res. 42, 4563–4576 (2014).
Liu, P. et al. The function of small RNA in Pseudomonas aeruginosa. PeerJ 10, e13738 (2022).
Sonnleitner, E. et al. The small RNA PhrS stimulates synthesis of the Pseudomonas aeruginosa quinolone signal. Mol. Microbiol. 80, 868–885 (2011).
Reinhart, A. A. et al. The Pseudomonas aeruginosa PrrF small RNAs regulate iron homeostasis during acute murine lung infection. Infect. Immun. 85. https://doi.org/10.1128/iai.00764-16 (2017).
Carver, T., Harris, S. R., Berriman, M., Parkhill, J. & McQuillan, J. A. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469 (2012).
Liu, J. H. et al. Bacmethy: a novel and convenient tool for investigating bacterial DNA methylation pattern and their transcriptional regulation effects. Imeta 3, e186 (2024).
Pettersen, E. F. et al. UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Yan, Y. M., Zhang, D., Zhou, P., Li, B. T. & Huang, S. Y. HDOCK: a web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 45, W365–W373 (2017).
Seeliger, D. & de Groot, B. L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aid Mol. Des. 24, 417–422 (2010).
Cai, Y. M. et al. The c-di-GMP Phosphodiesterase PipA (PA0285) regulates autoaggregation and Pf4 bacteriophage production in Pseudomonas aeruginosa PAO1. Appl. Environ. Microbiol. 88, e0003922 (2022).
Belon, C. et al. A macrophage subversion factor is shared by intracellular and extracellular pathogens. PLoS Pathog. 11, e1004969 (2015).
Ortori, C. A. et al. Simultaneous quantitative profiling of N-acyl-L-homoserine lactone and 2-alkyl-4(1H)-quinolone families of quorum-sensing signaling molecules using LC-MS/MS. Anal. Bioanal. Chem. 399, 839–850 (2011).
Chao, Y. J. et al. In vivo cleavage map illuminates the central role of RNase E in Coding and Non-coding RNA pathways. Mol. Cell 65, 39–51 (2017).
Winsor, G. L. et al. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 44, D646–D653 (2016).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Zou, L., Susko, E., Field, C. & Roger, A. J. Fitting nonstationary general-time-reversible models to obtain edge-lengths and frequencies for the barry-hartigan model. Syst. Biol. 61, 927–940 (2012).
Zhang, Y. F. et al. Probing the sRNA regulatory landscape of P. aeruginosa: post-transcriptional control of determinants of pathogenicity and antibiotic susceptibility. Mol. Microbiol. 106, 919–937 (2017).
Chao, Y., Papenfort, K., Reinhardt, R., Sharma, C. M. & Vogel, J. An atlas of Hfq-bound transcripts reveals 3’ UTRs as a genomic reservoir of regulatory small RNAs. Embo J. 31, 4005–4019 (2012).
Gill, E. E. et al. High-throughput detection of RNA processing in bacteria. BMC Genom. 19, 223 (2018).
Stover, C. K. et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964 (2000).
Van Gennip, M. et al. Inactivation of the rhlA gene in Pseudomonas aeruginosa prevents rhamnolipid production, disabling the protection against polymorphonuclear leukocytes. Apmis. 117, 537–546 (2009).
Flemming, H. C. et al. Biofilms: an emergent form of bacterial life. Nat. Rev. Microbiol. 14, 563–575 (2016).
Li, J. et al. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect. Dis. 6, 589–601 (2006).
Duan, X. et al. A bacterial Quorum sensing regulated protease inhibits host immune responses by cleaving death domains of innate immune adaptors. Adv. Sci. 10, e2304891 (2023).
Bisht, K., Baishya, J. & Wakeman, C. A. Pseudomonas aeruginosa polymicrobial interactions during lung infection. Curr. Opin. Microbiol. 53, 1–8 (2020).
Coleman, J. P. et al. Pseudomonas aeruginosa PqsA is an anthranilate-coenzyme A ligase. J. Bacteriol. 190, 1247–1255 (2008).
Zhang, Y. M., Frank, M. W., Zhu, K., Mayasundari, A. & Rock, C. O. PqsD is responsible for the synthesis of 2,4-dihydroxyquinoline, an extracellular metabolite produced by Pseudomonas aeruginosa. J. Biol. Chem. 283, 28788–28794 (2008).
Drees, S. L. et al. PqsBC, a condensing enzyme in the biosynthesis of the Pseudomonas aeruginosa Quinolone signal: crystal structure, inhibition, and reaction mechanism. J. Biol. Chem. 291, 6610–6624 (2016).
Schertzer, J. W., Brown, S. A. & Whiteley, M. Oxygen levels rapidly modulate Pseudomonas aeruginosa social behaviours via substrate limitation of PqsH. Mol. Microbiol. 77, 1527–1538 (2010).
Cao, H. et al. A quorum sensing-associated virulence gene of Pseudomonas aeruginosa encodes a LysR-like transcription regulator with a unique self-regulatory mechanism. Proc. Natl Acad. Sci. USA 98, 14613–14618 (2001).
McGrath, S., Wade, D. S. & Pesci, E. C. Dueling quorum sensing systems in Pseudomonas aeruginosa control the production of the Pseudomonas quinolone signal (PQS). FEMS Microbiol. Lett. 230, 27–34 (2004).
Diggle, S. P. et al. The Pseudomonas aeruginosa quinolone signal molecule overcomes the cell density-dependency of the quorum sensing hierarchy, regulates rhl-dependent genes at the onset of stationary phase and can be produced in the absence of LasR. Mol. Microbiol. 50, 29–43 (2003).
Goh, Y. F. et al. Associational resistance to predation by protists in a mixed species biofilm. Appl. Environ. Microbiol. 89, e0174122 (2023).
Campbell, E. A. et al. Structural mechanism for rifampicin inhibition of bacterial rna polymerase. Cell 104, 901–912 (2001).
Ramirez-Peña, E., Treviño, J., Liu, Z., Perez, N. & Sumby, P. The group A Streptococcus small regulatory RNA FasX enhances streptokinase activity by increasing the stability of the ska mRNA transcript. Mol. Microbiol. 78, 1332–1347 (2010).
Pfeiffer, V., Papenfort, K., Lucchini, S., Hinton, J. C. & Vogel, J. Coding sequence targeting by MicC RNA reveals bacterial mRNA silencing downstream of translational initiation. Nat. Struct. Mol. Biol. 16, 840–846 (2009).
Mackie, G. A. & RNase, E. at the interface of bacterial RNA processing and decay. Nat. Rev. Microbiol. 11, 45–57 (2013).
Turbant, F. et al. Interactions and insertion of Escherichia coli Hfq into outer membrane vesicles as revealed by infrared and orientated circular dichroism spectroscopies. Int. J. Mol. Sci. 24. https://doi.org/10.3390/ijms241411424 (2023).
Turbant, F. et al. Unraveling membrane perturbations caused by the bacterial riboregulator Hfq. Int. J. Mol. Sci. 23. https://doi.org/10.3390/ijms23158739 (2022).
Zavan, L. et al. The mechanism of Pseudomonas aeruginosa outer membrane vesicle biogenesis determines their protein composition. Proteomics 23, e2200464 (2023).
Cornforth, D. M. et al. Pseudomonas aeruginosa transcriptome during human infection. Proc. Natl Acad. Sci. USA 115, E5125–e5134 (2018).
Rossi, E., Falcone, M., Molin, S. & Johansen, H. K. High-resolution in situ transcriptomics of Pseudomonas aeruginosa unveils genotype independent patho-phenotypes in cystic fibrosis lungs. Nat. Commun. 9, 3459 (2018).
Qu, J. et al. Persistent bacterial coinfection of a COVID-19 patient caused by a genetically adapted Pseudomonas aeruginosa chronic Colonizer. Front. Cell Infect. Microbiol. 11, 641920 (2021).
Whiteley, M., Diggle, S. P. & Greenberg, E. P. Progress in and promise of bacterial quorum sensing research. Nature 551, 313–320 (2017).
Valentini, M., Gonzalez, D., Mavridou, D. A. & Filloux, A. Lifestyle transitions and adaptive pathogenesis of Pseudomonas aeruginosa. Curr. Opin. Microbiol. 41, 15–20 (2018).
Ferrara, S. et al. The small RNA ErsA plays a role in the regulatory network of Pseudomonas aeruginosa pathogenicity in airway infections. mSphere 5 (2020).
Reinhart, A. A. et al. The prrF-encoded small regulatory RNAs are required for iron homeostasis and virulence of Pseudomonas aeruginosa. Infect. Immun. 83, 863–875 (2015).
Goodman, A. L. et al. A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev. Cell 7, 745–754 (2004).
Moscoso, J. A., Mikkelsen, H., Heeb, S., Williams, P. & Filloux, A. The Pseudomonas aeruginosa sensor RetS switches type III and type VI secretion via c-di-GMP signalling. Environ. Microbiol. 13, 3128–3138 (2011).
Chua, S. L. et al. Dispersed cells represent a distinct stage in the transition from bacterial biofilm to planktonic lifestyles. Nat. Commun. 5, 4462 (2014).
Hör, J., Matera, G., Vogel, J., Gottesman, S. & Storz, G. Trans-acting small RNAs and their effects on gene expression in Escherichia coli and Salmonella enterica. EcoSal Plus 9. https://doi.org/10.1128/ecosalplus.ESP-0030-2019 (2020).
Jagodnik, J., Brosse, A., Le Lam, T. N., Chiaruttini, C. & Guillier, M. Mechanistic study of base-pairing small regulatory RNAs in bacteria. Methods 117, 67–76 (2017).
Hui, M. P., Foley, P. L. & Belasco, J. G. Messenger RNA degradation in bacterial cells. Annu. Rev. Genet. 48, 537–559 (2014).
Djapgne, L. et al. The Pseudomonas aeruginosa PrrF1 and PrrF2 Small Regulatory RNAs Promote 2-Alkyl-4-Quinolone Production through Redundant Regulation of the antR mRNA. J. Bacteriol. 200. https://doi.org/10.1128/jb.00704-17 (2018).
Heidrich, N., Moll, I. & Brantl, S. In vitro analysis of the interaction between the small RNA SR1 and its primary target ahrC mRNA. Nucleic Acids Res. 35, 4331–4346 (2007).
Cao, P. et al. A Pseudomonas aeruginosa small RNA regulates chronic and acute infection. Nature 618, 358–364 (2023).
Mugunthan, S. et al. RNA is a key component of extracellular DNA networks in Pseudomonas aeruginosa biofilms. Nat. Commun. 14, 7772 (2023).
Aytar Çelik, P. et al. Bacterial membrane vesicle functions, laboratory methods, and applications. Biotechnol. Adv. 54, 107869 (2022).
Flemming, H. C. et al. The biofilm matrix: multitasking in a shared space. Nat. Rev. Microbiol. 21, 70–86 (2023).
Pérez-Cruz, C., Briansó, F., Sonnleitner, E., Bläsi, U. & Mercadé, E. RNA release via membrane vesicles in Pseudomonas aeruginosa PAO1 is associated with the growth phase. Environ. Microbiol. 23, 5030–5041 (2021).
Lin, J. et al. A Pseudomonas T6SS effector recruits PQS-containing outer membrane vesicles for iron acquisition. Nat. Commun. 8, 14888 (2017).
Kulp, A. & Kuehn, M. J. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu. Rev. Microbiol. 64, 163–184 (2010).
Zhao, Z. et al. Regulation of the formation and structure of biofilms by quorum sensing signal molecules packaged in outer membrane vesicles. Sci. Total Environ. 806, 151403 (2022).
Li, Y. et al. Self-organized canals enable long-range directed material transport in bacterial communities. Elife 11. https://doi.org/10.7554/eLife.79780 (2022).
Koeppen, K. et al. A novel mechanism of host-pathogen interaction through sRNA in bacterial outer membrane vesicles. PLoS Pathog. 12, e1005672 (2016).
Moriano-Gutierrez, S. et al. The noncoding small RNA SsrA is released by Vibrio fischeri and modulates critical host responses. PLoS Biol. 18, e3000934 (2020).
Sahr, T. et al. Translocated Legionella pneumophila small RNAs mimic eukaryotic microRNAs targeting the host immune response. Nat. Commun. 13, 762 (2022).
Acknowledgements
The authors would like to thank Prof. Chris Soon Heng Tan from the Department of Chemistry, Southern University of Science and Technology for help with the LC-MS/MS analysis and Prof. Lei Dai from Shenzhen Institutes of Advanced Technology for discussing the possibility of math models. TEM data were obtained using equipment maintained by Southern University of Science and Technology Core Research Facilities. This work was supported by the National Key Research and Development Program of China (2022YFC2304700); the National Natural Science Foundation of China (32300068, 32270196, 91951204, 32200155, 32200053, and 32300060); Guangdong Basic and Applied Basic Research Foundation (2019A1515110640 and 2020A1515010316); Shenzhen Science and Technology Program (KQTD20200909113758004); and Guangdong Pearl River Talent Plan.
Author information
Authors and Affiliations
Contributions
T.J. and L.Y. designed the study. T.J., X.B., M.L., C.Z., S.L., and A.R. performed experiments. T.Z., Y.Z., Y.L., X. L., B.L., Y.D., and G.L. analyzed the results. T.J. and L.Y. drafted and revised the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jia, T., Bi, X., Li, M. et al. Hfq-binding small RNA PqsS regulates Pseudomonas aeruginosa pqs quorum sensing system and virulence. npj Biofilms Microbiomes 10, 82 (2024). https://doi.org/10.1038/s41522-024-00550-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41522-024-00550-4
This article is cited by
-
A Pseudomonas aeruginosa quorum-sensing metabolite manipulates macrophage ferroptosis through a methylation pathway
Nature Communications (2025)
