
Abstract
During the coronavirus disease 2019 (COVID-19) pandemic, the exploration of microecology has been essential for elucidating the intricacies of infection mechanisms and the recovery of afflicted individuals. To decipher the interplay of microorganisms between the intestinal and respiratory tracts, we collected sputum and throat swabs and feces from COVID-19 patients and explored the mutual migration among intestinal and respiratory microorganisms. Using next-generation sequencing (NGS) technology, we investigated intestinal and respiratory microorganism intermigration in two patients with severe COVID-19 during their hospitalization. Notably, we observed an expedited recovery of microecological equilibrium in one patient harboring Mycobacterium avium. Comparative analyses between 32 healthy controls and 110 COVID-19 patients with different disease severities revealed alterations in predominant microorganisms inhabiting the respiratory and intestinal tracts of COVID-19 patients. Among the alterations, intestinal Bacteroides vulgatus (BV) was identified as a noteworthy microorganism that exhibited marked enrichment in patients with severe COVID-19. BV, when highly abundant, may inhibit the transitional growth of Escherichia coli/Enterococcus, indirectly prevent the overgrowth of salivary streptococci, and maintain lung/intestinal microecology stability. In summary, this study elucidates the bidirectional microbial intermigration between the intestinal and respiratory tracts in COVID-19 patients. These findings are expected to provide new ideas for the treatment and management of COVID-19, underscoring the essential role of microecology in infectious diseases. Nevertheless, a systematic study of the roles of BV in recovery from infection is required to gain a deeper understanding of the mechanisms of microbial migration.
Similar content being viewed by others


Introduction
In the third year of the ongoing coronavirus disease 2019 (COVID-19) pandemic, the enduring presence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) necessitates continual vigilance. COVID-19 manifests most commonly with respiratory and digestive symptoms, and in severe cases, respiratory failure can lead to fatality. Empirical data indicate that the incidence of severe and critical COVID-19 ranges from 5% to 20% across nations1,2,3. While the proportion of severe cases is relatively modest, the mortality rate among patients necessitating mechanical ventilation, which is indicative of severe or critical disease, escalates significantly to 88.1%, notably higher than the 11.7% mortality rate observed in nonsevere cases2. Consequently, the intricacies surrounding severe and critical COVID-19, along with their contributing factors and mechanisms, demand heightened attention and scrutiny from healthcare practitioners.
Previous studies investigated the impact of SARS-CoV-2 infection on respiratory microbiomes, emphasizing microbiome diversity and metabolic functions in COVID-19. Analysis of transcriptional metagenomic data reveals dysbiosis in bacterial, archaeal, and viral compositions, with COVID-19 samples exhibiting higher proportions of opportunistic species. Additionally, SARS-CoV-2 infection significantly alters the nasopharyngeal mycobiome, increasing fungal populations and diversity, particularly opportunistic pathogens. These findings suggest a potential for microbiome-based diagnostics and therapeutics for COVID-19 and respiratory diseases, highlighting the importance of understanding microbiome dynamics in disease progression and management.
Several factors have been shown to influence the severity of COVID-19, among which one notable consideration is the dysregulation of the microbiome within the gut and/or lungs, given that the susceptibility of these primary target organs facilitates SARS-CoV-2 invasion4,5. Early in the pandemic, a report by Siew C. Ng et al.6 highlighted a correlation between fecal microbiome changes, fecal SARS-CoV-2 levels, and the severity of COVID-19. Subsequent investigations reinforced this connection, emphasizing the impact of the gut microbiome on the severity of COVID-197,8. Additionally, our previous research delineated associations between alterations in the respiratory microbiome and the severity of COVID-199. Notably, SARS-CoV-2-induced disruptions in the gut microbiome have been found to perturb immune cell balance and recruitment within the lungs via the “gut-lung axis,” thereby contributing to respiratory tract infections. Concurrently, studies proposed the oral cavity as a potential reservoir for microorganisms, potentially instigating lung coinfections and contributing to gut dysbiosis in COVID-19 patients5,10. However, critically, most studies conducted have been cross-sectional and potentially impacted by interindividual demographic disparities5. Furthermore, studies focusing on the longitudinal assessment of coordinated variations within the lung and gut microbiomes among long-term-hospitalized COVID-19 patients are rare.
Hence, during the pandemic period, we actively enrolled COVID-19 patients who required extended hospitalization and meticulously collected comprehensive clinical data and specimens, encompassing sputum and fecal matter. Subsequently, we conducted a rigorous investigation employing metagenomic next-generation sequencing (NGS) techniques to analyze and understand the synchronized fluctuations within the lung and gut microbiomes of these long-term-hospitalized individuals.
Methods and materials
Subject recruitment
This study was conducted in strict adherence to the Declaration of Helsinki, with all participating patients providing explicit written consent. Following the guidelines outlined by the Chinese Center for Disease Control and Prevention and the World Health Organization (WHO)11,12, our recruitment efforts focused on confirmed COVID-19 patients admitted to several esteemed medical institutions, specifically the First Affiliated Hospital of Guangzhou Medical University, Guangdong Second Provincial General Hospital, and Guangzhou Eighth People’s Hospital in Guangzhou, China, during the period from January to May 2020. Consistent with our prior research protocols13, all confirmed COVID-19 patients underwent quantitative reverse transcription PCR (RT-qPCR) analysis of nasopharyngeal swabs, confirming the presence of SARS-CoV-2 according to hospital procedures. Healthy individuals (controls) were recruited through open recruitment, ensuring that they exhibited no clinical signs of infection, such as fever or cough, and that laboratory-confirmed negative results for SARS-CoV-2 were obtained via RT-qPCR.
Moreover, our classification of confirmed COVID-19 patients involved discerning five distinct subgroups based on diverse clinical presentations, guided by criteria derived from the Chinese Government Diagnosis and Treatment Guideline (trial eighth version)11 and corroborative insights from additional research14. These subgroups were as follows: (1) asymptomatic individuals who tested positive for SARS-CoV-2 but were devoid of any clinical manifestations or pneumonia symptoms; (2) those with mild clinical features but lacking signs of pneumonia; (3) individuals classified as having common symptoms characterized by fever, respiratory tract manifestations, and detectable imaging features indicative of pneumonia; (4) patients with severe disease presenting with respiratory distress, a respiratory rate ≥30 times/min, resting-state mean oxygen saturation ≤93%, or arterial blood oxygen partial pressure (PaO2)/oxygen concentration (FiO2) ≤ 300 mmHg (1 mmHg = 0.133 kPa); and (5) critically ill patients exhibiting respiratory failure necessitating mechanical ventilation, experiencing shock, or requiring intensive care unit (ICU) admission.
Concurrent with patient recruitment, a cohort of healthy volunteers devoid of any pathogenic infection was enrolled. Within this study framework, the patient cohort was divided into three distinct subgroups, namely, asymptomatic patients (Group 1), patients with nonsevere manifestations comprising mild and common illness presentations (Group 2), and patients with severe and critical illness presentations (Group 3), along with a control group for comparative analysis.
Specimen collection
Clinical specimens, including respiratory tract specimens (throat swab (TS) and sputum), gastrointestinal tract specimens (stool), and serum, were obtained following the WHO and Chinese guidelines11,12. TS specimens were collected with synthetic fiber swabs. Each swab was then placed into a separate sterile tube containing 3 mL of viral transport medium. Sputum and/or stool specimens were collected in sterile specimen containers. The serum was collected in a separator tube and then centrifuged. The specimens were stored between 2 °C and 8 °C until they were ready for shipment to the standardized and accredited laboratory of the Guangdong Centers for Disease Control and Prevention and the standard laboratory of our hospital. Specimens from the COVID-19 patients were collected at different times. Briefly, for severe and/or critical cases, respiratory and gastrointestinal tract specimens were collected every 3–4 days when the virus test was positive; then, collection was performed approximately once weekly until the virus test was negative and up to two weeks after discharge. Serum specimens were collected once per week. However, for asymptomatic, mild, and common cases, the collection was performed only once during hospitalization due to the short length of hospitalization. The specimens from healthy volunteers were collected only once (Fig. 1).

The plot presents specific details concerning the research cohort, encompassing the number of donors within each cohort and the number of specimens categorized by the three specimen types. The x axis on the right denotes temporal distance, representing the duration of patient admission to the hospital, measured in weeks. Each row corresponds to an individual donor, and the plotted dots symbolize specimens collected at various time points. The term ‘initial’ designates the first specimen obtained within three days before hospital admission. The numerical values in brackets on the left denote the number of initial specimens. Additionally, the shaded gray interval depicted in the right panel signifies the designated time span for tallying ‘initial’ specimens.
DNA extraction and sequencing
DNA was extracted and purified from 0.1 g of homogenized stool sample using a commercial QIAamp Fast DNA Stool Mini Kit (Qiagen, 51604, USA). Swab and sputum samples were processed with a TIANamp Micro DNA Kit (TIANGEN, DP316, China). DNA, the total amount of which was >100 ng for each sample, was used to generate 350 bp libraries according to the Illumina standard operating procedure (SOP). After removing samples that were abnormal due to sampling or experimental reasons, each sample was used to generate an average of 6.49 ± 1.54 Gb of raw paired-end 150 bp reads using the NovaSeq sequencing system.
Compositional and functional analysis
Clean reads were obtained after the following preprocessing steps were performed on the raw reads. Using the NGS toolkit (v2.3.3), low-quality and adapter-contaminated sequences were removed. Then, Bowtie2 (v2.3.1) was used to remove contaminating human reads based on the reference genome GRCh38.p13.
Microbiome profiling was conducted by employing MetaPhlAn3 (v3.0.4), which facilitated the mapping of clean reads to clade-specific markers. Simultaneously, metabolic pathways were annotated using Humann3 (v3.0.0.alpha.3)15, leveraging databases such as MetaCyc and KEGG. Considering that MetaPhlAn3 primarily focuses on the classification of reads aligning with the marked database and excludes low-abundance fungi, we employed Kraken (v2.1.1) for fungal species diversity estimation16, supplemented by confirmation through ITS2 amplicon sequencing. This comprehensive approach ensured a more nuanced assessment of the fungal diversity within the microbiome.
Statistical and bioinformatic analysis
The primary statistical analyses and mapping procedures were executed within the R programming environment (v3.6.2), harnessing essential packages such as maaslin2, stats, ggplot2, vegan, and ade4 to ensure robust data interpretation. Complementary to these analyses, JMP13 and STAMP software were used to bolster the statistical assessment of the dataset, enhancing the depth of analysis. For the liquid chromatography-mass spectrometry raw data analysis, Profinder 10.0 (Agilent, USA) was utilized. The data were processed via peak identification, alignment, filtering, and normalization procedures to then construct a comprehensive three-dimensional dataset comprising sample information, peak intensities, peak retention times, and mass-to-charge ratios (m/z). This multifaceted approach facilitated a comprehensive evaluation of the data landscape.
Results
COVID-19 patient enrollment and sample collection
In this study, we collected clinical samples from 98 patients with COVID-19 confirmed by a positive SARS-CoV-2 RT-qPCR test, including 12 with critical illness, 2 with severe illness, 5 with common illness, 47 with mild illness, and 32 who were asymptomatic; 34 healthy individuals were used as controls. The patients were stratified into distinct categories, asymptomatic cases (Group 1), nonsevere cases encompassing mild and common illness (Group 2), severe cases encompassing severe and critical illness (Group 3), and a control group. Notably, the duration of viral shedding significantly differed among these groups, with Group 3 exhibiting a markedly longer duration than Group 2 or Group 1 (53.6 ± 23.7 days vs. 11.9 ± 5.7 days vs. 6.6 ± 3.8 days, respectively). Clinical symptom analysis revealed fever as the predominant symptom in COVID-19 patients, while shortness of breath was the predominant symptom in Group 3, in contrast with the rarity of shortness of breath in Group 1. In addition, primarily Group 3 patients experienced lymphocytopenia and thrombocytopenia. Antibiotic treatment was extensively administered in Group 3 (85.7%), which was significantly less prevalent in the other groups and notably rare in Group 1 (6.3%).
Following rigorous quality control procedures, a total of 351 metagenomic data samples were deemed suitable for further analysis. A total of 188 sputum samples were chosen for respiratory microbiome analysis, along with 53 TS samples and 110 stool samples to capture the characteristics of the gut microbiome. The samples collected across different tissues would facilitate the investigations on microbial migration between the respiratory and digestive tracts.
Alterations in sputum and fecal microecology in patients with severe COVID-19 during hospitalization
We employed metagenomic data to compute the microbial diversity indices across all samples and subsequently conducted correlation analyses between these indices and clinical data. These findings are consistent with those of previous studies. For example, in patients with severe COVID-19 (Group 3), a higher fungal Shannon diversity was observed within sputum samples. Additionally, the viral load, as indicated by the SputumCt, exhibited a significant relationship with the Shannon index. Notably, antibiotic administration in patients with severe COVID-19 alleviated the risk of decreasing Shannon diversity (Supplementary Fig. 1).
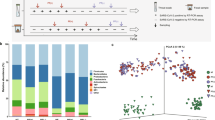
We examined the microecological discriminations among samples using the multidimensional scaling (MDS) method, and samples positioned closer together had more similar microbial structures (Fig. 2A). The control group sputum and TS samples, both representing respiratory tract specimens, exhibited similar MDS coordinates. Moreover, a pronounced distance boundary existed between the respiratory tract and stool specimens (denoted by the gray region in Fig. 2). Transitioning from Group 1 to Group 3, an increasing number of samples was manifest in the boundary region, with this effect being particularly prominent in Group 3. Notably, in terms of temporal dynamics (weeks), a substantial number of samples showed an increase during hospitalization, primarily originating from two critically ill patients who did not receive antibiotic treatment: C102 and C107. Figure 2B–D illustrates the evolving microecosystems observed in the sputum and stool samples of patients C102 and C107. In the case of patient C102, persistent Burkholderia multivorans infection spanned the hospitalization period from 200223 to 200428, marked by distinct transitions in the predominant infection sites between 200226–200306 and 200407–200414. Conversely, patient C107 exhibited concurrent infections involving Enterococcus faecium and Staphylococcus epidermidis throughout their hospital stay, with a single transition in the infection site occurring between 200306 and 200313. These cases displayed similarities while also presenting nuanced differences. Notably, the absence of antibiotic administration during hospitalization suggested that the observed shifts in microbial composition were primarily attributed to the dynamics of environmental competition.

A Multidimensional scaling (MDS) plots illustrating the dynamic changes across four distinct cohorts. B–D Microecological shifts observed in two Group 3 donor patients, C102 and C107, during their hospitalization. The plots present the predominant microorganisms, each accounting for >5% of the relative abundance in at least one sample. The column length corresponds to the relative abundance of these top microorganisms, with the upper half representing sputum samples and the lower half corresponding to stool samples. The X axis denotes the sampling time points. C Statistical outcomes concerning the top DS identified in the ‘initial’ specimen. E Model assessment featuring the abundance of Rothia mucilaginosa. The left histogram is organized by abundance levels, while the right histogram depicts the ROC curve. The triangular data points signify TS specimens, while the circular points represent sputum specimens. Group 1/Group 2 and other group distinctions were defined based on the linear value of abundance. The AUC values are also shown. F Abundance variations in the Bacteroides genus in the control and Group 3 stool cohorts. Each plotted point corresponds to a specific specimen, with the red dotted line indicating the reference average of Bacteroides vulgatus abundance in the control group. The right line portrays the fitted changes in the average abundance of each microorganism.
Across multiple samples, we identified microorganisms with high abundances in patients but seldom in healthy individuals, and we defined these microorganisms as indicative of severe infection. Coinciding with microbial migration (indicated by the red and blue arrows in the figure), the patients’ microbial ecosystems conformed to this situation. In patient C102, two transition events occurred. At the second time point, the predominant species (DSs) detected in sputum and stool were Herpesvirus 4 and Escherichia coli, which are conventionally associated with pathogenic infections. In contrast, patient C107 experienced a single transition event, with the DSs at the final time point comprising Prevotella oralis and Bacteroides, which are typically considered environmental commensals. The microbial migration between the respiratory and digestive tracts is probably associated with the clinical severity of COVID-19 patients.
Changes in the DSs in sputum and fecal microecology
A normal and stable microecological environment is a community structure dominated by a few colonized microorganisms. In the gut, the structural pattern of microecology is known as the enterotype17, and a similar dominant community structure is observed in oral microecology18. Multiple secondary infections recur in patients with COVID-19, leading to changes in the DSs in the microecology community. With increasing disease severity, rare microorganisms were more prevalent and exhibited a very high abundance (Fig. 2) compared to their abundance in healthy controls. For both patients C102 and C107, changes in the dominant microorganism species not only occurred multiple times within the same sample type but also occurred between the two types of samples, sputum and stool.
DSs were defined as the most abundant microorganisms in the microecology community, and the number and frequency of DSs in the different groups were statistically analyzed. Considering the multiple sampling steps taken for some patients with severe COVID-19, the first specimen of the same type taken within three days before admission and was marked as the initial time point (gray range in Fig. 1). Using only specimens sampled at the initial time point, the number of donors in each subgroup was counted, and the type of DS observed, the ratio of DS to donor, the weighted information entropy, and the frequency of detection of each DS were calculated (Fig. 2C). If a DS was observed fewer than 3 times in any subgroup sample, it was considered an auxiliary DS, and the DS with the highest frequency was considered the top DS (Fig. 2). According to the detection times of the top DS, the contingency table for the two groups was calculated on the basis of the Fisher test. Considering the small number of samples in some groups, all results with P values <0.1 were included (Table 1).
Neisseria is considered a representative species of oral samples, and was detected as the DS in at least half of the sputum specimens from the control group. The detection frequency of Neisseria unclassified as the DS in the sputum from the four groups was 30.8%, 20.4%, 8.5%, and 12.5%, and the detection frequency for the control group was significantly greater than that for Group 2, with contingency table values of 8, 18, 4, and 43. According to the comparison results, Rothia mucilaginosa was the most frequently occurring DS species and was present in mainly the respiratory tract specimens (sputum and TS) of Group 1 and Group 2, with detection frequencies of 3.8%, 32.1%, 23.4%, and 12.5% and 0%, 44.4%, 58.3%, and 0%, respectively.
Bacteroides and Prevotella are considered among the dominant colonizing bacteria in the healthy human intestinal tract. In the feces of Group 3, Prevotella was generally absent, and Bacteroides vulgatus was the DSs with the fastest recovery and highest abundance compared with other Bacteroides species (Table 2). The detection frequencies in the four groups were 7.7%, 0%, 22.2%, and 33.3%, and the contingency table values were 2, 24, 3, and 6. These findings indicated that the microbial migration was caused by the changes in the dominant bacteria in the respiratory and digestive tracts.
Associations between respiratory and intestinal microecology
To further understand the changes in the DSs and their role in the transformation of the respiratory and intestinal tracts, 74 individuals with both sputum and feces specimens at the same sampling time points were selected, and their specimens were applied for association analysis of the respiratory tract and digestive tract microecology. Among the 74 participants, 21 were healthy controls, and 53 were in Group 3. Microorganisms from sputum were marked as ““p:”” and those from stool were marked as ““t:”” Welch” t test revealed 17 significantly differentially abundant microorganisms between Group 3 and the control group (Fig. 3A). Among these differentially abundant microorganisms, 7 were sputum microorganisms, and 10 were stool microorganisms. The microorganisms st: Bacteroides vulgatus and st: Escherichia coli exhibited the most significant increase in average abundance within Group 3. Spearman correlation coefficients were calculated between the most abundant microorganisms (abundances greater than 1%) in the control group and Group 3, and their significant correlations are depicted in a network diagram (Fig. 3C). In the control group, many positive and negative relationships were roughly balanced, forming a complex network. In contrast, in Group 3, the number of red lines representing negative relationships was low, mainly centered around st: Bacteroides vulgatus, which suppressed st: Escherichia coli, st: Enterococcus faecium, and st: Klebsiella pneumoniae abundance in the intestine, and negatively regulated sp: Streptococcus salivarius and sp: Streptococcus parasanguinis abundance in the sputum.

To designate the microorganisms in the sputum specimens and stool specimens, sp and st were used as prefixes, respectively. A Comparative difference in statistics portraying the associated cohort. The left histogram displays the mean percentage differences between the control group and Group 3, while the right histogram illustrates the difference in the mean percentage. The presented values denote p values calculated using t tests. B An association clustering heatmap representing the relationships of the top microorganisms. This heatmap shows the abundance across all samples, displaying Spearman correlation coefficients for the top microorganisms with a degree >1%. In the heatmap, red indicates a negative correlation, blue signifies a positive correlation, and asterisks denote significance levels lower than 0.05. C Correlation network diagram showing the associations of the top microorganisms. The green and red nodes denote microorganisms significantly enriched in the control and Group 3 groups, respectively. Node size correlates with relative abundance. Spearman correlation coefficients between the top microorganisms were calculated within the cohort. Positive and negative relationships with P values <0.05 are indicated by gray and red connecting lines, respectively.
Changes in the abundances of sp: Streptococcus salivarius, st: Escherichia coli, and st: Bacteroides vulgatus were compared between the control group and Group 3 (Fig. 4). The reference abundances (90% quantile) of these three microorganisms in the normal control group were 3.8%, 5.7%, and 16.9%, respectively. In this study, samples with abundances above this value were temporarily labeled as being in an overgrowth state, while samples below this value were considered in a normal state. In Group 3, there was a positive correlation between st: Bacteroides vulgatus and st: Escherichia coli abundance when both microorganisms were in a normal state. However, as their abundance continued to increase to the overgrowth state, the relationship between them became negative, indicating a suppressive effect. Among the six patients in Group 3 who had more than five samples, the abundance of Bacteroides vulgatus and several other infectious microorganisms was negatively correlated. The alterations of the correlations between Bacteroides vulgatus in the respiratory tract and Escherichia coli in the digestive tract would reflect the severity of microbial migration and the clinical severity of the COVID-19 patients.

A, B Depiction of the relationships between the abundances of Ec and Sa and among Bv, Ec, and Sa, respectively. The dashed line represents the 90% abundance threshold of each microorganism in the control group. Notably, the statistical sample excludes 0 values. The x axis illustrates robust linear fitting across specimens with varying abundance intervals. The blue and red notations represent the normal and overgrowth states, respectively. C Relationships between changes in the abundances of Bv and three other pathogenic bacteria observed across the six patients in Group 3.
Changes of fungus in sputum and fecal microecology
After the fungi annotation, we detected the fungal diversity among the groups (Fig. 5). At the genus level, Saccharomyces was enriched in the patients underlying antibiotic treatment, while Malassezia and Candida were enriched in the patients without antibiotic administration (Fig. 6A). With the LEFSE analysis (Fig. 6B, C), Saccharomyces was significantly enriched in the patients with antibiotic treatment. From previous reports, we learned that the bacteria were diminished after antibiotic application, and Saccharomyces would overgrow in such an environment without any inhibition.

We applied t test to compare the fungal diversity between the different groups in sputum and stool specimens with the Simpson index. *P < 0.05; **P < 0.01; ***P < 0.001.

A Heatmap of fungal abundances in samples applied or not applied antibiotics. The red and blue bars on the right side represented the sample applied or not applied antibiotics, respectively. The fungal abundances of 0%, 10%, and 100% were marked with blue, green, and red, respectively. B, C plots exhibited the LEFSE analysis results on fungi between the used and unused antibiotics samples.
Discussion
Rothia mucilaginosa, which usually colonizes the mouth19, is sensitive to diet, and enrichment occurs in a few mild diseases20,21,22. In a recent report, elevated Rothia mucilaginosa levels in both the oral and intestinal tracts were hypothesized to be associated with the SARS-CoV-2 viral load, and elevated Rothia mucilaginosa levels in fecal samples seemed to be further associated with disease severity17. In our study, we did not observe proliferation of this bacterium in stool samples. Rothia mucilaginosa, serving as a top DS in the respiratory microbiome, exhibited notable potential as a marker in both the sputum and TS samples, showing sensitivity to mild and moderate disease status. A simple linear model established using Rothia mucilaginosa abundance effectively discriminated whether the sample was from a patient in a relatively mild disease state (Group 1 and Group 2). The area under the curve (AUC) values for sputum and TS reached 0.747 and 0.8899, respectively (Fig. 3E). Determining the levels of Rothia mucilaginosa, an environmentally sensitive microorganism, in the microbiome could assist clinicians in assessing the early progression of this disease, suggesting that this microbial marker is highly promising.
With respect to the Shannon index, we assessed the diversity of DSs detected in different types of samples using the ratio of DSs to host and weighted entropy. From the control group to Group 3, the diversity of DSs showed an upward trend, with the most significant inflection point occurring in Group 1 for stool samples and Group 3 for sputum. Although the TS samples had fewer data points, their trends were consistent with those of the stool samples. According to the statistical analysis of the DS, TSs exhibited earlier responses than sputum samples and were more similar to stool samples; moreover, we were able to detect DS changes in less severe donor groups. The throat may thus be a better sample site for respiratory microbiome detection studies.
In some models, Bacteroides vulgatus has been demonstrated to robustly coexist with other members of the microbial community, enabling the establishment of a foothold even in environments where other microorganisms dominantly occupy ecological niches3. In patient C107, Bacteroides vulgatus colonized the stool at the time point 200306, and subsequently, multiple Bacteroides species continued to increase in abundance in the stool and remained stable throughout all subsequent observation periods. The infectious bacteria Enterococcus faecium and Staphylococcus epidermidis migrated from the stool to the sputum. At the second time point, at 200320, the oral cavity-colonizing bacterium Prevotella oralis appeared in the sputum and persisted at all subsequent observation points. During this period, a Granulicatella adiacens infection occurred, but no further sputum-stool microbial migration events were observed. Correlation analysis revealed that under severe disease conditions, Bacteroides vulgatus enrichment was accompanied by a decreased abundance of pathogenic bacteria (positive correlation), assuming a dominant position in the ecological niches of most overgrowth bacteria when it reached high abundance, thus promoting the stabilization of the microbial community structure, and further preventing the recurrent migration of respiratory infection-causing microorganisms.
As one of the colonizing microbiome constituents in healthy individuals, Bacteroides vulgatus tends to proliferate under disease conditions, such as in the presence of high blood sugar levels, insulin deficiency, depression, and polycystic ovary syndrome. On the other hand, Bacteroides vulgatus has shown some clinical application value; for instance, Chul-Su Yang et al. proposed that Bacteroides vulgatus is a novel therapeutic candidate for colitis. Huajing Teng and colleagues reported that Bacteroides vulgatus gavage protects cancer cells from 5-fluorouracil treatment or irradiation18.
In the present study, B. vulgatus proliferated in Group 3 patients with severe disease and played a central role in the microbiome correlation network. During the treatment of six patients in Group 3, the proliferation of st: Escherichia coli and st: Enterococcus faecium was suppressed. As the earliest restored colonizing bacteria, B. vulgatus might mediate the stability of the gut microbiome, helping to prevent overgrowth bacteria from migrating from the lung to the gut and indirectly supporting the stability of the respiratory microbiome. Employing therapeutic approaches such as administration of microbial capsules or performance of fecal microbiome transplantation surgeries to promote the stabilizing effect of B. vulgatus on the microbiome in advance could contribute to the recovery of critically ill patients.
In COVID-19 cases, the translocation of B. vulgatus and E. coli from gastrointestinal to respiratory tracts can occur during prolonged hospitalization, impacting the microbial balance. B. vulgatus, a dominant gut bacterium, may overgrow and lead to respiratory dysbiosis. In addition, the replacement of DSs by pathogenic bacteria like E. coli signifies infection. These translocations highlight shifts in microbial states, potentially complicating clinical treatment. The impact is illustrated in patient data, emphasizing the need to monitor and address such changes to mitigate complications and optimize treatment outcomes in long-term hospitalized COVID-19 patients.
Bacterial and fungal infections significantly complicate COVID-19, particularly in severe cases. Immune dysregulation from SARS-CoV-2, prolonged ICUs stay with invasive procedures, and immunosuppressive therapies increase susceptibility to these infections. Common bacterial pathogens, including S. aureus, S. pneumoniae, and E. coli, can lead to pneumonia, sepsis, and bloodstream infections, occurring in ~15% of ICU COVID-19 patients. Fungal infections, though less occurred, are also significant in severe SARS-COV-2 infection cases. Invasive pulmonary aspergillosis caused by Aspergillus and Mucorales are particularly concerning. The incidence of aspergillosis ranges from 19% to 33% in ICU COVID-19 settings, with infections causing severe respiratory complications. Factors contributing to secondary infections include immune dysregulation from COVID-19, prolonged hospitalization, and the use of corticosteroids and other immunosuppressive treatments. Addressing these complications is crucial to improving outcomes in COVID-19 patients.
Lucie Bernard-Raichon et al. analyzed blood culture results testing for secondary microbial bloodstream infections and discovered that bacteria may translocate from the gut into the systemic circulation of COVID-19 patients1. In the case of pathogens infection and tissue barrier damage, microorganisms would probably pass through the blood system and transfer from the respiratory tract to the intestinal tract. Similar microbial translocation phenomena may also occur in patients with hypertension, chronic diseases, and cancer. However, as an important environmental factor, ventilator-associated pneumonia (VAP) cannot be ignored during the clinical treatment of SARS-COV-2 infection. Ventilator support is an important clinical strategy for COVID-19 patients, and VAP is a common complication of mechanical ventilation with an incidence ranging from 9 to 70% and averaging ~25%6. Some evidence exhibited that the frequency of VAP was closely related to the gastrointestinal microbial load7,8, and appropriate antibiotic treatment could reduce the infection rate and mortality rate of VAP. These discoveries also corresponded to the fact that the patients with microbial translocations in our study were treated without antibiotics.
This study has several limitations. There were differences in the management and treatment of different patients. Due to the heterogeneity of the patients, illness severity, ICU admission, underlying diseases, etc., the choice of treatment and management were affected. We could reduce the difference only through artificial grouping and continuous testing. Determining how much patient heterogeneity affects experimental results is a common challenge in observational studies. However, due to the strict artificial classification of patients, we obtained a partial picture of the natural progression of microecology with this disease. Although some of the results contradict those of existing studies, the data are reliable.
Since the COVID-19 pandemic began, the pathophysiological process of SARS-CoV-2 infection has been thoroughly studied in terms of respiratory or intestinal microecology6,23. The intestinal microbial network of COVID-19 patients is significantly weakened and becomes bare, and the diversity of the intestinal microbial community decreases. In terms of the severity and presentation of the disease, the gut microbiome of COVID-19 patients is characterized by the enrichment of opportunistic bacteria, fungi, and eukaryotic viruses. In this study, considering the role of antibiotics, we found that the changes in the Shannon index and fungal Shannon index were different among the groups. The fungal Shannon index showed an increasing trend in patients who used antibiotics but a decreasing trend in COVID-19 patients who did not use antibiotics. Antibiotics may kill bacteria, creating a fungus-friendly environment, which carries a risk and was noted in earlier reports as increasing susceptibility to fungal infection24.
Conclusion
This study explored the changes in respiratory and gut microecology in COVID-19 patients from a new perspective. This was the first study to describe the interaction between respiratory and intestinal dominant organisms in severely and critically ill COVID-19 patients and revealed new microecological markers, Rothia mucilaginosa and Bacteroides vulgatus, for asymptomatic and severe and critical cases, respectively. This paper attempts to explain that the mechanism of microorganism suitability as a marker may be the result of microbial competition from a metabolic and microecological perspective. The importance of the stability of intestinal microecology for respiratory microecology is discussed. These findings reveal the microecological changes in the respiratory tract and gut of patients with COVID-19 and will provide new ideas regarding the pathophysiology of this disease, potential microbial markers, and treatment strategies for COVID-19.
Data availability
The raw sequence data for the respiratory and gut microbiomes in this study were deposited in the Genome Sequence Archive in the Beijing Institute of Genomics (BIG) Data Center, Chinese Academy of Sciences, under BioProject PRJCA009091 with accession ID CRA006729, which is publicly accessible at https://bigd.big.ac.cn/gsa. All the other data are available from the authors upon reasonable request.
References
Wu, Z. & McGoogan, J. M. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China: summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA 323, 1239–1242 (2020).
Richardson, S. et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA 323, 2052–2059 (2020).
Livingston, E. & Bucher, K. Coronavirus disease 2019 (COVID-19) in Italy. JAMA 323, 1335 (2020).
Saleh, J., Peyssonnaux, C., Singh, K. K. & Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 54, 1–7 (2020).
Zhang, F. et al. Gut microbiota in COVID-19: key microbial changes, potential mechanisms and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 20, 323–337 (2023).
Zuo, T. et al. Alterations in gut microbiota of patients with COVID-19 during time of hospitalization. Gastroenterology 159, 944–955.e948 (2020).
Patel, P. & Roper, J. Gut microbiome composition is associated with COVID-19 disease severity. Gastroenterology 161, 722–724 (2021).
Yeoh, Y. K. et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 70, 698–706 (2021).
Li, Z. et al. Alteration of the respiratory microbiome in COVID-19 patients with different severities. J. Genet. Genomics 49, 258–261 (2022).
Parrot, T. et al. MAIT cell activation and dynamics associated with COVID-19 disease severity. Sci. Immunol. 5, eabe1670 (2020).
Prevention, C. C. f. D. C. A. The guideline of diagnosis and treatment of COVID-19 (the eighth edition). (2020).
World Health Organization. Clinical management of severe acute respiratory infection when novel coronavirus (nCoV) infection is suspected. (2020).
Li, Z. et al. Longitudinal virological changes and underlying pathogenesis in hospitalized COVID-19 patients in Guangzhou, China. Sci. China Life Sci. https://doi.org/10.1007/s11427-020-1921-5 (2021).
Li, Y. et al. Multi-platform omics analysis reveals molecular signature for COVID-19 pathogenesis, prognosis and drug target discovery. Signal Transduct. Target. Ther. 6, 155 (2021).
Beghini, F. et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 10, e65088 (2021).
Wood, D. E. & Salzberg, S. L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, R46 (2014).
Wu, Y. et al. Altered oral and gut microbiota and its association with SARS-CoV-2 viral load in COVID-19 patients during hospitalization. NPJ Biofilms Microbiomes 7, 61 (2021).
Liu, L. et al. Bacteroides vulgatus attenuates experimental mice colitis through modulating gut microbiota and immune responses. Front. Immunol. 13, 1036196 (2022).
Utter, D. R., Borisy, G. G., Eren, A. M., Cavanaugh, C. M. & Mark Welch, J. L. Metapangenomics of the oral microbiome provides insights into habitat adaptation and cultivar diversity. Genome Biol. 21, 293 (2020).
Amer, A., Galvin, S., Healy, C. M. & Moran, G. P. The microbiome of potentially malignant oral leukoplakia exhibits enrichment for Fusobacterium, Leptotrichia, Campylobacter, and Rothia Species. Front. Microbiol. 8, 2391 (2017).
Hansen, T. H. et al. Impact of a vegan diet on the human salivary microbiota. Sci. Rep. 8, 5847 (2018).
Sato, N. et al. Metagenomic analysis of bacterial species in tongue microbiome of current and never smokers. NPJ Biofilms Microbiomes 6, 11 (2020).
Ren, L. et al. Dynamics of the upper respiratory tract microbiota and its association with mortality in COVID-19. Am. J. Respir. Crit. Care Med. 204, 1379–1390 (2021).
Zuo, T., Wu, X., Wen, W. & Lan, P. Gut microbiome alterations in COVID-19. Genomics Proteom. Bioinform. https://doi.org/10.1016/j.gpb.2021.09.004 (2021).
Acknowledgements
We would like to thank the patients, along with the nurses and clinical staff who provided patient care, the staff at the hospital respiratory medicine departments, the staff at the hospital clinical laboratories, and the technical staff of the State Key Laboratory of Respiratory Disease for their excellent assistance and the staff at Guangdong Centers for Disease Control for aiding the diagnosis of COVID-19. Furthermore, we would like to thank the AJE team for polishing the English language of this manuscript. This work was funded by the Guangzhou Institute of Respiratory Health Open Project (funds provided by the China Evergrande Group—project No. 2020GIRHHMS14), the Guangzhou Science and Technology Planning Project and Zhongnanshan Medical Foundation of Guangdong Province (ZNSA-2020003), and the Guangzhou Institute of Respiratory Health.
Author information
Authors and Affiliations
Contributions
All the authors fulfilled the contribution requirements per the International Committee of Medical Journal Editors’ role of authors and contributor guidelines. All the authors helped conceptualize and design the study and critically reviewed and revised the manuscript. Z.T.L., J.C., Y.H.L., H.Q.W., and F.Y. carried out the analyses and wrote the first draft of the manuscript. L.H.L., Y.Q.Z., J.S.Y., S.Q.L., X.N.M., X.D.W., Y.M.L., Y.X.L., R.L.S., and W.P.C. carried out the clinical sample and information collection. J.C., Y.H.L., X.D.W., Y.Q.M., X.Z., and J.W. carried out the measurements and analyses. Z.T.L., J.C., and Y.H.L. extracted the data and carried out the analyses. Z.T.L., J.C., and F.Y. oversaw the manuscript development.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
This study conformed to the provisions of the Helsinki Declaration of 1975 (as revised in 2008) concerning human rights and was approved by the ethics committee at the First Affiliated Hospital of Guangzhou Medical University, China (ethical approval number: 2020-36). All patients provided written informed consent and volunteered to participate in the scientific research.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Z., Chen, J., Li, Y. et al. Impact of SARS-CoV-2 infection on respiratory and gut microbiome stability: a metagenomic investigation in long-term-hospitalized COVID-19 patients. npj Biofilms Microbiomes 10, 126 (2024). https://doi.org/10.1038/s41522-024-00596-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41522-024-00596-4
