
Abstract
The emergence of a broad spectrum of microbiome-based therapies has triggered changes in European regulatory frameworks. The first part of the review describes these innovative therapies. The second part provides an overview of the current framework and key changes introduced by the Regulation on substances of human origin (SoHO) for the development of microbiome-based therapies, highlighting the need of microbiome regulatory science to unlock the full potential of microbiome-based therapies.
Similar content being viewed by others

Introduction
Recent scientific advances have revealed the central role of the human microbiome (a complex community of microorganisms living in and on the human body) in maintaining human health, influencing disease development and progression, and even offering new therapeutic avenues1. These discoveries have brought the microbiome into the spotlight as a fertile ground for biotechnology and pharmaceutical innovation. Microbiome-based products represent a wide range of products, from food to medicinal products, including food supplements, foods for special medical purposes, cosmetics or medical devices. The different regulatory statuses of these microbiome-based products are governed by different legislative texts (Table 1). These regulatory frameworks are crucial for developers, prescribers and consumers, as the regulatory status of a product specifies the restrictions, standards and requirements to reach the market. A single substance (including micro-organisms) can be developed and marketed under different regulatory statuses, depending on the envisaged type of finished product, the target effect or targeted population. Microbiome-based products are no exception to that rule.
The intended use of a finished product, regardless of the substance it contains, is a key determinant of the product’s regulatory status. The FDA defines the concept of “intended use” as “the objective intent of the persons legally responsible for the labelling of an article […]. The intent may be shown by such persons’ expressions, the design or composition of the article, or by the circumstances surrounding the distribution of the article […]. This objective intent may, for example, be shown by labelling claims, advertising matter, or oral or written statements by such persons or their representatives”2.
Products intended for the prevention or treatment of disease are to be registered as medicinal/drug products (see definition for Medicinal Products in Table 1). The FDA specifies that a product’s intended use(s) is of high importance, because it can affect how the product will be regulated: it can make the product a drug, or not, regardless of its ingredients and whether or not it is considered a drug3.
A drug candidate’s quality, safety and efficacy within the intended population is assessed by drug competent authorities. In the European Union (EU) there is complementarity between two European bodies: (1) The European Directorate for the Quality of Medicines (EDQM), elaborating binding standards for controlling the quality of pharmaceutical ingredients and drugs and (2) The European Medicines Agency (EMA), supplemented by national drug authorities, who are competent in terms of risk/benefit assessment. In the US, the respective equivalents are the US Pharmacopeia (USP) and the US Food and Drug Administration (FDA).
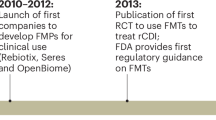
This review will focus on the development of microbiome-based therapies (MbT), including microbiota transplantations (MT) and microbiome-based medicinal products (MMPs). Indeed, these therapies are now increasingly being investigated or recognized as prophylactic and therapeutic approaches for diseases where traditional drugs fail or have severe side effects. The recent marketing approval of the first MMPs underscores this potential and marks a transformative shift in how we approach treatment and prevention. In November 2022, RebyotaTM (a liquid mix of trillions of live microbes sourced from the stool of qualified human donors) became the first MMP approved by the FDA for the prevention of recurrent Clostridioides difficile infections (rCDI)4. This first approval was followed by the approval of oral capsules intended to be used for rCDI: VOWST®5. Other MMPs are currently under clinical evaluation (a non-exhaustive list is provided in Table 2). To date, only MMPs intended to be used for rCDI have received marketing authorisation.
As MbTs gain traction, the regulatory landscape is evolving to address the unique challenges and opportunities they present. Current regulatory frameworks are not fully adapted to the assessment of safety, efficacy, and quality of these new types of therapies as it is the case for all innovative products. This discrepancy has catalysed the emergence of ‘regulatory science’, a field dedicated to developing new tools, standards and methodologies for the evaluation and approval of innovative regulated products6,7. According to the EMA definition, regulatory science refers to the range of scientific disciplines that are applied to the quality, safety and efficacy assessment of medicinal products and that inform regulatory decision-making throughout the lifecycle of a medicine. It encompasses basic and applied biomedical and social sciences and contributes to the development of regulatory standards and tools8. Regulatory bodies, such as the FDA and the EMA, are actively working to refine and adapt guidelines that address innovations, while promoting a balance between ensuring patient safety and fostering scientific progress and innovation.
In the first part of this review, we will be presenting an overview of the large spectrum of MbTs currently under development or recently placed on the market. We will then summarise the current changes in the European regulatory landscape, in response to these new therapies, and discuss future regulatory challenges that still need to be addressed to facilitate the development and approval of future MbTs. By examining these aspects, we aim to provide a comprehensive understanding of the opportunities and hurdles in this rapidly evolving field.
The spectrum of microbiome-based therapies
Microbiome-based therapies represent a large and diverse range of innovations that can be viewed as a continuum from MT, rationally-designed microbial ecosystems (co-culture of various strain for their synergistic activities), all the way to live biotherapeutic products (LBPs – Single strains or mixtures of multiple strains grown separately and are then blended in the appropriate amounts), non-living biotherapeutic products or phage therapies (Fig. 1). It is important to insist that considerable overlap can exist between area of the MbT continuum and therefore each product on the continuum should be assessed based on its specific characteristics and intended use.

Microbiome-based therapies represent a large and diverse range of innovations and can be viewed as a continuum, ranging from microbiota transplantation (on the left), whole ecosystem-based medicinal products, rationally-designed microbial ecosystems (co-culture of various strains for their synergistic activities) all the way to live biotherapeutic products (LBPs – Single strains or mixtures of multiple strains), non-living biotherapeutic products or phage therapies (on the right). For the therapies on the left of the spectrum such as microbiota transplantation and whole ecosystem-based medicinal products, the donor/origin of the microbiome sample has a major importance in the determination of the benefit/risk ratio as these therapies are only partly characterized and controlled. However, when moving to the right of the spectrum, the impact of donor/origin of the microbiome sample on the benefit/risk ratio decreases while the degree of characterization and control of the products themselves increases.
Depending on the type of therapies, the donor/origin of the microbiome sample, used to produce the therapeutic end product, can have a greater or lesser importance in the risk-benefit balance. For therapies such as MT, the donor/origin of the microbiome sample will have a major importance in the determination of the benefit/risk ratio, as these therapies are only partly characterized and controlled. Indeed, there is currently, no analytical method able to fully characterize these complex microbiome samples. However, for therapies with a higher degree of characterization and control (e.g., a single bacterial strain), the impact of donor/origin of the microbiome sample on the benefit/risk ratio will decrease. When specific microorganisms are isolated from humans or from other microbiome samples, such as food or environmental samples, the impact of the donor/origin of the microorganism(s) might become less important in terms of risk, because of the required level of characterization and the reduced complexity of the product. The microorganisms’ origin, however, must always be documented, as clarified in the FDA guidance9,10.
Microbiota transplantation
Microbiota transplantation can pragmatically be defined as “the transfer of biologic material containing a minimally manipulated community of microorganisms from a human donor to a human recipient (including autologous use), with the intent to beneficially affect the microbiota of the recipient”11. However, there is no consensus on a scientific or legal definition of MT at the European Union (EU) level. The preparations (e.g. from faecal material) used during MT procedures, may be associated with a higher risk of pathogen transmission and with the risk of transferring a microbiome inducing potential long-term negative health outcomes for the recipient12,13,14. Even if most of the currently performed MTs are faecal or intestinal MTs, data showing the potential benefit of vaginal and skin MTs are arising in the literature15,16.
Donor-derived microbiome-based medicinal products
Since (again) no official definition exist, these products could be referred to as “faecal microbiota-based medicinal products” or, more precisely, “human intestinal microbiome whole-ecosystem-based medicinal products” as proposed by a consortium of European based companies17. These products consist of whole or highly complex ecosystems, for which the starting materials are human microbiome samples and which differ from MT preparations as they are “manipulated” or “industrially manufactured”9,17. However, there remains a pressing need to clarify and harmonize the terminology for these products to ensure consistency and reduce confusion between the regulators, academia and industry18.
To our best knowledge, whole ecosystems-based products currently only exist for intestinal microbiota-based products and vaginal microbiota-based products are currently in development and early clinical phase (Table 2). In the near future, the concept could potentially be extended to other microbiomes, such as the skin or lung microbiomes.
Donor-independent microbiome-based medicinal products
When increasing the level of manipulation and going towards the selection of specific strains and/or microbial functions, developers may move away from the “whole-ecosystem-based medicinal products” to enter another area of the MbT continuum, referred to within the Pharmabiotic Research Institute as “rationally designed ecosystem-based medicinal products”.
“Rationally designed ecosystem-based medicinal products” are obtained by selecting microbial strains with the purpose to produce a desired ecosystem within the product. The objective is to shape a “controlled ecosystem” able to synthesize metabolites of interest and/or to re-establish targeted microbial functions, identified as desirable within the host. These products can contain dozens or, potentially in the future, even hundreds of different microbial strains, produced during a unique co-fermentation process (Table 2). In contrast to the “whole-ecosystem-based medicinal products”, these “rationally designed ecosystem-based medicinal products” are produced from clonal cell banks and not directly from a human donor microbiome sample.
With increased manipulation levels of the microbiome sample, including the isolation of specific strain(s) and their preservative banking, the impact of the donor/origin of the microbiome sample on the benefit-risk ratio assessment is nearly completely eliminated. Nonetheless, the impact of the processes applied must still be considered, because, due to the complexity of the product, risks may arise from the lack of control of these early manipulations or from the (co-)fermentation steps as well as culture stabilization procedure. In conclusion, the production process, as a whole, needs to be validated (process qualification).
As the product is designed based on targeted functional characteristics of the ecosystem, characterization of the composing microbial strains must be thorough, including potency tests as well as critical quality attributes related to safety and efficacy. Appropriate levels of quality control and batch-to-batch consistency are crucial in order to obtain marketing authorization. Nevertheless, batch-to-batch consistency may remain a challenge due to the complexity of (co-)fermenting multiple strains and the different impacts that the downstream processing may have on the different microbial components of the product.
Another area of the MbT continuum consists of products produced from clonal cell banks via the fermentation of a single microorganism. These products are referred to as Live Biotherapeutic Products (LBPs). The origin of the isolated micro-organism can be broad, including, for example, the human, food, environmental or animal microbiomes. These products can contain only one strain or a mixture of strains. In the latter case, it is important to note that, in the case of LBPs, the different strains are grown separately and then blended in the right amounts. These strains can be from the same origin or from different origins/ donors. The different strains are highly characterized through genotypic and phenotypic characterization and manufacturing process, dealing with the production from the drug substance (DS) all the way to the final drug product (DP), is subjected to very high levels of control. In this context, the donor/origin of the strain has a low impact on the benefit/risk ratio. However, information relating to strain isolation, banking and manufacturing become major parameters impacting this benefit-risk assessment.
From a regulatory perspective, LBPs are the only MMPs with a legal definition, both in Europe and the USA. In the USA, LBPs are defined by the guidance for industry on “Early clinical trials with live biotherapeutic products: chemistry, manufacturing and control information” as “a biological product that: 1) contains live organisms, such as bacteria; 2) is applicable to the prevention, treatment, or cure of a disease or condition of human beings; and 3) is not a vaccine”10. In Europe, LBPs are defined by the European Pharmacopoeia general monograph 3053 as “medicinal products containing live microorganisms (bacterial or yeasts) for human use”19. This monograph mainly addresses quality requirements for LBPs administrated orally or vaginally. However, it is important to note that new products with other delivery routes are under development, such as products with topical administration or for systemic injection20. These products will create new regulatory challenges regarding their manufacturing process controls and quality assessment, but also for safety and efficacy demonstration. A roadmap for safety assessment of LBPs has already been proposed, highlighting the importance of science-driven benefit/risk analysis to demonstrate a positive benefit/risk ratio within a specific intended use and target population9.
In addition to the products containing live microorganisms, medicinal products containing microorganisms which are intentionally rendered ‘non-living’, are also emerging in the spectrum of MbT21. Within the Pharmabiotic Research Institute, we referred to these products as “Non-living biotherapeutic products”. These products are associated with regulatory challenges linked to the characterization of the product, the enumeration of the “non-living cells” in the final drug product, as well as the safety assessment related to the presence or not of remaining living cells. This means that the inactivation step should be an integrated part of the production process. Relevant key parameters and controls have to be defined and confirmation of the “non-living” state of the cells should be provided. Importantly, the “non-living” state of these microorganisms does not guarantee the safety of these products. In addition, Qualified Presumption of Safety (QPS) or Generally Recognized As Safe (GRAS) status of the progenitor microorganisms is not sufficient to demonstrate the safety of the non-living biotherapeutic end product, as GRAS or QPS designations are established for food products, in the context of the general population, an intended use which is very different from a medicinal product intended for a diseased or vulnerable population.
Phage therapy-based medicinal products (PTMPs) represent a rather particular type of medicinal product, based on bacteriophages, targeting and infecting specific bacterial micro-organisms. High specificity can be obtained through appropriate phage selection from environmental or human samples. PTMPs can be a way to safely modulate microbiomes22. Thanks to their specificity, PTMPs can be associated with a high level of safety. Therefore, it is essential that the ecological and functional role of the bacterial target that PTMPs will eliminate from the microbiome is well known. PTMPs are also developed to fight the increasing problem of antibiotic resistance23,24. Due to the specific isolation and characterization process, the influence of donor/origin is less significant in the risk analysis of PTMPs, while the long-term implications of microbiome modulation by phage therapy may require a more in-depth and long-term safety assessment. Other challenges in developing PTMPs may reside in the more “personalized” approach of this type of therapy, requiring a regulatory difficult case-by-case approach25.
The diversity of MbT is striking, with a wide range of innovations emerging rapidly (Table 2). As is often the case, innovation tends to precede regulation, which leads to significant regulatory challenges when considering breakthrough innovations such as microbiome science-based products. Once more products and innovations are in late-stage development and are becoming available, hence when regulators have been confronted with a diversity of products, this process finally results in the needed regulatory adaptation. These changes are currently reshaping the EU regulatory landscape and are discussed in the following section.
The regulatory landscape for microbiome-based therapies
Regulatory framework for microbiota transplantations
Current regulatory framework
In the USA, the result of a stool preparation procedure, suitable for faecal microbial transplantation (FMT), is designed as “FMT product”. Being considered a biological product (drug product), all FMT products require an investigational new drug (IND) application and are consequently subject to FDA approval for clinical use in humans. There is an exception for the FMT used to treat Clostridioides difficile (C. difficile) infection not responsive to standard therapies, when the FMT product is not obtained from a stool bank and when the other conditions described in the FDA guidance (such as appropriate consent from the patient; screening and testing of the stool donor and stool sample) are met26.
In Europe, the regulatory framework is substantially different. Currently, there is a clear lack of harmonization regarding the regulatory status of preparations administered during MTs. EU Member States have taken different positions as the regulatory status of any preparation is in the remit of the National Competent authorities. A recent report published by the Heads of Medicines Agencies shows that FMT can be regulated a) as a medicinal product or equivalent; b) as a therapeutic intervention; c) as a tissue and cell preparation or d) on a case-by-case basis27. The lack of regulatory harmonization is reinforced by the lack of clarity on the “FMT” terminology18. Many stakeholders use the term “FMT” (the procedure) to refer to “stool-derived preparations” (the preparation administered during the procedure), or, in analogy, use the term “VMT” to refer to “vaginal microbiota-derived preparations”27. There is a good chance that this lack of harmonization regarding the regulatory status of FMT will be solved by the new EU ‘Regulation on standards of quality and safety for substances of human origin (SoHO) intended for human application’, as discussed in the next section.
Future regulatory framework
On 17 July 2024, the new EU SoHO regulation was published in the Official Journal of the EU (Regulation (EU) 2024/1938)28. The new Regulation came into force on 7 August 2024 and will apply from 7 August 2027, after a transition period of 3 years. This new Regulation will repeal the existing EU legislation on blood, tissues and cells (Directive 2002/98/EC on safety and quality of human blood and blood components and Directive 2004/23/EC on safety and quality of human tissues and cells)29,30 and aims to improve harmonisation, ensuring a uniform level of protection for SoHO donors and SoHO recipients across the EU, while at the same time facilitating the cross-border exchange of- and access to- SoHO therapies. Indeed, the Regulation is expected to reduce the disparities in the implementation of the rules by different Members States for all types of SoHOs. In the microbiome field, this regulation will introduce a major change, as human microbiomes will fall under the scope of this new SoHO Regulation, whereas they are not expressly specified in tissues and cells legislation. Indeed, ‘SoHO’ is defined in the regulation as “any substance collected from the human body, whether it contains cells or not and whether those cells are living or not, including SoHO preparations resulting from the processing of such substance” and ‘SoHO preparation’ as “a type of SoHO that: (a) has been subjected to processing and, where relevant, one or more other SoHO activities; (b) has a specific clinical indication; and (c) is intended for human application to a SoHO recipient or is intended for distribution”. Based on these definitions, it is clear that all microbiome samples collected from the human body will fall under the scope of this text.
This new SoHO regulation shall apply to: a) SoHO intended for human application (both in the context of clinical research and clinical practice) and SoHO used to manufacture products regulated by other European Union legislation (such as medicinal products or medical devices) and intended for human application; (b) SoHO donors, SoHO recipients and offspring from medically assisted reproduction; (c) SoHO activities that have a direct impact on the quality, safety or effectiveness of SoHO. The Regulation considers the following SoHO activities (Fig. 2): (i) SoHO donor registration; (ii) SoHO donor history review and medical examination; (iii) testing of SoHO donors; (iv) collection; (v) processing; (vi) quality control; (vii) storage; (viii) release; (ix) distribution; (x) import; (xi) export; (xii) human application and (xiii) clinical outcome registration.

The SoHO Regulation will apply to SoHO activities that have a direct impact on the quality, safety or effectiveness of SoHO. These SoHO activities are mentioned in Fig. 2. The SoHO Regulation defines a SoHO entity as an organization legally established in the European Union and carrying out one or more SoHO activities. These SoHO entities must be registered. However, some specific SoHO activities have to be carried out by a SoHO establishment, which will be authorised by the competent authority. Only registered SoHOs entities and authorised SoHO establishments will be permitted to carry out SoHO activities and release SoHO preparations.
In the context of the microbiome research, numerous actors are thus concerned by this new regulation, as they will need to be registered as “SoHO entities”. A SoHO entity means “an organization legally established in the European Union, carrying out one or more SoHO activities”. In addition, some specific SoHO activities must be carried out by a SoHO establishment that has been authorized to do so by the Competent Authority (Fig. 2). One of the key implications of this new Regulation is that only registered SoHO entities and authorised SoHO establishments will be permitted to perform SoHO activities and to release SoHO preparations. However, for SoHO used exclusively in the context of in vitro or animal research, the only requirement set out in this Regulation is to comply with the standards concerning voluntary and unpaid donation, in order to ensure a consistently high level of protection for SoHO donors.
Another major implication of the new SoHO regulation is the requirement for formal approval by a SoHO competent authority of a SoHO preparation. For instance, authorisation will be required for a SoHO preparation intended to be used for MT. This authorisation will involve a review of all SoHO activities performed for that SoHO preparation and that might influence the quality, safety and effectiveness of that SoHO preparation. The assessment of a SoHO preparation will be carried out by SoHO competent authorities, based on all scientific evidence and clinical data regarding the expected benefit and risk provided by the applicant SoHO entity. If scientific evidence and clinical data is not sufficient, or if the risk is more than negligible, further clinical studies may be required. The extent of the clinical monitoring plan will depend on the level of risk.
Regulatory framework for medicinal products
In the EU, medicinal products for human use are governed by the Directive 2001/83/EC. This directive specifies that “a biological medicinal product” is “a product, the active substance of which is a biological substance”. A biological substance is “a substance that is produced by or extracted from a biological source and that needs, for its characterization and the determination of its quality, a combination of physico-chemical-biological testing, together with the production process and its control”. MMPs are biological medicinal products for which the active substances are microorganism(s) coming from microbiome samples (such as food, environmental or human microbiome samples) (Fig. 3).

In the European Union, medicinal products for human use are governed by the Directive 2001/83/EC. Biological medicinal products comprise several diverse product types, including blood and human plasma-derived medicinal products, immunological medicinal products (i.e. vaccines, toxins, serums and allergens) and Advanced therapy medicinal products (ATMPs). By their nature, microbiome-based medicinal products are considered as biological medicinal products but currently they do not have a “separate status” and are not referred to in any of the legislations. For some of these biological medicinal products, such as the blood and plasma-derived medicinal products, but also the ATMPs, the starting material can be tissues and cells from human origin. In this case, an additional piece of legislation is to be taken into account. For the EU, until August 2027, this additional piece of legislation consists of the blood directive (2002/98/EC) and the tissues and cells directive (2004/23/EC) (together designated as the BTC directives). In 2027, the regulation on standards of quality and safety for substances of human origin (SoHO) intended for human application (so called “SoHO Regulation”) will replace the BTC directives. The SoHO Regulation includes human microbiomes within the scope of this regulation, meaning that human microbiomes used as starting material for the production of microbiome-based medicinal products will have to follow the standards and requirements set by this new SoHO Regulation, in addition to the one set by the pharmaceutical legislation.
Generally speaking, for all medicinal products using starting materials from human origin, there is an additional layer of legislation to consider. For example, this is notably the case for advanced-therapy medicinal products (ATMP) produced from human cells and tissues where regulatory interplay between the ATMP legislation and the tissues and cells legislation is clearly defined in Regulation 1394/2007 on ATMP. The new SoHO regulation will by 2027 harmonize practices in Europe by including human microbiomes in the broader concept of “substances of human origin” in its extended scope, and clearly envisages that SoHO (including the human microbiomes) can be collected for the purpose of manufacturing medical devices (regulated by Regulation (EU) 2017/745), medicinal products (regulated by Directive 2001/83/EC), advanced therapy medicinal products (regulated by Regulation (EC) No 1394/2007) or investigational medicinal products (regulated by Regulation (EU) No 536/2014). However, the interplay with the other regulatory frameworks is intentionally not specified in the SoHO regulation and will be laid out in the other regulatory frameworks. In this context, it is important to note that a revision of the EU general pharmaceutical legislation is also ongoing31. The proposal published by the European Commission (EC), after going through parliamentary process and final approval by the European Parliament and the Council, will replace the existing EU pharmaceutical legislation (Directive 2001/83/EC). Among the main changes expected, are clarifications of some definitions, and proposals for new ones, such as “SoHO-derived medicinal product other than ATMPs”. Thanks to this new definition proposal, the interplay between the pharmaceutical legislation and the SoHO regulation will probably be further clarified in the context of microbiome samples and MbTs.
Regarding the application of phages to humans, there are currently no phage-specific provisions in the EU legislation currently in force. So far, EMA has only stated that some principles introduced in the “Guideline on the evaluation of medicinal products indicated for treatment of bacterial infections” can also be applied to phages32. The EC’s proposal for a new Directive for medicinal products for human use also clearly addressed the question of “phage-containing medicinal products”, considering it “as a category of products which may in some instances require adapted rules to fully take account of their specific characteristics”31. This is why “phage-containing medicinal products” (PTMPs) are currently the only medicinal products mentioned in Annex VII of the proposal, as an area in need for an adapted framework, covering products that will be subject to specific scientific or regulatory requirements, due to the characteristics or methods inherent to that medicinal product31. Other regulatory developments regarding PTMPs are also ongoing, with e.g. the publication by the European Pharmacopoeia of a draft chapter on “Phage therapy active substance and medicinal products for human and veterinary use”33. Finally, in December 2023, the EMA launched a process to prepare a guideline on the development and manufacture of human medicinal products specifically designed for phage therapy34.
All these regulatory updates will need to be monitored by stakeholders wanting to develop and/or distribute microbiome-based therapies in Europe, ensuring their development plans reflect tomorrow’s regulatory reality.
Conclusion
The regulatory landscape for microbiome-based therapies is currently evolving rapidly and significantly. This progress is marked by the recent approval in the US of some microbiome-based medicinal products intended to be used for rCDI. These approvals represent the formal recognition of the prophylactic and therapeutic potential of the human microbiome, through products derived from the human microbiome. In line with these developments, a new regulatory framework, the new SoHO regulation, including human microbiomes, is currently being implemented in the EU. In contrast, the regulatory framework for microbiome-based therapies in low- and middle-income countries (LMICs) is currently underdeveloped and varies significantly across regions. Many LMICs lack specific guidelines or policies addressing the development, approval, and oversight of these innovative treatments35,36. Microbiome stakeholders have to deal with a new, but rapidly evolving global market, and could gain great benefit in anticipating harmonization across countries/regions to avoid hampering the global development and patients access to MbT.
Despite this encouraging progress, there is still an urgent need for more robust regulatory science activities in the field. A number of challenges continue to impede the development and approval of microbiome-based therapies. These include the complexity of designing clinical studies targeting the human microbiome, dealing with numerous confounding factors that can affect the safety and efficacy of new MbT candidates. In addition, there is a lack of validated analytical methods that can accurately assess and characterize the composition and functionalities of the microbiome. This gap is not only critical to the discovery of new candidates and ensuring the safety and efficacy of these products but also represents a major limiting factor in the qualification of microbiome-based biomarkers, a tool for accelerating clinical studies and drug development37. The lack of validated analytical methods can also limit the development of IVD microbiome testing and, thus, the integration of microbiome data in clinical practice38. Another major challenge is the lack of consensus on key definitions. For example, there is currently a need to define what characterizes a “healthy” versus a “dysbiotic” microbiome39. The lack of a consensus definition makes it difficult to develop standardized guidelines and benchmarks for microbiome-related research and product development. The complexity of pharmacodynamic and pharmacokinetic assessments for microbiome-based therapies adds another layer of difficulty, as traditional models may not fully capture the dynamic interactions between the microbiome, host, and the therapeutic agents tested. Translating preclinical data into clinical settings also presents additional hurdles. Inter-individual variability in microbiome composition makes it difficult furthermore to predict clinical outcomes based on preclinical studies.
While the recent regulatory developments and product approvals are promising, continued efforts are needed to overcome the remaining challenges. Ongoing initiatives (such as IHMCSA, Human Microbiome Action project; or MMHP, Million Microbiome of Humans Project) are important steps towards achieving this goal, and their success will depend on continued collaboration and commitments from all stakeholders. Advancing regulatory science and fostering innovation in microbiome research are thus necessary steps to unlock the full potential of microbiome-based therapies and improve health outcomes for patients worldwide.
Data availability
No datasets were generated or analysed during the current study.
References
Gilbert, J. et al. Current understanding of the human microbiome. Nat. Med. 24, 392–400 (2018).
FDA. Meaning of Intended Use. Code of Federal Regulations vol. 21CFR201.128 (2023).
Regulations Regarding “Intended Uses”. Federal Register https://www.federalregister.gov/documents/2020/09/23/2020-20437/regulations-regarding-intended-uses (2020).
Khanna, S. et al. Efficacy and Safety of RBX2660 in PUNCH CD3, a Phase III, Randomized, Double-Blind, Placebo-Controlled Trial with a Bayesian Primary Analysis for the Prevention of Recurrent Clostridioides difficile Infection. Drugs 82, 1527–1538 (2022).
Feuerstadt, P. et al. SER-109, an Oral Microbiome Therapy for Recurrent Clostridioides difficile Infection. N. Engl. J. Med. 386, 220–229 (2022).
FDA. Advancing Regulatory Science. https://www.fda.gov/science-research/science-and-research-special-topics/advancing-regulatory-science.
Piniero, S. et al. One health microbiome research across fda centers.
Regulatory science strategy | European Medicines Agency (EMA). https://www.ema.europa.eu/en/about-us/how-we-work/regulatory-science-strategy (2020).
Rouanet, A. et al. Live Biotherapeutic Products, A Road Map for Safety Assessment. Front. Med. 7, 237 (2020).
FDA. Early Clinical Trials with Live Biotherapeutic Products: Chemistry, Manufacturing, and Control Information; Guidance for Industry. 20 (2016).
Hoffmann, D. E., Palumbo, F. B., Ravel, J., Rowthorn, V. & von Rosenvinge, E. A proposed definition of microbiota transplantation for regulatory purposes. Gut Microbes 8, 208–213 (2017).
Research, C. for B. E. and. Safety Alert Regarding Use of Fecal Microbiota for Transplantation and Risk of Serious Adverse Events Likely Due to Transmission of Pathogenic Organisms. FDA (2020).
Keitel, S., European Directorate for the Quality of Medicines & Healthcare, & European Committee (Partial Agreement) on Organ Transplantation. Guide to the Quality and Safety of Tissues and Cells for Human Application. (2017).
Porcari, S. et al. Fine-tuning the gut ecosystem: the current landscape and outlook of artificial microbiome therapeutics. Lancet Gastroenterol. Hepatol. 9, 460–475 (2024).
Han, Y., Liu, Z. & Chen, T. Role of Vaginal Microbiota Dysbiosis in Gynecological Diseases and the Potential Interventions. Front. Microbiol. 12, 643422 (2021).
Callewaert, C., Knödlseder, N., Karoglan, A., Güell, M. & Paetzold, B. Skin microbiome transplantation and manipulation: Current state of the art. Comput. Struct. Biotechnol. J. 19, 624–631 (2021).
Mikkelsen, T. A., McIlroy, J. R., Mimiague, M., Rouanet, A. & Sterkman, L. Towards an EU-wide suitable regulatory framework for faecally derived, industrially manufactured medicinal products. United Eur. Gastroenterol. J. 205064062091031 https://doi.org/10.1177/2050640620910313 (2020).
Olle, B., Ranallo, R. & McDonald, L. C. Correspondence to ‘Escape Velocity-the Launch of Microbiome Therapies’. J. Infect. Dis. jiae575 https://doi.org/10.1093/infdis/jiae575 (2024).
EDQM (European Pharmacopoeia). 3053E General Monograph on Live Biotherapeutic Products Published.Pdf. (2019).
Vargason, A. M. & Anselmo, A. C. Live Biotherapeutic Products and Probiotics for the Skin. Adv. NanoBiomed Res. 1, 2100118 (2021).
Mosca, A. et al. The clinical evidence for postbiotics as microbial therapeutics. Gut Microbes 14, 2117508 (2022).
Federici, S., Nobs, S. P. & Elinav, E. Phages and their potential to modulate the microbiome and immunity. Cell. Mol. Immunol. 18, 889–904 (2021).
Uyttebroek, S. et al. Safety and efficacy of phage therapy in difficult-to-treat infections: a systematic review. Lancet Infect. Dis. 22, e208–e220 (2022).
Pirnay, J.-P. et al. Personalized bacteriophage therapy outcomes for 100 consecutive cases: a multicentre, multinational, retrospective observational study. Nat. Microbiol. 9, 1434–1453 (2024).
Huys, I. et al. Paving a regulatory pathway for phage therapy. Europe should muster the resources to financially, technically and legally support the introduction of phage therapy. EMBO Rep. 14, 951–954 (2013).
Food and Drug Administration (Center for Biologics Evaluation and Research). Enforcement Policy Regarding Investigational New Drug Requirements for Use of Fecal Microbiota for Transplantation to Treat Clostridioides Difficile Infection Not Responsive to Standard Therapies; Guidance for Industry. (2022).
Heads of Medicines Agencies & European Medicines Agency. Faecal microbiota transplantation EU-IN Horizon Scanning report. (2022).
Regulation - 2024/1938 - EN - EUR-Lex. https://eur-lex.europa.eu/eli/reg/2024/1938/oj.
The European Parliament and the Council of the European Union. Directive 2002/98/CE Setting Standards of Quality and Safety for the Collection, Testing, Processing, Storage and Distribution of Human Blood and Blood Components. Official Journal of the European Union (2003).
The European Parliament and the Council of the European Union. Directive 2004/23/EC on Setting Standards of Quality and Safety for the Donation, Procurement, Testing, Processing, Preservation, Storage and Distribution of Human Tissues and Cells. (2004).
Proposal for a DIRECTIVE OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL on the Union Code Relating to Medicinal Products for Human Use, and Repealing Directive 2001/83/EC and Directive 2009/35/EC. (2023).
Faltus, T. The Medicinal Phage-Regulatory Roadmap for Phage Therapy under EU Pharmaceutical Legislation. Viruses 16, 443 (2024).
EDQM (European Directorate for the Quality of Medicines and healthcare). 5.31. Phage Therapy Active Substances and Medicinal Products for Human and Veterinary Use. (2023).
Development and manufacture of human medicinal products specifically designed for phage therapy - Scientific guideline | European Medicines Agency (EMA). https://www.ema.europa.eu/en/development-and-manufacture-human-medicinal-products-specifically-designed-phage-therapy-scientific-guideline (2023).
McGrath, E. et al. A WHO remit to improve global standards for medical products of human origin. Bull. World Health Organ. 102, 707–714 (2024).
Labuschaigne, M. et al. The ethicolegal framework relevant to human faecal microbiota transplants in South Africa: Part 1. A legal vacuum. South Afr. Med. J. Suid-Afr. Tydskr. Vir. Geneeskd. 110, 812–815 (2020).
Rodriguez, J. et al. State of the art and the future of microbiome-based biomarkers: a multidisciplinary Delphi consensus. Lancet Microbe 100948 https://doi.org/10.1016/j.lanmic.2024.07.011 (2024).
Rodriguez, J. et al. Microbiome testing in Europe: navigating analytical, ethical and regulatory challenges. Microbiome 12, 258 (2024).
Joos, R. et al. Examining the healthy human microbiome concept. Nat. Rev. Microbiol. https://doi.org/10.1038/s41579-024-01107-0 (2024).
Author information
Authors and Affiliations
Contributions
J.R., M.C.S., and C.D. drafted the first version of the manuscript. All authors revised and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
J.R., M.C.S., and C.D. are employees of the Pharmabiotic Research Institute. B.P. is a board Member of the Pharmabiotic Research Institute. B.P. is an employee of Yakult Europe BV.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rodriguez, J., Cordaillat-Simmons, M., Pot, B. et al. The regulatory framework for microbiome-based therapies: insights into European regulatory developments. npj Biofilms Microbiomes 11, 53 (2025). https://doi.org/10.1038/s41522-025-00683-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-025-00683-0
