
Abstract
Mass spectrometry (MS)-based proteomics is widely used for quantitative protein profiling and protein interaction studies. However, most current research focuses on single-species proteomics, while protein interactions within complex microbiomes, composed of hundreds of bacterial species, remain largely unexplored. In this study, we analyzed peptide abundance correlations within a metaproteomics dataset derived from in vitro cultured human gut microbiomes subjected to various drug treatments. Our analysis revealed that peptides from the same protein or taxon exhibited correlated abundance changes. By using t-SNE for visualization, we generated a peptide correlation map in which peptides from the same taxon formed distinct clusters. Furthermore, peptide abundance correlations enabled genome-level taxonomic assignments for a greater number of peptides. For instance, 1880 (48.9%) of the 3845 peptides initially assigned only to the family Bacteroidaceae could now be assigned to a specific genome. In species representative genome subsets, peptide correlation networks based on taxon-normalized peptide abundance (TNPA) linked functionally related peptides and provided insights into uncharacterized proteins. Altogether, our study demonstrates that analyzing peptide abundance correlations enhances both taxonomic and functional analyses in human gut metaproteomics research.
Similar content being viewed by others


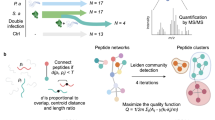
Introduction
Despite many studies linking the human gut microbiome with human health and disease1,2,3,4,5, elucidating the mechanisms underlying the impact of the microbiome on host biology remains challenging, largely due to the complexity and the dark matter in gut microbial communities6,7. Different omics technologies, including metagenomics, metatranscriptomics, metaproteomics, and metabolomics, have become important tools for studying the human gut microbiome8. One of these techniques, metaproteomics, measures the presence and abundance of proteins that drive biological functions within microbial communities, providing a direct glimpse of gut microbiome pathways9.
Proteins involved in the same biological pathways tend to be co-regulated10. These functionally related proteins typically have similar expression profiles, leading to positive abundance correlations. Conversely, proteins involved in mutual inhibition, feedback regulation, or competition for binding sites tend to display negative abundance correlations11,12. Given accurate quantification using mass spectrometry, abundance correlations between proteins could reveal functional linkages, even for proteins that do not physically interact or colocalize, thus enabling the prediction of unknown protein functions13. Protein abundance correlations have been applied to predict the functions of uncharacterized proteins in various model organisms, such as Escherichia coli14, Saccharomyces cerevisiae15, and Homo sapiens13. In addition, a study of proteome variation across thousands of bacterial species constructed functional and physical protein interaction networks, revealing the emergence of complex bacterial phenotypes16. However, these studies have been largely limited to single-species proteomics. In complex microbial communities, like the gut microbiome, protein abundance correlations remain unexplored. A previous study applied gene abundance correlation in metagenomics to predict microbial functional organization17, but since proteins directly execute biological processes, studying protein abundance correlations is expected to more faithfully reflect functional dynamics in microbial communities. Given that over 40% of proteins from the human gut microbiome remain functionally uncharacterized18, and that some of these unknown proteins have been implicated in disease development19,20, understanding their functions is critical for advancing our knowledge of host-microbiome interactions.
Studying protein abundance correlations is more challenging in metaproteomics compared to single-species proteomics due to the difficulties in protein inference21. In complex microbial communities, peptides can be shared among homologous proteins from different species, making it hard to assign them to their actual proteins of origin. Our previous studies showed that a large number of identified peptides were shared by multiple species, complicating taxonomic and protein source assignments22. Moreover, even after protein inference, it remains debatable whether it is reasonable to aggregate peptide quantities into a single protein quantity, as peptides from the same protein-coding sequence could express different quantitative responses23. Recently, peptide-centric metaproteomics analysis, which directly links peptides to their taxonomic and functional annotations, provides an alternative approach24. Since mass spectrometry directly measures peptides instead of proteins, peptide-centric analysis is inherently reasonable and has been shown to be more sensitive and uncover features that are masked at the protein level25.
Given the challenges in protein inference and the inherent advantages of peptide-centric analysis, we focus on peptide abundance correlations in this study. By leveraging peptide-centric analysis, we aim to overcome the biases associated with protein inference and provide a more accurate representation of functional and taxonomic relationships in microbial communities.
In this study, we analyzed peptide abundance correlations in a metaproteomics dataset derived from in vitro cultured human gut microbiomes subjected to over 100 different drug treatments, also referred to as perturbations. These drugs cover 12 Anatomical Therapeutic Chemical (ATC) level-1 classes and were previously shown to potentially impact the human gut microbiome in an initial metaproteomics screening26. We explored the biological foundations of these correlations and visualized them using a peptide correlation map, which was generated by calculating pairwise correlation coefficients of peptide abundance profiles and embedding the resulting matrix into a low-dimensional space using the t-SNE algorithm. Additionally, we applied peptide abundance correlations to improve the taxonomic assignment of peptides. Focusing on single-species representative genome subsets, we calculated taxon-based normalized peptide abundance (TNPA) and constructed peptide abundance correlation networks, where peptides are represented as nodes and edges connect peptide pairs with high correlation in their abundance profiles. These networks revealed functional linkages, providing new insights into the roles of previously uncharacterized microbial proteins.
Results
Peptide abundance correlations in the metaproteomics dataset
Figure 1 summarizes the overall experimental design and research framework. Briefly, 6 individual microbiomes were individually exposed to 107 drugs and five controls using the RapidAIM assay27, which is a 96-well plate assay that maintains the composition and function of the microbiome. The in vitro model used showed a good ability in maintaining microbial taxon-function stability, with pre-post culture correlations of taxon-function-coupled profiles reached an average of r = 0.83 ± 0.03, and also preserved the ability to show microbiome responses to drugs similar to in vivo microbiome28. The metaproteomes were extracted from each well and analyzed by metaproteomics. The 107 selected drugs were shown to have potential impacts on the human gut microbiome in the previous study26. These drugs span a wide range of ATC level-1 categories, with the largest groups being Nervous System (N, 29 drugs), Alimentary Tract and Metabolism (A, 14 drugs), Anti-infectives for Systemic Use (J, 10 drugs), and Cardiovascular System (C, 10 drugs). Peptide identification and quantification results were acquired as previously described26. After acquiring peptide quantification results, peptides with non-zero values in \(\ge 20 \%\) of samples from each individual were selected. Peptide abundance correlations for all peptide pairs from each individual were calculated using Spearman correlation coefficients (SCCs) for log2-transformed abundance fold changes from all 112 samples against the control group. Global peptide abundance correlation maps created with t-SNE of all six individuals, colored with family-level taxonomic annotation of peptides, showed a clear clustering of peptides from the same family (Fig. 2A). In addition, these maps revealed substantial inter-individual variation in the taxonomic composition of microbiomes, which was further supported by peptide-based taxonomic profiling for each individual (Supplementary Fig. 1). Among all six individuals, individual V52 has the largest number of quantified peptides (Fig. 2B) and was selected as an example in the main text. For 21,363 peptides with non-zero values in \(\ge 20 \%\) of samples from individual V52, SCC calculation yielded a total of 228,178,203 peptide pairs with an average SCC of 0.13 ± 0.35 (Fig. 3A). The selection of a 20% threshold for peptides with non-zero values across samples aims to strike a balance between retaining a sufficient number of peptides and ensuring enough non-zero values to make the calculation of SCC statistically meaningful (Supplementary Fig. 2).

Gut microbiomes from six individuals were extracted from human stool samples. The microbiomes were then treated with 107 various drugs, resulting in 112 samples per individual, and in vitro cultured using RapidAIM. After culturing, samples were loaded to LC–MS/MS for metaproteomics analysis. Raw files were searched against the IGC database using the MaxQuant workflow from MetaLab V2.3, yielding a peptide abundance table. To measure peptide abundance correlations, we calculated the Spearman correlation coefficient (SCC) of peptide abundance fold changes relative to the control sample for all peptide pairs. We then created a peptide correlation map to visualize these correlations. Furthermore, we utilized these peptide abundance correlations to assign peptide taxonomic sources, connect peptides from functionally related proteins, and predict the functions of previously uncharacterized proteins.

A Peptide correlation maps were generated using t-SNE of all six individuals, colored by peptide family-level taxonomic annotations. B Number of peptides identified from different individuals. For each individual, peptides identified in \(\ge\)20% of total samples were used for abundance correlation analysis and displayed in the peptide correlation map.

A Distribution of pairwise Spearman correlation coefficients (SCC) of peptide abundance fold changes for all peptide pairs. B Comparison of the SCC of peptide pairs with both peptides from the same protein and peptide pairs of peptides from different proteins. C Comparison of the SCC of peptide pairs of both peptides from the same taxon (genome) and peptide pairs of peptides from different taxa (genomes). D Comparison of peptide pairs of both peptides from the same genome and same protein (SGSP), peptides from the same genome but different proteins (SGDP), and peptides from different genomes and different proteins (DGDP). E Comparison of SCCs between peptide pairs from the same COG category and peptide pairs from different COG categories. F Comparison of SCCs between peptide pairs from the same COG family and peptide pairs from different COG families. **** indicates statistical significance at the p \(\le\) 0.0001 level by two-sided Mann–Whitney U-test.
Upon assigning both protein sources and taxonomic sources to the peptides, we observed that peptides from the same protein and peptides from the same taxon exhibited higher abundance correlation. First, peptide pairs derived from the same protein (7407 pairs) exhibited higher SCCs (0.63 ± 0.22) compared to peptide pairs from different proteins (8,781,121 pairs; SCC = 0.18 ± 0.32, with p \(\le\) 0.0001 by Mann–Whitney U-test and a large effect size with Vargha and Delaney’s A of 0.88, Fig. 3B). To be more intuitive, we selected peptides from the three proteins with the highest total peptide abundances to visualize peptide abundance profiles across samples (Supplementary Fig. 3). Principal coordinates analysis (PCoA) revealed that peptides derived from the same protein tend to cluster together based on their abundance profiles across samples (Supplementary Fig. 3). Consistent with this observation, PERMANOVA analysis based on Euclidean distances showed that peptide abundance profiles for these three proteins were significantly associated with their protein source (R2 = 0.240, F = 10.76, p = 0.001), indicating that peptides derived from the same protein tend to show more similar abundance changes across samples. In addition, peptide pairs derived from the same genome (457,957) had an average SCC of 0.60 ± 0.22, while those from different genomes (8,330,571) averaged an SCC of 0.16 ± 0.31 (Fig. 3C, p \(\le\) 0.0001 by Mann–Whitney U-test and a large effect size with Vargha and Delaney’s A of 0.88). It is worth noting that even after excluding peptide pairs from the same protein, we found that peptides from different proteins within the same genome (450,550 pairs; SCC = 0.60 ± 0.22) still had higher SCCs than those from different genomes (8,330,571 pairs; SCC = 0.16 ± 0.31, p \(\le\) 0.0001 by Mann–Whitney U-test, Vargha and Delaney’s A of 0.88), indicating that sourcing from the same genome contributes significantly to higher SCCs of peptide abundance changes (Fig. 3D). We also assigned functional annotations to the peptides, however, the difference in SCCs between peptides from the same functional category and those from different functional categories was relatively minor at both high-level COG category (Fig. 3E) and refined COG family level (Fig. 3F), with negligible effect size, Vargha and Delaney’s A of 0.51 and 0.56, respectively.
Higher SCC of peptides from the same protein and peptides from the same taxon collectively suggested that studying peptide abundance correlations in metaproteomics datasets is biologically meaningful and can provide valuable insights into metaproteomics analysis.
A global peptide abundance correlation map
The SCC matrix of selected 21,363 peptides from individual V52 recorded how strongly or weakly each peptide is correlated with all other peptides. Although it is theoretically possible to be represented as a peptide interaction network with edges indicating strong correlations, even selecting only peptide pairs with the top 1% SCCs resulted in an extremely dense network (2,281,783 edges among 12,549 peptides), which was not informative (Supplementary Fig. 4). To address this, we visualized all 228,178,203 peptide-peptide correlations from 21,363 peptides as a global peptide correlation map using the t-SNE algorithm by embedding the peptides in a low-dimensional space (Fig. 4A). In this map, the distance between peptides reflects the similarity in their abundance changes under various drug treatments. Notably, the global map was generated based on all pairwise peptide abundance changes under different perturbations in the metaproteomics datasets per individual, without any filtering on the SCC.

A Peptide correlation map colored by family-level taxonomic annotations, showing that the map broadly corresponds to taxa sources of peptides. The areas highlighted in panels (B–E) are boxed with dashed lines. B–E Zoomed-in sections of the peptide correlation map for peptides from the different families: B Burkholderiaceae, C Eggerthellaceae, D Bacteroidaceae, and E Lachnospiraceae. Peptides are colored by genome-level taxonomic annotations. F Peptide correlation map colored by functional annotations (COG categories), showing no overall clustering based on functional associations.
The peptide correlation map revealed a strong correlation between peptide abundance changes and their taxonomic sources. Peptides from the same taxon tend to cluster together in the correlation map across various taxonomic levels (Fig. 4A and Supplementary Fig. 5). Specifically, at the family level, distinct clusters emerged, particularly for families such as Burkholderiaceae, Eggerthellaceae, and Enterobacteriaceae. Zooming into these families, most peptides with genome-level annotations were either assigned to a specific genome or grouped into sub-clusters corresponding to different genomes (Fig. 4B, C). For other families, such as Bacteroidaceae and Lachnospiraceae, the cluster of peptides was less obvious. However, peptides were still grouped at the genome level in the zoomed-in maps (Fig. 4D, E). This clustering trend was also observed in the map colored using peptide taxonomic annotations obtained from Unipept (Supplementary Fig. 6). However, fewer peptides received family-level annotations (10,532 peptides from Unipept versus 15,720 peptides from our own annotation pipeline), and a few differences were observed in taxonomic annotations, likely due to differences in taxonomic naming systems. It is worth noting that all current analyses applied all calculated SCCs without filtering on the p-values of calculated SCCs. However, by only keeping SCCs with Bonferroni-adjusted p-values < 0.01, the global correlation map showed a similar trend of clear taxonomic clusterings (Supplementary Fig. 6).
Although there is a clear clustering of peptides from the same taxon, the global peptide abundance correlation map showed less obvious clustering patterns based on protein function (Fig. 4F and Supplementary Fig. 7). However, when zooming into a single species, more distinct clusters emerged, representing peptides from proteins with similar or related functions (Supplementary Fig. 8). This suggests that studying protein functional linkage through peptide abundance correlations may be more effective at the single-species level, as inter-species functional correlations are not easily discernible in the global map.
Overall, this global peptide correlation map provides an overall trend of peptide abundance correlations between different peptides and indicates that changes in taxa abundance are the primary driver of peptide abundance correlations across different perturbations.
Peptide abundance correlations provide additional information on assigning the peptide taxonomic source
Our results have shown that peptides from the same family clustered together in the global peptide correlation map (Fig. 4A). However, within each family-level cluster, only a small proportion of peptides were annotated to specific genomes, while a larger proportion were only annotated at the family level, leaving their genome-level sources unclear (Fig. 4B–E and Supplementary Fig. 5). Also as mentioned above, peptides from the same genome exhibit high SCCs in their abundance changes across different drug treatments (Fig. 3C). Given a specific sample from the family Bacteroidaceae, genome-distinct peptides from the same genome were not grouped into a single cluster in the heatmap of the peptides SCC matrix. However, these peptides can still be grouped into several visually discernible modules, defined as regions in the SCC heatmap where within-module correlations are clearly higher than between-module correlations (Fig. 5A). Similar genome cluster modules were also observed in other families, such as Eggerthellaceae, Lachnospiraceae, and Burkholderiaceae (Supplementary Figs. 9–11). This observation suggests that the SCC matrix can serve as a valuable input for machine learning models to assign peptide taxonomic sources.

A Heatmap of the SCC matrix for genome-distinct peptides from different genomes within the Bacteroidaceae family. B Distribution of predicted probabilities for peptides sourced from Genome A, focusing on genome-distinct peptides in the test dataset. C Distribution of predicted probabilities for peptides sourced from Genome A, focusing on peptides from other genomes within the same family. D Confusion matrix from the Random Forest model applied to the combined dataset of genome-distinct peptides in the test dataset and peptides from other genomes within the family. E Distribution of predicted probabilities for peptides sourced from Genome A, focusing on genome-unannotated peptides within the Bacteroidaceae family. F Heatmap of the SCC matrix for both genome-distinct peptides and genome-unannotated peptides within the Bacteroidaceae family. Genome-unannotated peptides were colored gray in the color strip. Genome A, MGYG000002281; Genome B, MGYG000000243.
Using the trained Random Forest model for the family Bacteroidaceae, we found that most genome-distinct peptides from Genome A (MGYG000002281, 71 out of 78, 91.0%) and Genome B (MGYG000000243, 61 out of 66, 92.4%) in the test set were correctly classified with probabilities exceeding 90% (Fig. 5B). Specifically, the model assigned these peptides to their respective genome sources with high confidence. In contrast, when applying the same 90% probability threshold to peptides from other genomes within the same family and peptides from other families, the model did not tend to attribute most of the peptides from other genomes within the same family (166 out of 230, 72.2%) and the peptides from other families (438 out of 500, 87.6%) to either Genome A or Genome B (Fig. 5C and Supplementary Fig. 12). The confusion matrices generated from the two test sets demonstrated that the trained model achieved high sensitivity (Fig. 5D and Supplementary Fig. 12). A 5-fold cross-validation demonstrated the strong and consistent ability of our model to assign genome-specific peptides to their respective genomes while effectively rejecting peptides from other sources. To be specific, the model achieved high classification accuracy, precision, recall, and F1-score for peptides from Genome A and Genome B (Supplementary Fig. 13), and exhibited stable False Discovery Rates (FDR) and Rejection Rates (RR) for peptides from both other families and from the same family but different genomes (Supplementary Fig. 13). For peptides without genome-level annotations from the same family, the model classified a large amount of these peptides (48.9%) into either Genome A or Genome B (Fig. 5E). This classification was further supported by the SCC heatmap, where unannotated peptides clustered closely with genome-distinct peptides from a specific genome (Fig. 5F). In addition, models trained using the same method also effectively predicted the genome sources of peptides lacking genome-level annotations in other families, such as Eggerthellaceae, Lachnospiraceae, and Burkholderiaceae (Supplementary Figs. 9–11).
Functionally related peptides were connected in peptide abundance correlation networks for species representative genomes
Our results revealed limited abundance correlations between peptides from proteins with the same or related functions (Fig. 3E, F, and Supplementary Fig. 7). Additionally, we found that taxa-abundance changes are the major driver of peptide abundance changes across different perturbations or drug treatments. Consequently, peptides from proteins with the same or related functions were less likely to be correlated across different taxa compared to peptides originating from the same taxon. Therefore, it is more appropriate to analyze peptide functional linkages at the single-species level by focusing on single-species representative genome subsets of the metaproteomics dataset.
The strong effect of taxonomic abundance changes could cause peptides from the same taxon to show high correlations, even if they are involved in different functions (Supplementary Fig. 14). To minimize this effect and better study the correlations between functionally related peptides, we calculated taxon-based normalized peptide abundance (TNPA) from original peptide abundance (OPA) for the top 10 species/genomes with the highest number of identified peptides, as described in the methods section. TNPA was mostly moderately or weakly positively correlated with OPA in most studied species (Supplementary Fig. 15), and we observed that some peptides with high SCCs for OPA log2-FC exhibited low SCCs for TNPA log2-FC (Fig. 6A and Supplementary Fig. 16). This suggests that many peptide abundance correlations driven by taxa-abundance changes were not significantly correlated at the TNPA level, as shown in Supplementary Fig. 14.

A Correlation between SCCs of peptide log2-FC based on TNPA and SCCs based on OPA (original peptide abundance) in the top ten genomes from individual V52. B, C Higher modularity and lower clustering coefficient of peptide correlation networks constructed with SCC of TNPA log2-FC than networks constructed with SCC of OPA log2-FC. D Comparison of representative peptide correlation networks for species representative genomes constructed with TNPA and OPA. Nodes are colored according to their modularity class from Gephi. E Proportion of peptide pairs from the same protein in peptide correlation networks for species representative genomes compared to the proportion in all peptide pairs from the corresponding species representative genome. F Proportion of peptide pairs from proteins potentially located near each other in the genome (\(\le\)10 gene ID difference) in peptide correlation networks for species representative genomes compared to the proportion in all peptide pairs from the corresponding species representative genomes. ** indicates statistical significance at the p \(\le\) 0.01 level by two-sided Mann–Whitney U-test. G, H Peptide abundance correlation networks for species representative genomes MGYG000004769 and MGYG000002528. Peptides from proteins with only one peptide in the network were removed. Peptides from the top eight proteins with the most peptides in each network are annotated with distinct colors, and peptides from other proteins are colored gray.
To reveal functional linkages, peptide abundance correlation networks for species representative genomes were constructed using peptide pairs with the top 5% SCC from each of the top 10 species representative genomes with the most identified peptides. Two sets of networks were constructed using SCCs derived from OPA and TNPA, respectively (Supplementary Figs. 17 and 18). Networks constructed with TNPA had higher modularity (Fig. 6B) and lower clustering coefficients (Fig. 6C), indicating that these networks had dense connections within modules but sparse connections between different modules. The lower clustering coefficients also suggested that TNPA-based networks were less tightly connected as shown in the Fig. 6D. By selecting peptide pairs with top 5% SCC, each species representative genome have an average threshold of 0.60 for SCC derived from TNPA (Supplementary Fig. 19), and selecting peptide pairs with top 5% SCC kept a moderate amount of peptides left in the constructed networks for each species representative genome (Supplementary Fig. 19). Most of these selected pairs to construct networks (91.6 ± 14.4% across the selected 10 genomes) also had a significant Bonferroni-adjusted p-value (<0.01) on their original abundances.
Peptide abundance correlation networks for species representative genomes constructed with TNPA revealed functional linkages. Peptides from the same protein and those from proteins potentially located near each other in the genomes (with \(\le\)10 gene ID differences in the UHGG) were considered functionally related. Peptide pairs in the network showed a higher percentage of these types of relationships compared to all peptide pairs (Fig. 6E, F). It is worth noting that gene ID differences do not always perfectly reflect physical genome distance; however, they generally indicate proximity when genes are located on the same contig. Here, we provide examples of connections between peptides from functionally related proteins. In the peptide correlation network of genome MGYG000004769 (Phascolarctobacterium faecium), peptides from MGYG000004769_01826 (Methylmalonyl-CoA mutase large subunit) and MGYG000004769_01827 (Succinyl-CoA: coenzyme A transferase) were densely connected (Fig. 6G). Genes encoding these two proteins are located near each other in the genome, with adjacent gene ID numbers and only a 218 bp intergenic region on the same contig. These proteins also have closely related functions in the TCA cycle. Methylmalonyl-CoA mutase catalyzes the conversion of methylmalonyl-CoA to succinyl-CoA, while Succinyl-CoA:coenzyme A transferase then catalyzes the reversible reaction: succinyl-CoA + L-malate \(\rightleftharpoons\) succinate + L-malyl-CoA. In the same network, peptides from MGYG000004769_00624 (Molecular chaperone DnaK) and MGYG000004769_02173 (Chaperonin GroEL), which have similar functional annotations, were also connected. In another peptide correlation network of genome MGYG000002528 (Anaerostipes hadrus), peptides from proteins MGYG000002528_01044 (NAD-dependent dihydropyrimidine dehydrogenase subunit PreA) and MGYG000002528_01048 (Allantoate amidohydrolase) were densely connected (Fig. 6H). These proteins are also encoded by genes with adjacent gene numbers, which are separated by a 2717 bp intergenic region. Similar functional linkages were also observed in networks with Unipept-derived peptide functional annotations (Supplementary Fig. 20). For example, in the network of the genome MGYG000004769, peptides annotated with GO:0006083 (acetate metabolic process) were densely connected to peptides annotated with GO:0004494 (methylmalonyl-CoA mutase activity) (Supplementary Fig. 20).
In summary, peptide abundance correlation networks for species representative genomes constructed using TNPA revealed connections between functionally related peptides. These connections demonstrated the potential for using peptide abundance correlations to predict protein functions.
Applying peptide correlation networks of species representative genomes for predicting unknown protein functions
An average of 5.2% of peptides in the 10 constructed peptide correlation networks for species representative genomes originated from proteins of unknown functions (PUFs). This percentage was comparable to the overall proportion of peptides from unannotated proteins in each species (Fig. 7A). To predict functions for PUFs, we calculated peptide abundance correlations between peptides from PUFs and peptides from annotated proteins. The proportion of connections between peptides from PUFs and peptides from annotated proteins varied across species (Fig. 7B). For genome MGYG000002478 (Phocaeicola dorei), the proportion reached 24%, while in other genomes, the lowest was 2%. Here, we provide examples from the top two genomes with the highest percentage of connections between peptides from PUFs and peptides from annotated proteins to show the feasibility of predicting microbial protein functions using peptide abundance correlations.

A Proportion of peptides from PUFs in each peptide correlation network for the species representative genome and the proportion of peptides from PUFs in all identified peptides from the corresponding species representative genome. NS. indicates statistical significance at the p > 0.05 level by two-sided Mann–Whitney U-test. B Distribution of the proportions of edges between peptides from PUFs and peptides from proteins with known function in the peptide correlation network of each species' representative genome. C, D Peptide abundance correlation networks for the two species representative genomes, MGYG000002478 and MGYG000002281, with the highest percentage of edges between peptides from PUFs and peptides from proteins with known function. Networks were constructed based on peptide pairs with the top 5% SCCs from each species. Peptides from proteins with only one peptide in the network were removed. Peptides from the top eight COG families in each network are annotated with distinct colors, and peptides from other COG families are colored gray.
In the network of genome MGYG000002478 (Fig. 7C), Phocaeicola dorei, peptides from uncharacterized protein MGYG000002478_00658 had the largest number of connections (31) to annotated peptides in the network. Of these, 10 connections were to peptides annotated to COG1629 (Outer-membrane receptor protein, Fe transport), with 7 linked to peptides from MGYG000002478_00657, an iron complex outer-membrane receptor protein (K02014). This indicates that the uncharacterized protein is likely to relate to transmembrane transport. This prediction was further supported by the InterProScan of the uncharacterized protein, showing it has a SusD-like domain, a typical outer-membrane protein feature. In addition, most parts of the protein were assigned to a non-cytoplasmic domain, also indicating its extracellular location.
In the network of genome MGYG000002281 (Fig. 7D), Bacteroides faecis, peptides from uncharacterized protein MGYG000002281_00118 had the largest number of connections (52) to annotated peptides in the network. Excluding 9 peptide connections to functionally ambiguous COG0457 (Tetratricopeptide repeat protein), the second-largest group comprises 7 connections to COG3525 (N-acetyl-beta-hexosaminidase), including 3 connections to MGYG000002281_02589 (hyaluronoglucosaminidase, K01197), 2 to MGYG000002281_04635 (hexosaminidase, K12373), and 2 to MGYG000002281_01369 (hexosaminidase, K12373). These connections suggest the uncharacterized protein is likely to be involved in carbohydrate metabolism. An InterProScan analysis of the protein revealed a non-cytoplasmic domain, indicating that it is a membrane-bound protein predicted to be outside the membrane, possibly acting as a signal protein for carbohydrate metabolism. In contrast, searching this protein in the AlphaFold Protein Structure Database29,30 matched UniProt ID A0A1E9BZD4 (100% identity, HSP score of 1197), an uncharacterized protein without a known biological function. These findings highlight the value of peptide correlation networks in providing complementary insights for predicting the potential biological role of PUFs.
Discussion
We analyzed peptide abundance correlations in a metaproteomics dataset of in vitro cultured human gut microbiomes with different drug treatments/perturbations, demonstrating the feasibility of applying peptide abundance correlations for peptide taxonomic assignments as well as revealing protein functional linkages in subsets for species representative genomes.
Protein abundance correlations have been applied to study protein functional linkage and to predict functions of functionally unknown proteins in single-species proteomics datasets13,14,15. In these single-species studies, functionally associated proteins have coordinated changes in abundance across perturbations13,14,15, and proteins with identical subcellular localization also exhibited coordinated abundance changes13. However, in our metaproteomics dataset, the abundances of peptides from proteins of the same taxon, instead of peptides from proteins with associated functions, were more likely to have correlated abundance changes.
Peptides from the same taxon showing high abundance correlation are largely due to the significant impact of environmental disturbances on the composition of microbial communities31. This is supported by the observed variation in the relative abundance of peptides assigned to different genera across samples from individual V52 (Supplementary Fig. 21). In contrast, the limited correlation of peptides from proteins with related functions can be attributed to the high functional redundancy of human gut microbial communities32. Specifically, a decrease in peptides from proteins with a particular function could lead to an increase in peptides from proteins performing a similar function in a phylogenetically unrelated species. This compensatory mechanism helps maintain the overall functional stability of the microbial community, resulting in limited correlations among peptides from proteins with related functions. The microbial community’s proteome-level functional redundancy (FRp) of the studied samples was calculated in our previous study26. It was found that some compounds transited the microbiome from its original state of high FRp to an alternative state of low FRp, showing the inherent high functional redundancy and the perturbations caused by drug treatments. Notably, a study focused on thousands of single-bacterium proteomes applied a method called SCALES (Spectral Correlation Analysis of Layered Evolutionary Signals), which progressively revealed phylogeny, pathways, and protein complexes from proteome data variance16. However, our metaproteomic data focus on specific microbial communities, in which global functional associations were not directly evident. Novel analytical approaches may offer new perspectives for exploring complex functional interactions within metaproteomics data.
Our study is the first to investigate peptide abundance correlations in a gut microbiome metaproteomic dataset. However, similar studies have been conducted using metagenomic data, such as genetic correlation networks from soil metagenomes, which have revealed a hierarchical functional structure17, similar to findings in single-species cellular genetic correlation networks33. In contrast, our analysis of peptide abundance correlations primarily showed a clear clustering of peptides from the same taxon, with a less obvious functional hierarchy. This underscores that functional insights derived from protein abundance measurements are different from those inferred from genetic materials through metagenomics or metatranscriptomics34.
Our global peptide abundance correlation map also provided clues for studying microbial responses to drugs. In the constructed peptide correlation maps (Fig. 2A), peptides from the family Enterobacteriaceae were usually separated from other taxa, indicating unique response patterns for this taxonomic group. This is supported by previous experimental studies. For example, psychotropic drugs like fluoxetine have shown antimicrobial activity against Escherichia coli (family Enterobacteriaceae), while the abundance of other human gut species increased in the in vitro microbiome after the treatment with the same drug35. In addition, microbial genera have been classified into three distinct taxonomic clusters, each representing a different pattern of drug response26. Patterns resembling these taxonomic clusters can also be observed in our peptide correlation maps. For instance, peptides from Parasutterella (Burkholderiaceae family) and Eggerthella (Eggerthellaceae family), which belong to two clusters exhibiting distinct response patterns, are also well separated in our peptide correlation map (Fig. 2A and Supplementary Fig. 5).
Higher correlations of peptides from the same taxon also provide a solution for peptide taxonomic source assignment, which remains a challenge for peptide-centric metaproteomics analysis. Although most of the in-silico digested peptides were genome-distinct peptides36, most of the peptides identified from real metaproteomics datasets were shared by different genomes22. In this study, even after refining the potential genome sources, a large number of peptides could only be assigned to a family-level LCA, and the actual taxonomic sources of these peptides remain unknown (Fig. 4B–E). By investigating peptide abundance correlations, additional information was provided for peptide taxonomic source assignment. This is expected to provide a more accurate microbial biomass profile, which quantifies species biomass contributions to the microbial community using metaproteomics data37. However, in this study, peptides could only be assigned to a species representative genome, which represents a cluster of genomes potentially from different strains18. With the current workflow, the contribution of individual strains within a species cannot be resolved. Further analysis using a species-specific database containing all possible strains may help assess the contribution of different strains to specific species.
Although taxa-abundance changes were major contributors to peptide abundance correlations, to reveal peptide functional linkages, peptide abundance correlations were further analyzed at the species representative genomes level by extracting subsets of genome-distinct peptides of each genome from the metaproteomics dataset. In addition, to reduce the impact of taxa-abundance changes, TNPA was calculated. The principle of the calculation of TNPA was similar to a normalization method named LFQRatio38, which divided protein LFQ intensity by the sum of all protein LFQ intensities for its respective strain in a microbial coculture system. LFQRatio has shown its ability to transform absolute protein quantification data into accurate and biologically meaningful protein abundance values for samples with multiple species at variable cell ratios. Similarly, in our study, we also found that taxa-based normalized peptide abundance (TNPA) showed a good ability to reveal functional linkages (Fig. 6E, F). Peptides from proteins with related functions were connected in the peptide abundance correlation network for species representative genomes constructed with TNPA.
A limitation of this study comes partly from metaproteomics itself. Current metaproteomics has limited proteome coverage39,40. It is estimated that around 200 bacterial species reside in the human gut microbial community41. However, bacterial species with an abundance lower than 0.5% were hard to detect with current metaproteomics techniques42, and the number of peptides and proteins identified in low-abundant species is limited40. In contrast, most metagenomics studies with moderate sequencing depth (10 Gbases) have the ability to detect species with relative abundances down to approximately 0.01% in a given sample43. With ultra-deep metagenomics sequencing, it is possible to reconstruct metagenomic-assembled genomes (MAGs) of species with extra-low abundance (<0.1%), enabling more accurate and deep comparative metagenomics analysis44. Moreover, metatranscriptomics presents additional challenges for detecting low-abundance species due to the wide dynamic range of transcript expression45. As a result, a 0.1% relative abundance threshold is commonly applied in metatranscriptomics to discard ultra-low abundance species that likely arise due to limitations of short-read-based taxonomic classification46. Considering the limited proteome coverage of current metaproteomics, only the top ten genomes were investigated to study intra-species functional relations in this work. Even in these genomes, the number of peptides was still limited. This results in the limited scale of constructed peptide correlation networks for species representative genomes. A lot of peptides from proteins with interesting or unknown functions were not identified and were not included in the network. In addition to considering proteome coverage, it is also worth noting that a high-resolution LC–MS/MS for identification and quantification is also needed. Our analysis in another metaproteomics dataset from Q Exactive mass spectrometer with the same procedure showed a weak abundance correlation of same taxon peptides (Supplementary Fig. 22). Fortunately, DIA-based metaproteomics has proven to be able to significantly increase the number of identified peptides as well as to improve quantification reproducibilities47,48, which has the potential to expand and strengthen applying peptide abundance correlations to reveal more interesting findings. Furthermore, protein sizes, proteome sizes across bacterial species, and differences in cell numbers across samples could impact the interpretation of metaproteomics results. These factors may warrant consideration in future analyses.
Another aspect that needs improvement is distinguishing true functional correlations from incidental ones in peptide correlation networks for species representative genomes. First, the number of samples used to calculate Spearman correlation coefficients should be carefully considered, as these coefficients require sufficient sample sizes to achieve high confidence in the correlations detected49. Additionally, applying a strict threshold or employing more refined methods to filter out functionally related interactions from correlations driven by other factors is crucial, since abundance correlations do not always indicate functional relationships50. It is also worth noting that in this study, for each individual, the abundance of peptides under a specific drug treatment was measured only once. Although the large number of drug treatments may mitigate the impact of random fluctuations in peptide abundance, the inclusion of biological replicates would enhance the reliability of measuring individual-specific microbiome responses to drugs and could potentially improve the robustness of peptide abundance correlation analyses. And the in vitro culture system used in this study might have a systemic impact on the microbiome, potentially affecting peptide abundance correlation patterns. This limitation should be taken into consideration in future studies. Ultimately, the field would benefit from larger, more reproducible datasets and refined protein association reference methods to advance the study of peptide/protein abundance correlations.
In summary, fluctuations in peptide abundance contain wealthy information on the microbiome’s response to perturbations. These fluctuations can be harnessed to assign peptides to their protein and taxon of origin, as well as to predict function for functionally unknown proteins. In the metaproteomics dataset, peptides from the same taxon were clustered in the peptide correlation map, suggesting that microbiome taxonomic abundance change is the major contributor to peptide abundance changes. We anticipate that the concept of peptide/protein abundance correlations needs further investigation to deepen taxonomic and functional understanding of metaproteomics data.
Methods
Metaproteomics dataset
In this study, we analyzed a metaproteomics dataset comprising 672 raw files from human gut microbiomes of six individuals subjected to various drug treatments (112 samples per individual). Specifically, stool samples were treated with 109 different compounds—107 drugs, two DMSO samples as negative controls, and three kestose samples as positive controls (detailed in Supplementary Table S1)—and then in vitro cultured using the RapidAIM assay27, a culture- and metaproteomics-based rapid method for studying individual microbiome responses to drugs. Detailed information on sample collection, preparation, and LC–MS/MS analysis has been documented in a separate study26. It is worth noting that an equal volume of each sample was applied for protein digestion and loaded for LC–MS/MS metaproteomics analysis26,27. Metaproteomics raw files obtained from LC–MS/MS were searched against the IGC (Integrated Gene Catalog) database51 with MetaLab2.352 with the MaxQuant53 workflow, using default settings. Peptide quantification results were extracted from the “peptides.txt” file generated by MetaLab for subsequent analysis.
Peptide abundance correlation calculation
Pre-processing and peptide filtering. Peptide abundance correlations across different perturbations were calculated separately for each individual. First, peptide identification and quantification results were extracted individually. Peptides were filtered by retaining only those with non-zero intensity in at least 20% of the samples for each individual (≥23 samples). The quantification results of peptides that passed this filtering step were used for further analysis.
Peptide abundance log2-fold change (log2-FC) calculation. To avoid zero values, 1 was added to peptide raw intensity values. The log2-fold change in peptide abundance for each sample was calculated by dividing the peptide intensity under each treatment by the average intensity of the two DMSO-treated control samples, followed by log2 transformation.
Spearman correlation coefficients (SCC) calculation. Peptide abundance correlations across different treatments were calculated for all peptide pairs using Spearman correlation coefficients (SCCs) of the log2-FC values across the treatments. The SCCs were computed using the cor function in R.
Statistical assessment of calculated SCC. To estimate the statistical significance of each pairwise correlation in a computationally feasible manner, we used a standard approximation based on the t-distribution:
where ρs is the Spearman correlation coefficient (SCC) and n is the number of samples. This t-value was then used to compute two-tailed p-values.
Peptide annotations
To enable meaningful biological analysis, peptides were assigned to their protein sources and annotated with taxonomic and functional information using the following procedures.
Protein source refinement. We first generated a genome-level taxonomic profile for each individual to refine peptide protein source assignments and improve peptide annotation resolution. All identified peptides from each individual were mapped to their taxonomic sources by aligning them against MetaPep22 records. Genome-distinct peptides, defined as peptides exclusively present in a single bacterial genome, were utilized to enhance the accuracy of assignments. In the subsequent step, only genomes with at least three genome-distinct peptides identified across all samples were considered for peptide protein source assignments.
Peptide protein sources annotation. Next, for peptide protein source assignments, protein sequences from the 4744 UHGG representative genomes18 were in-silico digested using DeepDigest54 with the following parameters: miscleavage = 2, minimum peptide length = 7, maximum peptide length = 47 (default settings), and trypsin as the protease. Subsequently, all identified peptides were mapped to their unique protein source or multiple protein sources.
Peptide taxonomic and functional sources annotation. Each peptide was taxonomically annotated by assigning it to a specific genome source or the lowest common ancestor (LCA) of the genomes corresponding to all its protein sources. For functional annotations, protein functional annotations were extracted from the UHGG18 database, and each peptide was functionally annotated based on the functional annotation of its source protein(s).
Additional annotation from Unipept24. All studied peptides from individual V52 were uploaded to the Unipept online metaproteomics analysis platform (https://unipept.ugent.be/mpa) to obtain additional peptide-level taxonomic and functional annotations (Gene Ontology [GO] terms and Enzyme Commission [EC] numbers) using the default settings.
Visualize peptide abundance correlations with global peptide correlation maps
t-SNE (t-distributed stochastic neighbor embedding) was applied to visualize the peptide abundance correlations. The SCC matrix of peptide pairs from each individual was used as the input for the Rtsne package55 with default parameters (perplexity = 30). In the peptide correlation map, each point represents a peptide, and the peptides were colored based on their taxonomic or functional annotations acquired as described in the previous section.
Applying a machine learning model to predict peptide genome source
Genome-distinct peptides from the two genomes with the highest number of such peptides within the same bacterial family were extracted. Seventy percent of these peptides were randomly selected as the training dataset, while the remaining 30% constituted the test dataset. Additionally, genome-distinct peptides from other genomes within the same family and 500 randomly selected peptides from other families were included for further evaluation of the model’s performance.
For the training dataset, the Spearman correlation coefficients (SCCs) of each peptide’s abundance changes relative to all peptides in the dataset were calculated and used as input features. A Random Forest classifier was then trained using the randomForest package56 in R with ntree = 500, importance = True. The model’s performance was assessed using confusion matrices calculated on two different combined test datasets: (1) genome-distinct peptides from the test dataset combined with genome-distinct peptides from other genomes of the same family, and (2) genome-distinct peptides from the test dataset combined with 500 randomly selected peptides from other families. Following these assessments, the trained model was used to predict genome sources of peptides from the same bacterial family that lacked genome-level taxonomic annotations.
Calculation of taxon-based normalized peptide abundance (TNPA)
To mitigate the impact of changes in taxon abundance on peptide abundance and to better study the effect of different functions on peptide abundance, taxon-based normalized peptide abundance (TNPA) was calculated based on the original peptide abundance (OPA), total peptide abundance of all peptides from the same taxon in the sample (TPATS, Total Peptide Abundance per Taxon in a Sample), and an average level of peptide abundance(\({10}^{8}\)) using the following formula:
The TNPA calculation was at the genome level, using genome-distinct peptides specific to each species’ representative genome. TNPA was calculated only for the top 10 genomes from each individual microbiome with the largest number of identified genome-distinct peptides. The subsequent peptide abundance correlation analysis for species representative genomes also focused on these genomes.
Peptide abundance correlation networks for species representative genomes
Two types of peptide abundance correlation networks for species representative genomes were constructed using the SCCs of the log2-FC of OPA and the SCCs of the log2-FC of TNPA. Peptide pairs with the top 5% SCCs were retained for network construction, which was carried out using the graph_from_adjacency_matrix function from the igraph package57 in R (with parameters: weighted = TRUE, mode = “undirected”, and diag = FALSE). Nodes and edges from each network were extracted with the same package, and all peptides in networks were mapped to their protein source annotations.
Peptide abundance correlation networks for species representative genomes were visualized using Gephi 0.10.1 with the Yifan Hu layout58. Network modularity and clustering coefficients were calculated using the modularity and transitivity functions from the igraph package, respectively. Networks constructed with TNPA were used to study the correlations between peptides with related functions and to predict the functions of previously uncharacterized proteins.
Data availability
Metaproteomics raw files used to compile analysis were deposited at the ProteomeXchange Consortium59 via the PRIDE60 partner repository as described in our previous study26 (https://doi.org/10.1101/2025.02.13.637346). These files will be publicly available upon publication. Additionally, another dataset generated using a Q Exactive mass spectrometer is available with the data set identifiers PXD012724.
Code availability
All codes to perform the analysis in this study are available on GitHub at https://github.com/northomics/Peptide_Abundance_Correlations.
References
Lynch, S. V. & Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 375, 2369–2379 (2016).
de Vos, W. M., Tilg, H., Van Hul, M. & Cani, P. D. Gut microbiome and health: mechanistic insights. Gut 71, 1020–1032 (2022).
Wu, G. et al. A Core Microbiome signature as an indicator of health. Cell 187, 6550–6565.e11 (2024).
Sanna, S. et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 51, 600–605 (2019).
Lloyd-Price, J. et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662 (2019).
Heintz-Buschart, A. & Wilmes, P. Human gut microbiome: function matters. Trends Microbiol. 26, 563–574 (2018).
Lobb, B., Kurtz, D. A., Moreno-Hagelsieb, G. & Doxey, A. C. Remote homology and the functions of metagenomic dark matter. Front. Genet. https://doi.org/10.3389/fgene.2015.00234 (2015).
Franzosa, E. A. et al. Sequencing and beyond: integrating molecular “omics” for microbial community profiling. Nat. Rev. Microbiol. 13, 360–372 (2015).
Kleiner, M. Metaproteomics: much more than measuring gene expression in microbial communities. mSystems 4, e00115-19 (2019).
Wang, J. et al. Proteome profiling outperforms transcriptome profiling for coexpression based gene Function Prediction. Mol. Cell. Proteom. 16, 121–134 (2017).
Brandman, O. & Meyer, T. Feedback loops shape cellular signals in space and time. Science 322, 390–395 (2008).
Garrido-Rodriguez, M., Zirngibl, K., Ivanova, O., Lobentanzer, S. & Saez-Rodriguez, J. Integrating knowledge and omics to decipher mechanisms via large-scale models of signaling networks. Mol. Syst. Biol. 18, e11036 (2022).
Kustatscher, G. et al. Co-Regulation map of the human proteome enables identification of protein functions. Nat. Biotechnol. 37, 1361–1371 (2019).
Mateus, A. et al. The functional proteome landscape of Escherichia coli. Nature 588, 473–478 (2020).
Messner, C. B. et al. The proteomic landscape of genome-wide genetic perturbations. Cell 186, 2018–2034.e21 (2023).
Zaydman, M. A. et al. Defining hierarchical protein interaction networks from spectral analysis of bacterial proteomes. eLife 11, e74104 (2022).
Ma, B. et al. Genetic correlation network prediction of forest soil microbial functional organization. ISME J. No. 12, 2492–2505 (2018).
Almeida, A. et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 39, 105–114 (2021).
Haynes, W. A., Tomczak, A. & Khatri, P. Gene annotation bias impedes biomedical research. Sci. Rep. 8, 1362 (2018).
Kustatscher, G. et al. Understudied proteins: opportunities and challenges for functional proteomics. Nat. Methods 19, 774–779 (2022).
Schiebenhoefer, H. et al. Challenges and promise at the interface of metaproteomics and genomics: an overview of recent progress in metaproteogenomic data analysis. Expert Rev. Proteom. 16, 375–390 (2019).
Sun, Z. et al. MetaPep: a core peptide database for faster human gut metaproteomics database searches. Comput. Struct. Biotechnol. J. 21, 4228–4237 (2023).
Plubell, D. L. et al. Putting Humpty Dumpty back together again: what does protein quantification mean in bottom-up proteomics?. J. Proteome Res. 21, 891–898 (2022).
Gurdeep Singh, R. et al. Unipept 4.0: functional analysis of metaproteome data. J. Proteome Res. 18, 606–615 (2019).
Simopoulos, C. M. A. et al. pepFunk: a tool for peptide-centric functional analysis of metaproteomic human gut microbiome studies. Bioinformatics 36, 4171–4179 (2020).
Li, L. et al. Systematic metaproteomics mapping reveals functional and ecological landscapes of human gut microbiota responses to therapeutic drugs. Preprint at bioRxiv https://doi.org/10.1101/2025.02.13.637346 (2025).
Li, L. et al. RapidAIM: a culture- and metaproteomics-based rapid assay of individual microbiome responses to drugs. Microbiome 8, 33 (2020).
Li, L. et al. An in vitro model maintaining taxon-specific functional activities of the gut microbiome. Nat. Commun. 10, 4146 (2019).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold Protein Structure Database in 2024: providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 52, D368–D375 (2024).
Nguyen, J., Lara-Gutiérrez, J. & Stocker, R. Environmental fluctuations and their effects on microbial communities, populations and individuals. FEMS Microbiol. Rev. 45, fuaa068 (2021).
Li, L. et al. Revealing proteome-level functional redundancy in the human gut microbiome using ultra-deep metaproteomics. Nat. Commun. 14, 3428 (2023).
Costanzo, M. et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 353, aaf1420 (2016).
Armengaud, J. Metaproteomics to understand how microbiota function: the crystal ball predicts a promising future. Environ. Microbiol. 25, 115–125 (2023).
Cussotto, S. et al. Differential effects of psychotropic drugs on microbiome composition and gastrointestinal function. Psychopharmacology 236, 1671–1685 (2019).
Mesuere, B. et al. Unipept: tryptic peptide-based biodiversity analysis of metaproteome samples. J. Proteome Res. 11, 5773–5780 (2012).
Kleiner, M. et al. Assessing species biomass contributions in microbial communities via metaproteomics. Nat. Commun. 8, 1558 (2017).
Shi, M., Evans, C. A., McQuillan, J. L., Noirel, J. & Pandhal, J. LFQRatio: a normalization method to decipher quantitative proteome changes in microbial coculture systems. J. Proteome Res. https://doi.org/10.1021/acs.jproteome.3c00714 (2024).
Lohmann, P. et al. Function is what counts: how microbial community complexity affects species, proteome and pathway coverage in metaproteomics. Expert Rev. Proteom. 17, 163–173 (2020).
Sun, Z., Ning, Z. & Figeys, D. The landscape and perspectives of the human gut metaproteomics. Mol. Cell. Proteom. 23, 100763 (2024).
Lloyd-Price, J., Abu-Ali, G. & Huttenhower, C. The healthy human microbiome. Genome Med. 8, 51 (2016).
Duan, H. et al. Assessing the dark field of metaproteome. Anal. Chem. 94, 15648–15654 (2022).
Blanco-Míguez, A. et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 41, 1633–1644 (2023).
Jin, H. et al. Hybrid, ultra-deep metagenomic sequencing enables genomic and functional characterization of low-abundance species in the human gut microbiome. Gut Microbes https://doi.org/10.1080/19490976.2021.2021790 (2022).
Zhang, Y. et al. Metatranscriptomics for the human microbiome and microbial community functional profiling. Annu. Rev. Biomed. Data Sci. 4, 279–311 (2021).
Spurbeck, R. R., Catlin, L. A., Mukherjee, C., Smith, A. K. & Minard-Smith, A. Analysis of metatranscriptomic methods to enable wastewater-based biosurveillance of all infectious diseases. Front. Public Health https://doi.org/10.3389/fpubh.2023.1145275 (2023).
Aakko, J. et al. Data-independent acquisition mass spectrometry in metaproteomics of gut microbiota—implementation and computational analysis. J. Proteome Res. 19, 432–436 (2020).
Zhao, J. et al. Data-independent acquisition boosts quantitative metaproteomics for deep characterization of gut microbiota. npj Biofilms Microbiomes 9, 4 (2023).
Bonett, D. G. & Wright, T. A. Sample size requirements for estimating Pearson, Kendall and Spearman correlations. Psychometrika 65, 23–28 (2000).
Chase, L. S. S. & Priya, S. S. I’m walking into spiderwebs: making sense of protein−protein interaction data. J. Proteome Res. 23, 2723–2732 (2024).
Li, J. et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 32, 834–841 (2014).
Cheng, K. et al. MetaLab 2.0 enables accurate post-translational modifications profiling in metaproteomics. J. Am. Soc. Mass Spectrom. 31, 1473–1482 (2020).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016).
Yang, J. et al. DeepDigest: prediction of protein proteolytic digestion with deep learning. Anal. Chem. 93, 6094–6103 (2021).
Jesse, K. & Laurens van der, M. Rtsne: T-distributed stochastic neighbor embedding using a Barnes-hut implementation. https://doi.org/10.32614/CRAN.package.Rtsne (2023).
Leo, B., Adele, C., Andy, L. & Matthew, W. randomForest: Breiman and Cutlers random forests for classification and regression. https://doi.org/10.32614/CRAN.package.randomForest (2024).
Gábor, C. et al. Igraph: network analysis and visualization. https://doi.org/10.32614/CRAN.package.igraph (2025).
Bastian, M., Heymann, S. & Jacomy, M. Gephi: an open source software for exploring and manipulating networks. ICWSM 3, 361–362 (2009).
Deutsch, E. W. et al. The ProteomeXchange Consortium in 2020: enabling `big data' approaches in proteomics. Nucleic Acids Res 48, D1145–D1152 (2020).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Acknowledgements
Substantial financial support was provided by the Natural Sciences and Engineering Research Council of Canada (NSERC) through the Discovery Grant (to D.F.). Z.S. and Q.W. were funded by a stipend from the NSERC CREATE in Technologies for Microbiome Science and Engineering (TECHNOMISE) Program.
Author information
Authors and Affiliations
Contributions
Z.S. curated the data, conducted formal analysis, investigation, and visualization, and wrote the original draft. Z.N. contributed to conceptualization, methodology, investigation, and drafting of the manuscript. Q.W. contributed to methodology, investigation, and manuscript review and editing. L.L. provided resources and contributed to manuscript review and editing. A.D. contributed to methodology, investigation, and manuscript review and editing. D.F. supervised the study, acquired funding, contributed to conceptualization, and reviewed and edited the original draft. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
D.F. is a co-founder of Biotagenics and MedBiome, both of which are clinical microbiomics companies. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sun, Z., Ning, Z., Wu, Q. et al. Peptide abundance correlations in metaproteomics enhance taxonomic and functional analysis of the human gut microbiome. npj Biofilms Microbiomes 11, 166 (2025). https://doi.org/10.1038/s41522-025-00801-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41522-025-00801-y
