
Abstract
Five to ten percent of ER+ metastatic breast cancer (MBC) tumors harbor somatic PTEN mutations. Loss of function of this tumor-suppressor gene defines a highly aggressive, treatment-refractory disease for which new therapies are urgently needed. This Phase I multipart expansion study assessed oral capivasertib with fulvestrant in patients with PTEN-mutant ER+ MBC. Safety and tolerability were assessed by standard methods. Plasma and tumor were collected for NGS and immunohistochemistry analyses of PTEN protein expression. In 31 eligible patients (12 fulvestrant naive; 19 fulvestrant pretreated), the 24-week clinical benefit rate was 17% in fulvestrant-naive and 42% in fulvestrant-pretreated patients, with objective response rate of 8% and 21%, respectively. Non-functional PTEN was centrally confirmed in all cases by NGS or immunohistochemistry. Comutations occurred in PIK3CA (32%), with less ESR1 (10% vs 72%) and more TP53 (40% vs 28%) alterations in fulvestrant-naive versus fulvestrant-pretreated patients, respectively. PTEN was clonally dominant in most patients. Treatment-related grade ≥3 adverse events occurred in 32% of patients, most frequently diarrhea and maculopapular rash (both n = 2). In this clinical study, which selectively targeted the aggressive PTEN-mutant ER+ MBC, capivasertib plus fulvestrant was tolerable and clinically active. Phenotypic and genomic differences were apparent between fulvestrant-naive and -pretreated patients.
Trial registration number for the study is NCT01226316.
Similar content being viewed by others


Introduction
Breast cancer (BC) is the most frequently diagnosed malignancy and the leading cause of cancer mortality in women1, with estrogen-receptor-positive (ER+), human epidermal growth factor receptor 2 negative (HER2–) BC being the most common BC subtype2. Within this heterogeneous subtype, 5–10% harbor somatic mutations in PTEN, a frequently mutated tumor-suppressor gene in human cancer3,4. Phosphatase and tensin homolog (PTEN) functions as a negative regulator of the PI3K/AKT/PTEN pathway; therefore, its loss results in pathway activation that drives tumor growth5. Loss of function of PTEN is associated with poor prognosis in BC6,7 and the ER+ HER2– subtype (Fig. 1) and has been implicated in resistance to endocrine therapy and CDK4/6 and PI3Kα inhibitors8,9,10,11,12. The development of effective therapeutic strategies after progression on these agents remains an unmet need in metastatic BC (MBC). To that end, an understanding of both de novo and acquired driver tumor genomic alterations may unlock precision medicine approaches for patients with this disease.

a Early-stage ER+ HER2– breast cancers (METABRIC data)19. b Metastatic ER+ HER2– breast cancers (MSK-IMPACT data)12. Kaplan–Meier survival analysis for ER+ HER2– breast cancer patients by PTEN status in early-stage and metastatic BC: a overall survival of patients with ER+ HER2– primary breast tumors (n = 1398) by PTEN mutation status, using the same criteria employed in this study for enrollment; b overall survival from time of metastatic recurrence of patients with metastatic ER+ HER2– breast cancer (n = 949) by PTEN status. A patient with multiple metastatic samples sequenced by next-generation sequencing was considered PTEN altered if at least one sample harbored an eligible PTEN alteration. Overall survival for the METABRIC data utilized univariable or multivariable Cox proportional hazards models to examine the association between mutations and survival. Breast cancer-specific survival was used as the endpoint. Patients with deaths from other or unknown causes were censored at the date of death, and all other patients were censored at the date of last contact19. Overall survival for the MSK-IMPACT data, as defined by time of metastatic recurrence until death or last follow-up, was analyzed utilizing the MSK cohort12 restricted to patients with metastatic ER+/HER2– disease (n = 949 patients). Univariate P values were calculated using the log-rank test. The models were further adjusted using left truncation methods37 for late entry when tumor sequencing to assess PTEN status was performed after metastatic recurrence. ER+ estrogen-receptor-positive, HER2– human epidermal growth factor receptor negative.
Capivasertib (AZD5363) is an oral, potent, selective, ATP-competitive pan-AKT kinase inhibitor13,14,15. In a multipart Phase I study (ClinicalTrials.gov, NCT01226316), capivasertib monotherapy and, subsequently, combination therapy with fulvestrant was tested in a number of genomically selected expansion cohorts with expected PI3K/AKT/PTEN pathway activation14. Here we report the Phase I expansion cohort evaluating capivasertib and fulvestrant in PTEN-mutant, ER+ MBC patients. Study objectives were to confirm14,16 safety and tolerability and assess preliminary antitumor activity of the combination therapy in this patient population, and to describe exploratory genomic biomarker analyses of collected circulating tumor DNA (ctDNA) and tumor samples.
Results
Patient demographics and disease characteristics
In total, 32 patients with PTEN-mutant ER+ MBC were enrolled across eight sites in six countries, of whom 31 patients ultimately received treatment with capivasertib in combination with fulvestrant (n = 12 fulvestrant naive and n = 19 fulvestrant pretreated). Patient demographics and disease characteristics are shown in Table 1 and Supplementary Table 1. Mean age of study participants was 53 years (range 31–70). Most (90%) patients had visceral disease at enrollment and were heavily pretreated, with a median of 7 (range 1–13) prior therapies. Although the median number (7) of prior anticancer regimens was equivalent in the cohorts, fulvestrant-naive patients received more prior lines of chemotherapy (median 4 vs 2) and fewer lines of endocrine therapy (median 2 vs 4) compared with fulvestrant-pretreated patients. The slightly higher rates of visceral involvement (100% vs 84%) and progesterone-receptor-negative status (17% vs 5%) observed in the fulvestrant-naive versus fulvestrant-resistant patients may also support this observation. Patients in the fulvestrant-pretreated group were also more likely to have received prior mTOR inhibitors (48% vs 25%), CDK4/6 inhibitors (63% vs 8%) and PI3K inhibitors (11% vs 0%) than fulvestrant-naive patients (Table 1).
Efficacy analyses
At data cut-off (June 2019), three patients remained on study treatment, the majority having discontinued because of disease progression (n = 24; Supplementary Fig. 1). Response Evaluation Criteria in Solid Tumors (RECIST) response data are presented in Fig. 2 and summarized by prior fulvestrant exposure in Table 2. Of 30 patients with available RECIST data at baseline and at least one follow-up assessment, 17 (57%) demonstrated target lesion shrinkage (Fig. 2). Objective response rate (ORR) was 8% (1/12, 95% confidence interval [CI] 0–39) in fulvestrant-naive patients and 21% (4/19, 95% CI 6–46) in fulvestrant-pretreated patients; clinical benefit rate at 24 weeks (CBR24) was 17% (2/12, 95% CI 2–48) and 42% (8/19, 95% CI 20–67), respectively. Median duration of response (DOR) was 169 days (95% CI not calculable) in the fulvestrant-naive cohort (n = 1) and 210 days (95% CI 43–670) in the fulvestrant-pretreated cohort (n = 4). Median progression-free survival (PFS) was 2.7 months (95% CI 2–4) in all patients (n = 31): fulvestrant naive 2.6 months (95% CI 1–4), fulvestrant pretreated 4.1 months (95% CI 2–7).

AF allele fraction, ER+ estrogen-receptor-positive, FFPE formalin-fixed paraffin-embedded, IHC immunohistochemistry, MAF mutant allele fraction, MBC metastatic breast cancer, NGS next-generation sequencing, RECIST Response Evaluation Criteria in Solid Tumors.
Safety and therapy exposure
Median treatment duration (at data cut-off) was 103 days (range 22–740) overall: 102.5 days (24–410) in fulvestrant-naive patients and 103 days (22–740) in fulvestrant-pretreated patients. All patients experienced adverse events (AEs; Table 3), the most common of which were diarrhea (n = 21, 68%) and nausea (n = 14, 45%). Grade ≥3 AEs occurred in 17 patients (55%), most frequently maculopapular rash (n = 3, 10%), headache and diarrhea (both n = 2, 7%). Twelve grade ≥3 AEs occurring in 10 patients were considered causally related to capivasertib, of which both diarrhea and maculopapular rash were seen in two patients. Serious AEs (Supplementary Table 2) were reported in 11 patients (36%), 3 (10%) of which were considered causally related to capivasertib (malaise, nausea, vomiting). Capivasertib dose reduction and discontinuation was required because of diarrhea (n = 1, 3%) and drug hypersensitivity (n = 1, 3%), respectively. No treatment-related or AE-attributable deaths in either cohort were observed.
Exploratory biomarker analyses
Thirty-one patients were enrolled based on detection of an eligible PTEN alteration by local testing of tumor tissue (n = 29) or ctDNA (n = 2; Fig. 2). The majority of detected PTEN alterations led to a premature stop codon (21, 68%; frameshift, n = 14; stop gain, n = 5; splice, n = 2). Actionable missense mutations were detected in three (10%) patients, and a PTEN gene deletion was detected in the remaining seven (23%). Although numbers were very small, there was no apparent correlation between the type of PTEN alteration and clinical response during the study. Central next-generation sequencing (NGS) of plasma and/or tissue detected the PTEN alteration in 28 of the 30 (93%) cases with evaluable samples. Central immunohistochemistry (IHC; n = 25) showed complete loss of PTEN protein in 21 cases (84%), with various levels of PTEN expression in the remaining four evaluable samples. Overall, evidence of non-functional PTEN by central testing was confirmed in all patients by NGS or IHC.
Interestingly, two PTEN-positive cases by IHC had a C124S PTEN mutation by NGS, a dominant-negative form of PTEN reported to inhibit phosphatase activity17. Intriguingly, one of these patients had a complete response (CR) to study therapy (Fig. 2). The two other PTEN-positive cases may have been related to potential heterogeneous expression of the PTEN mutations; for one patient, central NGS analysis of the PTEN-positive primary breast tumor did not identify a PTEN alteration, whereas the central ctDNA analysis identified the same PTEN alteration as detected by local NGS analysis of a liver metastasis. For the other patient, low-intensity PTEN protein expression was identified in only a small subset of tumor cells (30%).
Broader genetic profiling by NGS of baseline ctDNA samples revealed co-occurring alterations in PIK3CA in 9/28 (32%) patients with sufficient tumor DNA for analysis (Fig. 2), a slightly greater rate than that (19–20%) reported by The Cancer Genome Atlas and METABRIC analyses18,19,20. Three of the four cases with a partial response (PR) had a co-occurring PIK3CA mutation, but the association of co-occurring PIK3CA mutations with PFS did not reach statistical significance (P = 0.15) in this small cohort. As expected, there was a higher prevalence of co-occurring ESR1 mutations in the fulvestrant-pretreated compared with the fulvestrant-naive cohort (72% vs 10%, respectively). In contrast, co-occurring TP53 mutations were more prevalent in the fulvestrant-naive than in the fulvestrant-pretreated cohort (40% vs 28%, respectively). No other mutations in the PI3K pathway were identified. A closer look at the comparison of the PTEN mutant allele fractions (MAFs) with the median of the co-occurring mutation MAFs in each patient showed that PTEN was detected at or above the median MAF in almost all patients, suggesting PTEN as the dominant driver mutation in those patients (Fig. 2).
Discussion
This Phase I expansion study reported on the safety and efficacy of the pan-AKT inhibitor, capivasertib, in combination with the ER antagonist, fulvestrant, in a genomically selected advanced ER+ BC population harboring an eligible deleterious PTEN gene alteration in the tumor. PTEN mutations select for an aggressive genomic subtype of ER+ BC, with associated resistance to standard-of-care therapies.
Capivasertib plus fulvestrant had an acceptable safety profile that was consistent with prior data16 and demonstrated antitumor activity in this heavily pretreated patient cohort (median 7 prior therapies), including in those previously treated with fulvestrant. Although efficacy appeared marginally better in fulvestrant-pretreated than in fulvestrant-naive patients, there were notable phenotypic and genomic differences between these patient cohorts. Specifically, at enrollment, fulvestrant-naive patients had more visceral disease, received less prior endocrine and more chemotherapy, and, likely reflective of this prior therapy receipt, had a lower ESR1- and higher TP53-mutation rate, which indeed could also be indicative of a more aggressive disease biology at baseline21. Overall, however, and given the poor prognostic genomic subgroup selected for this study, reasonable efficacy (ORR 21%; CBR24 42%) was seen in the fulvestrant-pretreated cohort. On the whole, this clinical dataset supports prior observations7,22 suggesting PTEN as a negative prognostic biomarker in BC, given the relatively short median PFS (2.7 [95% CI 2–4]) duration observed across the study population.
In this multicenter international study, local testing was reliable, with central retrospective confirmation of non-functional PTEN status achieved in all patients. Importantly, PTEN did appear to be the dominant driver tumor mutation in this study population. In addition, the trial adds support for enrollment in genomically selected studies to be based primarily on local testing, thus avoiding delays to study accruals from the impact of central testing, particularly in early-phase signal-seeking studies such as these.
The incorporation of mTOR and CDK4/6 inhibitors into endocrine therapy has led to substantial improvements in patient outcomes23,24,25,26,27. Almost half of our PTEN-mutant study population had received prior CDK4/6 inhibitor therapy. This is of particular interest given recent data proposing PTEN inactivation as a mechanism of resistance to this therapeutic class as well as PI3Kα-selective inhibitors, lending support to direct AKT inhibitors in this setting9,11,28.
Notably, in the Phase I/II randomized FAKTION study, which demonstrated a PFS benefit with the addition of capivasertib to fulvestrant in a molecularly unselected, aromatase-inhibitor-resistant but fulvestrant-naive ER+ MBC population, a subgroup of patients with PIK3CA mutation (by digital droplet polymerase chain reaction) and/or PTEN loss (by IHC) did not appear to have any greater sensitivity to the combination than those without the predefined alterations. Importantly, however, no FAKTION participants had received previous CDK4/6 inhibitor therapy, and the rate of AKT1 and PTEN mutations in that study has not yet been reported29.
Our study had several important limitations, including the trial not being formally powered to compare efficacy across fulvestrant-naive and -pretreated cohorts, as well as the small patient numbers. It is also noteworthy that the fulvestrant-naive patients enrolled may have had a more aggressive disease phenotype and a poorer prognosis than the fulvestrant-pretreated cohort, although the sample size limited any formal comparisons. At the planned interim analysis, the fulvestrant-naive cohort did not meet its target value for CBR24, so recruitment was halted, resulting in a cohort of only 12 patients. Furthermore, the rarity of this biomarker led to slow accrual in the fulvestrant-pretreated cohort (approximately 29 months), with the result that this cohort was closed before reaching the target of 24 patients.
In conclusion, this study shows that capivasertib in combination with fulvestrant has clinical activity in heavily pretreated PTEN-mutant ER+ MBC patients, a poor prognostic BC subtype. PTEN was the dominant driver tumor mutation in these patients. Further analyses of patients pretreated with a CDK4/6 inhibitor in the ongoing Phase III study CAPItello-291 (NCT04305496), which is evaluating combination capivasertib with fulvestrant, will definitively inform PTEN’s role as a therapeutic target in BC. In the Phase I study reported here, this aggressive disease entity appeared to display unique biology with at least a subset dependent on AKT and ER for proliferation, an observation that may benefit therapeutically from using an AKT inhibitor combination.
Methods
Study design and participants
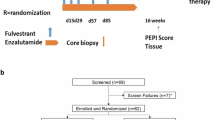
This was a Phase I, dose- and schedule-finding study of capivasertib with multiple expansion cohorts that included evaluation of capivasertib with fulvestrant (NCT01226316). Results from the earlier parts of the study presenting the dose-finding, recommended Phase II dose and pharmacodynamic evaluation, as well as efficacy in patients with advanced solid tumors and those with activating PIK3CA30 or AKT1 mutations, have previously been reported14,16. The final part of this study enrolled PTEN-mutant ER+ MBC patients into two subcohorts: fulvestrant naive and fulvestrant pretreated (with a maximum of 24 patients per cohort).
Inclusion criteria
Eligible patients ≥18 years old had histological or cytological confirmation of ER+ advanced or MBC refractory to standard therapies and confirmation of an eligible PTEN alteration in tumor tissue by local testing. Eligible PTEN alterations were defined as a deleterious mutation in PTEN or copy loss of the PTEN gene31. Further inclusion criteria were measurable disease by RECIST v1.1, WHO performance status 0–1, and minimum life expectancy of 12 weeks. Key exclusion criteria included active central nervous system metastases, prior treatment with catalytic AKT inhibitors (prior exposure to all other agents in the PI3K/AKT/mTOR pathway, including allosteric AKT inhibitors, was allowed), and clinically significant abnormalities of glucose metabolism [defined by any of the following criteria: i. diagnosis of diabetes mellitus type I or II (irrespective of management); ii. baseline fasting glucose value of ≥7 mmol/L (fasting is defined as no calorific intake for at least 8 h); and iii. glycated hemoglobin >8% (>64 mmol/mol)].
All patients provided written informed consent, the institutional review boards or independent ethics committees of all investigational sites approved the protocol, and the study was performed in accordance with the Declaration of Helsinki, Good Clinical Practice, and the AstraZeneca policy on bioethics32.
Procedures
Patients were treated with oral capivasertib 400 mg twice daily, 4 days on followed by 3 days off, weekly (cycle length: 21 days), and fulvestrant at the labeled dose, in accordance with the previously established recommended combination dose29. Antitumor activity was assessed by computed tomography or magnetic resonance imaging (RECIST v1.1) every 6 weeks for 24 weeks, then every 12 weeks. Safety was assessed throughout the study period and until day 28 after discontinuation of study treatment, according to the National Cancer Institute Common Terminology Criteria for Adverse Events v4.0. AEs were coded with the Medical Dictionary for Regulatory Activities v19.1.
For study enrollment, PTEN mutation (with known functional or therapeutic significance as described in Carr et al.33) status was determined in tissue/plasma by local NGS and involved a variety of assays in accordance with local standard practice without any specific threshold for positivity. PTEN status was also centrally evaluated in tumor tissue from either primary or metastatic disease (as indicated in Fig. 2) by FoundationOne34 testing, by IHC analyses of PTEN protein expression (using the CST138G6 PTEN antibody assay with Hscore ≤10 classified as PTEN protein deficient)35, and in ctDNA by using a hybrid capture-based panel covering 600 genes (AZ600) and low-pass whole-genome sequencing for cases with a PTEN gene deletion reported by the local test. PTEN was considered clonal if the PTEN MAF was greater than the median MAF of the remaining mutations identified in the plasma sample with MAF > 1%. These central analyses were conducted retrospectively.
Outcomes
Safety and tolerability were assessed by continual monitoring of AEs. Efficacy outcomes included: ORR, defined as a confirmed PR or CR; DOR, defined as the time from first objective response to disease progression or death (or censoring if neither outcome is observed); PFS, defined as the time from the first day of treatment to disease progression or death; and CBR24, defined as confirmed disease response (PR or CR) or stabilization for ≥24 weeks. Responses were investigator assessed in accordance with RECIST v1.1 and required a confirmatory scan. Exploratory biomarker analyses included mutation analysis of baseline tissue and ctDNA plasma samples (by NGS), along with analysis of PTEN protein expression in baseline tumor tissue (by IHC).
Statistical analysis
Although the primary endpoint throughout this multipart Phase I study remained safety and tolerability, the sample size of the Phase I expansion cohort reported here was determined with the aim of detecting a signal of efficacy, should one exist, using CBR24. The sample size was determined based on prespecified CBR24 target values of 65% and 40% for fulvestrant-naive and fulvestrant-pretreated patients, respectively. With 24 patients per cohort (Fig. 3), there would be a 90% chance of at least 13 and 7 clinical benefit responses, respectively. At the planned interim analysis, the fulvestrant-naive cohort did not meet its target (CBR24 of 65%), and recruitment was halted. The fulvestrant-pretreated cohort did meet its predefined boundary (CBR24 of 40%), and recruitment continued. However, subsequent to this and owing to the difficulty in recruiting this rare patient population, this latter cohort was also closed before the target of 24 patients was reached. It was considered that sufficient data were available from the 19 patients dosed in this fulvestrant-pretreated cohort to allow a reasonable chance of assessing any signal of efficacy. Final analyses were conducted when all patients had the opportunity to reach 24 weeks of treatment for assessment of CBR24. DOR and PFS were analyzed by Kaplan–Meier plots, and patients without a progression event at the analysis date were censored at the last known RECIST assessment. Exploratory biomarker analyses investigated the association between mutations and radiographic response. All analyses were conducted with SAS v9.04.

Up to 24 patients in each cohort. Interim analyses were carried out after 12 patients were followed up for 24 weeks or withdrawn from the study. Subsequent patients were recruited only if sufficient antitumor activity (assessed by CBR24) was observed at the interim analyses. CBR24 clinical benefit rate at 24 weeks, ER+ estrogen-receptor-positive, ORR objective response rate, PFS progression-free survival.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The raw sequencing data are not publicly available because of data privacy regulations and restrictions for use of such data, as stated in the study protocol and patient consent form. Individual-level data can potentially be accessed via a collaborative agreement with AstraZeneca Group. The authors declare that the clinical dataset analyzed here, including PFS and tumor response data, is available and may be obtained in accordance with AstraZeneca’s data sharing policy as part of an external collaborative request (https://astrazenecagroup-dt.pharmacm.com//DT/Home/Index/) or an external data access request (https://vivli.org/ourmember/astrazeneca/). The data generated and analyzed during this study are described in the following metadata record: https://doi.org/10.6084/m9.figshare.1419234536.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Gong, Y., Liu, Y. R., Ji, P., Hu, X. & Shao, Z. M. Impact of molecular subtypes on metastatic breast cancer patients: a SEER population-based study. Sci. Rep. 7, 45411 (2017).
Salmena, L., Carracedo, A. & Pandolfi, P. P. Tenets of PTEN tumor suppression. Cell 133, 403–414 (2008).
Bertucci, F. et al. Genomic characterization of metastatic breast cancers. Nature 569, 560–564 (2019).
Bose, S., Wang, S. I., Terry, M. B., Hibshoosh, H. & Parsons, R. Allelic loss of chromosome 10q23 is associated with tumor progression in breast carcinomas. Oncogene 17, 123–127 (1998).
Carbognin, L., Miglietta, F., Paris, I. & Dieci, M. V. Prognostic and predictive implications of PTEN in breast cancer: unfulfilled promises but intriguing perspectives. Cancers (Basel) 11, 1401 (2019).
Li, S. et al. Loss of PTEN expression in breast cancer: association with clinicopathological characteristics and prognosis. Oncotarget 8, 32043–32054 (2017).
Razavi, P. et al. Molecular profiling of ER+ metastatic breast cancers to reveal association of genomic alterations with acquired resistance to CDK4/6 inhibitors. J. Clin. Oncol. 37(15 Suppl), abst 1009, https://doi.org/10.1200/JCO.2019.37.15_suppl.1009 (2019).
Costa, C. et al. PTEN loss mediates clinical cross-resistance to CDK4/6 and PI3Kα inhibitors in breast cancer. Cancer Discov. 10, 72–85 (2020).
Fu, X. et al. Overcoming endocrine resistance due to reduced PTEN levels in estrogen receptor-positive breast cancer by co-targeting mammalian target of rapamycin, protein kinase B, or mitogen-activated protein kinase kinase. Breast Cancer Res. 16, 430 (2014).
Juric, D. et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 518, 240–244 (2015).
Razavi, P. et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 34, 427–438.e426 (2018).
Davies, B. R. et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol. Cancer Ther. 11, 873–887 (2012).
Hyman, D. M. et al. AKT inhibition in solid tumors with AKT1 mutations. J. Clin. Oncol. 35, 2251–2259 (2017).
Kalinsky, K. et al. AZD5363 in patients (pts) with tumors with AKT mutations: NCI-MATCH subprotocol EAY131-Y, a trial of the ECOG-ACRIN Cancer Research Group (EAY131-Y). Eur. J. Cancer 103(Suppl 1), e15 (2018).
Smyth, L. M. et al. Capivasertib, an AKT kinase inhibitor, as monotherapy or in combination with fulvestrant in patients with AKT1E17K-mutant, ER-positive metastatic breast cancer. Clin. Cancer Res. 26, 3947–3957 (2020).
Papa, A. et al. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell 157, 595–610 (2014).
Curtis, C. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012).
Pereira, B. et al. The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun. 7, 11479 (2016).
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Meric-Bernstam, F. et al. Survival outcomes by TP53 mutation status in metastatic breast cancer. JCO Precis. Oncol. 2018, PO.17.00245 (2018).
Lebok, P. et al. Partial PTEN deletion is linked to poor prognosis in breast cancer. BMC Cancer 15, 963 (2015).
Baselga, J. et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 366, 520–529 (2012).
Goetz, M. P. et al. MONARCH 3: abemaciclib as initial therapy for advanced breast cancer. J. Clin. Oncol. 35, 3638–3646 (2017).
Im, S. A. et al. Overall survival with ribociclib plus endocrine therapy in breast cancer. N. Engl. J. Med. 381, 307–316 (2019).
Turner, N. C. et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N. Engl. J. Med. 379, 1926–1936 (2018).
Finn, R. S. et al. Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 375, 1925–1936 (2016).
Razavi, P., Dickler, M. N. & Chandarlapaty, S. Alterations in PTEN and ESR1 promote clinical resistance to alpelisib plus aromatase inhibitors. Nat. Cancer 1, 382–393 (2020).
Jones, R. H. et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive breast cancer (FAKTION): a multicentre, randomised, controlled, Phase 2 trial. Lancet Oncol. 21, 345–357 (2020).
Banerji, U. et al. A Phase I open-label study to identify a dosing regimen of the pan-AKT inhibitor AZD5363 for evaluation in solid tumors and in PIK3CA-mutated breast and gynecologic cancers. Clin. Cancer Res. 24, 2050–2059 (2018).
Schmid, P. et al. Capivasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer: the PAKT trial. J. Clin. Oncol. 38, 423–433 (2020).
AstraZeneca. Global standard: bioethics. https://www.astrazeneca.com/content/dam/az/Sustainability/2018/Bioethics%20Policy%20final.pdf.
Carr, T. H. et al. Defining actionable mutations for oncology therapeutic development. Nat. Rev. Cancer 16, 319–329 (2016).
Foundation Medicine. FoundationOne® CDx. (2019). https://www.foundationmedicine.com/genomic-testing/foundation-one-cdx.
Ferraldeschi, R. et al. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur. Urol. 67, 795–802 (2015).
Smyth, L. M. et al. Metadata record for the article: Selective AKT kinase inhibitor capivasertib in combination with fulvestrant in PTEN-mutant ER-positive metastative breast cancer. https://doi.org/10.6084/m9.figshare.14192345 (2021).
Kalbfleisch, J. D. & Prentice, R. The Statistical Analysis of Failure Time Data. 2nd edn, Vol. 77 (John Wiley & Sons, Inc., 2002).
Acknowledgments
This work was supported by AstraZeneca. Capivasertib was discovered by AstraZeneca subsequent to a collaboration with Astex Therapeutics (and its collaboration with the Institute of Cancer Research and Cancer Research Technology Limited). U. B. acknowledges infrastructural funding from Cancer Research UK, Experimental Cancer Medicine Centre and Biomedical Research Centre grants, in addition to a National Institutes of Health Research Professorship award (RP-2016-07-028). All Memorial Sloan Kettering Cancer Center investigators wish to acknowledge the support of the NCI Cancer Center Support Grant (CCSG P30 CA08748). S. C. acknowledges support of the Breast Cancer Research Foundation. We thank the patients and their caregivers who participated in this study. Medical writing assistance was provided by Martin Goulding, DPhil, from Mudskipper Business Ltd, funded by AstraZeneca. Capivasertib (AZD5363) is an investigational medical product that is not currently approved.
Author information
Authors and Affiliations
Contributions
L. M. S., T. H. C., E. C. d. B., A. F., H. A., J. P. O. L., G. S., and D. M. H. contributed to the study design. H. A., T. H. C., E. C. d. B., C. S.-S., A. F., J. H., J. P. O. L., R. M., M. M., M N., G. S., P. R., and S. C. analyzed and interpreted the data. R. M. performed statistical analysis. L. M. S., G. B., F. M.-B., P. K., I. S., A. L., K. J., A. V., A. W., A. M. S., U. B., J. B., and D. M. H. recruited patients, collected samples, and assisted with data interpretation. L. M. S. wrote the manuscript. All authors made substantial contributions to the critical revision of the manuscript, and all authors provided their final approval of the manuscript.
Corresponding author
Ethics declarations
Competing interests
L. M. S. has acted in a consultancy or advisory role for AstraZeneca, Loxo Oncology at Lilly, Novartis, Pfizer, and Roche Genentech and has received research funding from AstraZeneca, Puma Biotechnology, and Roche Genentech, travel or accommodation expenses from Pfizer, Puma Biotechnology, and Roche Genentech, honoraria from AstraZeneca, Pfizer, and Roche Genentech, employment from Loxo Oncology at Lilly, and stock and other ownership interests from Lilly. G. B. has acted in a consultancy or advisory role for Roche. F. M.-B. has received commercial research grants from Abbvie, Aileron, AstraZeneca, Bayer, Calithera, Curis, CytoMx, Daiichi Sankyo, eFFECTOR, GlaxoSmithKline, Guardant Health, Jounce, Millennium, Novartis, PUMA Biotechnology, Seattle Genetics, Takeda, and Zymeworks, grants and travel-related fees from Debiopharm Group, Genentech, Pfizer, and Taiho, has acted in a consultancy role for Aduro, Dialectica, Jackson Laboratory, Kolon Life Science, OrigiMed, Parexel International, Pieris, Samsung Bioepis, Sumitomo Dainippon, Xencor, and Zymeworks, and in an advisory role for Darwin Health, GRAIL, Inflection Biosciences, Mersana, Seattle Genetics, and Spectrum. P. K. has performed contracted research for AstraZeneca, Eli Lilly, Genentech, Pfizer, Radius Health, and Sanofi. I. S. has received research funding from PUMA Biotechnology and travel expenses from BMS, Novartis, and Pfizer. K. J. has reported consultant and advisory board activities for Novartis, Spectrum Pharmaceuticals, ADC Therapeutics, Pfizer, Bristol-Myers Squibb, Jounce Therapeutics, and Taiho Oncology and research funding from Novartis, Clovis Oncology, Genentech, AstraZeneca, ADC Therapeutics, Novita Pharmaceuticals, Debio Pharmaceuticals, and Pfizer. A. V. has received research grants from AstraZeneca, BMS, Boehringer Ingelheim, Janssen Cilag, Merck, Novartis, Pfizer, Roche, and Sanofi and non-financial support from AstraZeneca, Bayer, BMS, Boehringer Ingelheim, Johnson & Johnson, Lilly, Medimmune, Merck, NH TherAGuiX, Pfizer, and Roche. P. R. has performed consultation and attended advisory boards for Novartis and received institutional research funding from Grail, Inc and Illumina. U. B. has received research grants from AstraZeneca, Chugai, and Onyx Pharmaceuticals and consultancy fees from Astex and Novartis. D. M. H. reports stock ownership in Fount Therapeutics, has acted in a consultancy or advisory role for AstraZeneca, Bayer, Boehringer Ingelheim, Chugai Pharma, Eli Lilly, Genentech, and Pfizer, and has received research funding from AstraZeneca, Bayer, Loxo Oncology, and Puma Biotechnology and travel or accommodation expenses from Chugai Pharma and Genentech. S. C. has received a research grant from Daiichi Sankyo and consultancy fees from BMS, Context Therapeutics, Eli Lilly, Novartis, Revolution Medicines, and Sermonix Pharmaceutical. H. A., T. H. C., E. C. d. B., C. S.-S., A. F., J. H., J. P. O. L., R. Maudsley, R. McEwen, M. M., M. N., G. S., and J. B. are employees of AstraZeneca. A. L., A. W., and A. M. S. declare no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Smyth, L.M., Batist, G., Meric-Bernstam, F. et al. Selective AKT kinase inhibitor capivasertib in combination with fulvestrant in PTEN-mutant ER-positive metastatic breast cancer. npj Breast Cancer 7, 44 (2021). https://doi.org/10.1038/s41523-021-00251-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41523-021-00251-7
This article is cited by
-
Evaluation of quantitative polymerase chain reaction for detecting BRCA1 or BRCA2 copy number loss in high-grade serous ovarian cancer
Scientific Reports (2026)
-
PTEN inactivating mutations are associated with hormone receptor loss during breast cancer recurrence
Breast Cancer Research and Treatment (2025)
-
Evolving treatment paradigms after CDK4/6 inhibitors in advanced breast cancer
memo - Magazine of European Medical Oncology (2025)
-
AKT kinases as therapeutic targets
Journal of Experimental & Clinical Cancer Research (2024)
-
Capivasertib: First Approval
Drugs (2024)
