
Abstract
The time of day of administration (chronotherapy) of certain medications can affect both their toxicity and efficacy. In this pragmatic, multicenter trial, women starting adjuvant endocrine therapy (ET) for hormone receptor-positive early-stage breast cancer were randomized (1:1) to either morning or evening administration. The primary endpoint was endocrine toxicity/tolerability measured by the change in total Functional Assessment of Cancer Therapy-Endocrine Subscale (FACT-ES) score from baseline to 12-weeks. Secondary endpoints included: endocrine toxicity/tolerability and quality of life (FACT-ES and FACT-B) from baseline to 4, 8, 12, and 52 weeks, non-persistence or non-adherence, and patient preference for timing of ET. Between June 30, 2021, and March 18, 2022, 245 eligible participants were randomized to either morning (122/245, 49.8%) or evening ET (123/245, 50.2%). In the overall population, there was no statistical difference in the change in total FACT-ES score from baseline to 12 weeks (p = 0.086). There were no statistically significant differences for any of the secondary endpoints between the two groups. The study provides evidence for the enthusiasm of patients and investigators to take part in chronotherapy studies. Additional prospective studies should be performed to assess how the timing of ET affects survival outcomes to ensure optimal patient care. Trial Registration: ClinicalTrials.gov, NCT04864405.
Similar content being viewed by others


Introduction
Despite the proven benefits of adjuvant endocrine therapy (ET) for patients with early breast cancer (EBC), ET adherence rates (defined as taking the medication at the prescribed dosage and frequency) in patients vary from 41 to 72% and, non-persistence rates (defined as rates of discontinuation of treatment that has been started), from 31 to 73%1. This is a major public health issue as non-adherence and non-persistence are associated with reduced disease-free survival and increased mortality2,3. A common cause of non-adherence and non-persistence is drug-related side effects4. ET can cause a range of side effects including hot flashes, joint and muscle pain, sleep disturbance, weight gain, memory or mood changes and vaginal symptoms. While adjunct medications are often prescribed to reduce these side effects, there is little evidence to show that they have a discernible impact on long-term adherence to ET5,6,7. Hence, there is a need to find simple interventions that will both reduce side effects and improve adherence to ET globally.
The concept that the timing of administration of medication in relation to the circadian rhythm can influence side effects and efficacy has led to the field of chronotherapy8. Timing of treatment administration has been shown to influence outcomes in many fields of medicine9,10. For instance, bedtime administration of anti-hypertensive treatment improves blood pressure control and reduces cardiovascular risk as compared to morning administration11. Some studies suggest that adherence is greater when medication is taken in the morning as opposed to the evening12. In oncology, clinical trials involving cytotoxic agents and immune checkpoint inhibitors have demonstrated a differential dose response and toxicity profile associated with time of administration9,13,14. In women with breast cancer (BC), it was shown that the generation of breast cancer circulating tumor cells highly prone to metastasize accelerates during sleep, and molecular evidence suggests an interplay between circulating estradiol and the expression of circadian clock genes15,16,17.
Anecdotal reports suggest that the timing of ET administration might influence side effects, and choosing the optimal time of day might offer a practical strategy to improve tolerability and adherence18,19. The best time of the day to take ET is a recurrent question from patients, we therefore designed the current trial to compare the effects of morning versus evening dosing of ET on a range of patient-reported outcomes (PROs).
Results
Participant characteristics
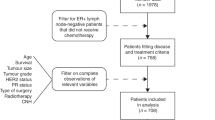
Between June 30, 2021, and March 18, 2022, 261 patients were approached, and 245 were eligible and consented to participate (Supplementary Fig. 1. Accrual rate). Of these 245 patients, 122 (49.8%) were randomized to an ET morning dose and 123 (50.2%) to an ET evening dose (Fig. 1. CONSORT Diagram). Patients baseline characteristics are presented in Table 1 and were well balanced between the two arms. All but one participant were women, and their median age was 61 (sd 12); 56 (23%) were pre-menopausal, 7 (3%) were perimenopausal (defined as the presence of irregular menstrual cycles due to declining ovarian function and up to a year after the last menstrual period), and 181 (74%) were post-menopausal. One participant was male. Chemotherapy was received prior to starting ET in 87 (35.5%) of participants. The planned ET type was: 187 (76%) tamoxifen, 42 (17%) anastrozole, and 16 (7%) letrozole. Of the 63 pre/peri-menopausal women, 22 received concurrent LHRH therapy. Of 201 patients who received radiotherapy, ET was started before or during radiotherapy for 49 (24%) patients and after radiotherapy for 152 (76%) patients.

CONSORT Diagram.
Health-related quality of life
Absolute FACT-ES scores are presented at each time point in Supplementary Table 1 and change in FACT-ES scores from baseline are presented in Supplementary Table 2. The mean (sd) for morning/evening dose patients were 148.0 (19.5)/144.7 (20.7) at baseline, 147.7 (21.5)/144.3 (22.2) at week 4, 146.2 (23.3)/142.8 (23.0) at week 8, 146.5 (22.6)/139.9 (24.1) at week 12, and 145.8 (22.7)/139.3 (23.4) at week 52. The differences in absolute scores were significant on univariate analysis for week 12 (p = 0.025) and week 52 (p = 0.041), but not at other time points, nor for any change score from baseline.
For the primary analysis using the mITT population, evaluating 12-week FACT-ES score adjusted for baseline FACT-ES and stratum, the effect of intervention (defined as morning vs evening dosing) was not statistically significant (p = 0.086) (Fig. 2). Results were similar in all supportive analyses when using the PP population (p = 0.081), or if the outcome was defined as the change from baseline to week 12 FACT-ES (p-value = 0.11 for both mITT and PP), or if a repeated measures mixed model was used to evaluate FACT-ES over time (p-value = 0.14 for intervention). Post hoc subgroup analyses found similar results based on use of tamoxifen or an aromatase inhibitor, however, women who were pre- or peri-menopausal had a statistically significant difference for intervention (p = 0.018). Among pre- and peri-menopausal women who had the morning dose, the week 12 FACT-ES score was a mean (sd) of 138.1 (25.9) for 30 women compared with 124.1 (22.7) for 31 women who were allocated to the evening dose. The 5-level EQ-5D (EQ-5D-5L) scores at baseline to 4 weeks, 8 weeks, 12 weeks, and 52 weeks for morning and evening arms are presented in Supplementary Table 3.

• Within group: negative mean change indicates deterioration from baseline. • Between groups: mean change favors morning ET dose. • Error bars denote standard deviation.
Adherence and discontinuation
Table 2 summarizes data collected regarding adherence to the assigned intervention. There was no statistical difference between the arms. At 12 weeks, 87% and 89% (p = 0.84) in the morning and in the evening arms respectively, were fully adherent. At 52 weeks, the rate of full adherence dropped to 79% in the morning arm and 76% in the evening arm (p = 0.63). Also, we evaluated the association between higher stage and adherence and found no statistically significant difference by stage, although there was a slight trend of higher adherence with higher stage (p-values 0.53 and 0.49 at 12 weeks and 52 weeks, respectively).
Time of ET administration
In the morning arm, the average time that patients took their ET was 8:15 AM (range 5 to 11 AM) and in the evening arm, the average time of administration was 8:15 PM (range 4 PM to midnight). (Supplementary Fig. 2).
Patient preference for the timing of ET administration
Patient preference regarding dose timing at baseline (after randomization) and 52 weeks is presented in Supplementary Table 4. At the beginning of the study, 50% (55/110) of patients randomized to the morning dose arm compared with 10% (11/111) randomized to evening dosing, strongly preferred the idea of morning dosing. Whereas 15% (16/110) of patients randomized to the morning dose arm compared with 34% (38/111) randomized to the evening dosing strongly preferred the concept of an ET evening dose. At week 52, 57% (60/110) in the morning arm vs 14% (14/111) in the evening arm strongly preferred an ET morning dose, whereas 8% (8/110) vs 42% (44/111) strongly preferred an ET evening dose. No significant preference changes were observed in patient-rated preference to time of administration from baseline to week 52 (p-value = 0.081).
Discussion
Notwithstanding the proven benefit of oral ET agents in treating EBC, major challenges in tolerability and adherence limit their large-scale impact. While multiple interventions, including adjunct medications, novel technologies (e.g., smart pill bottles, mobile applications), text communications, and verbal communications have demonstrated enhanced short-term adherence to ET there are no data indicating that these measures improve tolerability/quality of life, long-term adherence (>1 year) or persistence7. Seeking interventions to improve both tolerability and adherence to ET are critical to the global improvement of breast cancer survivorship. Based on evidence that circadian rhythms play a role in both tumorigenesis and effects of anti-cancer treatment14,15,19,20,21,22,23, we hypothesized that optimizing the time of the day at which ET is taken might offer a new practical strategy to improve EBC outcomes.
In this pragmatic, multicenter randomized, open-label, controlled trial comparing morning vs evening dosing of ET in patients with EBC, we found no difference in tolerability and adherence between the morning vs. the evening dose of ET. A subgroup analysis suggested a difference in the primary outcome in favour of morning administration for pre- and peri-menopausal women, but this is based on a post hoc subgroup analysis of a small number of patients and may be due to statistical variation. It is known that circadian rhythm could be dysregulated in cancer patients and in older people14,24. The difference observed based on menopausal status could potentially be explained by a greater influence of the circadian rhythm in a younger population, and additional studies should be performed to confirm this finding.
The main strengths of the study were its pragmatic design and patient-centered outcomes. Thus, our results are applicable and relevant to real-world practice. The limitations of the study included its small sample size, limiting future definitive efficacy endpoints results (e.g., disease-free survival and overall survival), and the lack of pre-planned analysis for specific subgroups (e.g., pre/peri-menopausal women, age). We cannot exclude that the absence of an effect of the time of intake on tolerability is due to the modest sample size of our study population, but we can certainly exclude a major difference in outcome. Thus, it is safe to allow women to take their ET at whichever time they prefer. Another potential limitation could be that many more postmenopausal women received upfront tamoxifen (76%) in a switch strategy context as opposed to an aromatase inhibitor (24%). This is based on the results of large RCTs and meta-analyses that show no difference in survival outcomes for post-menopause patients treated with the switch strategy versus an aromatase inhibitor alone25. Furthermore, adherence in this study appears higher than previously reported, this is likely due to frequent patient contact by the study team, which increased motivation, and the use of a patient questionnaire that may have underestimated nonadherence and treatment discontinuation.
The results of the current study are meaningful and of importance from a number of standpoints. This is the first and only report on a clinical trial that randomized patients with EBC to a morning dose or an evening dose of ET. The rapid rate of accrual demonstrates both patient and clinician enthusiasm for the study (Supplementary material S1). Furthermore, despite the patient’s own time preference for ET administration, more than 85% adhered to the assigned study arm, which supports the feasibility of chronotherapy trials in this population. While the best time of the day to take ET is an important and frequently asked question by patients to oncologists, there is a paucity of studies evaluating the optimal time of the day to take ET18,19. On this subject, the only other prospective study that we are aware of is the ancillary study of the UNIRAD trial23. In this study, ET timing was recorded for patients with high-risk HR + HER2- BC randomized to adjuvant ET with placebo or everolimus. Importantly, the timing of administering treatment was not randomized in this study and was based on patient and investigator choice. In the whole study population, the timing of ET intake was not associated with disease-free survival after a median of 42 months follow-up. However, the analysis according to the stratification factors revealed interaction between ET timing and ET agent (tamoxifen vs aromatase inhibitor), suggesting that tamoxifen intake in the evening or at night might be associated with a better DFS compared to morning or afternoon intakes. Altogether, the accrual success and the results of the REaCT-CHRONO study, as well as the hypothesis-generating research of Giachetti et al. lend support to a large, randomized study evaluating the optimal time of the day to take ET with efficacy and tolerability endpoints.
In conclusion, no significant differences in quality of life or adherence related to ET administration were demonstrated. The REaCT-CHRONO study provides evidence for the enthusiasm of patients to take part in chronotherapy prospective clinical trials. Further studies should be performed to evaluate the time-of-day effects of taking ET on endpoints such as disease-free and overall-survival, with a special focus on the pre/perimenopausal population.
Methods
Study design
The REaCT-CHRONO study is a pragmatic, multicenter, open-label, randomized trial. The protocol and procedures of this clinical trial were in compliance with the ethical standards of the Ontario Cancer Research Ethic Board (CTO 3594 approved April 2021), the 1964 Helsinki declaration and its later amendments, and with ethical standards of the institutions involved. This trial is registered with NCI ClinicalTrials.gov (NCT04864405) and the 3CTN trial portfolio in year 8 (April-Sept, 2021). The study report was prepared following the CONSORT statement- extension for pragmatic trials26.
Participants
The study was conducted in 2 Canadian cancer centers: the Ottawa Hospital Cancer Centre and Thunder Bay Regional Health Sciences Centre. All participants provided integrated verbal consent. Patients with hormone receptor-positive (HR + ) EBC who were planned to start adjuvant ET were eligible. Other inclusion criteria were: age 18 years or older, able to provide informed oral consent, and be willing and able to complete questionnaires as per study protocol. Exclusion criteria were: metastatic breast cancer, previous endocrine therapy for breast cancer, and planned to receive adjuvant abemaciclib. All patients provided oral consent using the integrated consent model.
Study procedures and endpoints
Eligible and consented women were randomized in a 1:1 ratio to either: an ET morning dose (Arm A: within 1 hour of the patient wake-up time) or an ET evening dose (Arm B: within 1 hour of patient bedtime). For randomization, a permuted block design with variable block sizes of 4 and 6 developed by the Ottawa Methods Centre was used and patients were stratified by center, type of endocrine therapy (tamoxifen yes/no), and if prior chemotherapy had been received (yes/no). Participants and investigators were not blinded to their treatment allocation. The type of ET, as well as the timing relative to radiation therapy (if given), was left to the discretion of the patient and the treating physician.
The primary endpoint was endocrine therapy tolerability measured by the change in total Functional Assessment of Cancer Therapy-Endocrine Subscale (FACT-ES) score from baseline to 12 weeks following the beginning of ET. This endpoint is patient-centered, clinically meaningful, and validated in women treated with ET27,28,29,30. The 12 week-time point coincides with a high incidence of endocrine symptoms27,28. Secondary endpoints included: endocrine toxicity/tolerability and quality of life measured respectively by the change in total score and individual items of FACT-ES and Functional Assessment of Cancer Therapy for patients with a Breast cancer (FACT-B) from baseline to 4, 8, 12, and 52 weeks following the beginning of ET. Other endpoints included rates of non-persistence (defined as rates of discontinuation of treatment that has been started) and rates of non-adherence (defined as one of the 3: not taking ET, ET interruption of ≥7 days or change in ET type).
Endpoint data were collected from patient-completed questionnaires on the secure online patient portal or on paper, if the patient preferred. The first portion of the FACT-ES and FACT-B questionnaires is the same, study participants completed the duplicate sections once at each time point. In addition, participants also completed a questionnaire on their preference for the time of taking their ET at baseline and 52 weeks. Women were evaluated at their usual clinic visits for study-related events, including adherence to dose timing, discontinuation of endocrine therapy, or switching from one type of ET to another. Additional study data were also obtained through review of the patient’s electronic medical record and emails to the treating physician at 12 weeks and 52 weeks. As this was a pragmatic trial, no additional study-mandated visits were required31.
Statistical approach
It was hypothesized that the dose timing (morning vs evening) of ET would impact toxicity and tolerability at 12 weeks compared to baseline using the FACT-ES. A ‘clinically meaningful change’ from baseline to 12 weeks was defined as a change of 4.25 or greater, based on Fallowfield et al., which showed that a clinically meaningful effect is observed if the change exceeded 0.5 of the standard deviation at baseline and the standard deviation at baseline was approximately 8.527. It was also assumed that 40% of patients would experience a clinically meaningful change in FACT-ES score, and prescribing the time of day for administration of treatment would be important if there was a difference in clinically meaningful change rates of 20% or more (i.e., 40% with a clinically meaningful change in one arm and 60% in the other). Using these assumptions and a two-sided, alpha = 0.05 Fisher’s exact test, 214 participants are required to achieve 80% statistical power. To account for potential loss to follow-up, and to account for stratification factors, the target sample size was inflated by 10% to a total of 235.
The primary analysis was based on a modified intent-to-treat (mITT) principle. Specifically, all patients with a baseline FACT-ES score and at least one FACT-ES score between weeks 4 and 12 were included in the primary outcome analysis. The last observation carried forward method was used to impute results for any patient who did not have a FACT-ES score measured at the 12-week timepoint. This is a conservative approach to imputation because it will likely result in smaller differences between groups than would actually be present (if there is an actual difference), under the assumption that increased effects will occur over time up to week 12. A supportive analysis of the primary outcome measure was conducted using the per-protocol population, which included only those patients who completed both the baseline and 12-week FACT-ES score. All other analyses of quality-of-life scores were based on the per-protocol population (i.e., those with available values at baseline and at the timepoint under study), while overall adherence and preference results were based on the mITT population. The FACT scores were calculated as per FACIT guidance. A higher FACT-ES score indicates better tolerability.
The primary analysis was conducted using a linear regression analysis with 12-week FACT-ES score as the outcome, with adjustment for stratification factors and baseline FACT-ES score. A supportive analysis evaluated FACT-ES scores across all time points using a repeated measures model. Univariable comparisons between the intervention and control groups at each time point were examined using a Chi-Squared test for categorical outcomes, Fisher’s exact test for dichotomous events, and a Wilcoxon rank sum test for continuous measures. Post hoc subgroup analyses based on whether the participant was prescribed tamoxifen or an aromatase inhibitor and menopausal status were also performed. Confidence intervals were constructed for outcomes of interest. All tests are two-sided, and statistical significance was defined at the α = 0.05 level.
Data availability
The de-identified study dataset is available upon request to the Principal Investigator (msavard@toh.ca) with permission from the Ontario Cancer Research Ethics Board (OCREB).
References
Murphy, C. C., Bartholomew, L. K., Carpentier, M. Y., Bluethmann, S. M. & Vernon, S. W. Adherence to adjuvant hormonal therapy among breast cancer survivors in clinical practice: a systematic review. Breast Cancer Res. Treat. 134, 459–478 (2012).
Hershman, D. L. et al. Early discontinuation and non-adherence to adjuvant hormonal therapy are associated with increased mortality in women with breast cancer. Breast Cancer Res. Treat. 126, 529–537 (2011).
Chirgwin, J. H. et al. Treatment adherence and its impact on disease-free survival in the Breast International Group 1-98 trial of Tamoxifen and Letrozole, alone and in sequence. J. Clin. Oncol. 34, 2452–2459 (2016).
Salgado, B. A. & Zivian, M. T. Aromatase inhibitors: side effects reported by 622 women. Breast Cancer Res. Treat. 100, S168–S168 (2006).
Garreau, J. R., DeLaMelena, T., Walts, D., Karamlou, K. & Johnson, N. Side effects of aromatase inhibitors versus tamoxifen: the patients’ perspective. Am. J. Surg. 192, 496–498 (2006).
Ekinci, E. et al. Interventions to improve endocrine therapy adherence in breast cancer survivors: what is the evidence?. J. Cancer Surviv. 12, 348–356 (2018).
Ganna S., Rahimi S., Lu A., Laborde K., Trivedi M. Interventions to improve oral endocrine therapy adherence in breast cancer patients. J. Cancer Surviv. https://doi.org/10.1007/s11764-023-01513-y (2024).
Selfridge, J. M. et al. Chronotherapy: Intuitive, sound, founded…but not broadly applied. Drugs 76, 1507–1521 (2016).
Lévi, F. A., Okyar, A., Hadadi, E., Innominato, P. F. & Ballesta, A. Circadian regulation of drug responses: toward sex-specific and personalized chronotherapy. Annu. Rev. Pharm. Toxicol. 64, 89–114 (2024).
Dallmann, R., Okyar, A. & Lévi, F. Dosing-time makes the poison: circadian regulation and pharmacotherapy. Trends Mol. Med. 22, 430–445 (2016).
Hermida R. C. et al. Bedtime hypertension treatment improves cardiovascular risk reduction: the Hygia Chronotherapy Trial. Eur. Heart J. https://doi.org/10.1093/eurheartj/ehz754 (2019).
Phillips L. A., Burns E., Leventhal H. Time-of-day differences in treatment-related habit strength and adherence. Annals Behav. Med. https://doi.org/10.1093/abm/kaaa042 (2020).
Ballesta, A., Innominato, P. F., Dallmann, R., Rand, D. A. & Lévi, F. A. Systems chronotherapeutics. Pharm. Rev. 69, 161–199 (2017).
Amiama-Roig, A., Verdugo-Sivianes, E. M., Carnero, A. & Blanco, J.-R. Chronotherapy: Circadian rhythms and their influence in cancer therapy. Cancers 14, 5071 (2022).
Diamantopoulou, Z. et al. The metastatic spread of breast cancer accelerates during sleep. Nature 607, 156–162 (2022).
Blakeman, V., Williams, J. L., Meng, Q.-J. & Streuli, C. H. Circadian clocks and breast cancer. Breast Cancer Res. 18, 89 (2016).
Hatcher, K. M., Royston, S. E. & Mahoney, M. M. Modulation of circadian rhythms through estrogen receptor signaling. Eur. J. Neurosci. 51, 217–228 (2020).
BREAST CANCER NOW. TAMOXIFEN n.d. https://breastcancernow.org/information-support/facing-breast-cancer/going-through-treatment-breast-cancer/hormone-therapy/tamoxifen (accessed August 4, 2020).
Beltran-Bless, A.-A. et al. Does the time of day at which endocrine therapy is taken affect breast cancer patient outcomes?. Curr. Oncol. 28, 2523–2528 (2021).
Landré, T. et al. Effect of immunotherapy-infusion time of day on survival of patients with advanced cancers: a study-level meta-analysis. ESMO Open 9, 102220 (2024).
Printezi, M. I. et al. Toxicity and efficacy of chronomodulated chemotherapy: a systematic review. Lancet Oncol. 23, e129–e143 (2022).
Shuboni-Mulligan, D. D., Breton, G., Smart, D., Gilbert, M. & Armstrong, T. S. Radiation chronotherapy—clinical impact of treatment time-of-day: a systematic review. J. Neurooncol. 145, 415–427 (2019).
Giacchetti, S. et al. Influence of hormone therapy timing intake on disease free survival (DFS) for patients with high-risk early breast cancer: Results of the UCBG-UNIRAD phase III randomized trial. J. Clin. Oncol. 41, 546–546 (2023).
Talamanca, L., Gobet, C. & Naef, F. Sex-dimorphic and age-dependent organization of 24-hour gene expression rhythms in humans. Science (1979) 379, 478–483 (2023).
Papakonstantinou, A. et al. Adjuvant endocrine treatment strategies for non-metastatic breast cancer: a network meta-analysis. EClinicalMedicine 81, 103116 (2025).
Zwarenstein, M. et al. Improving the reporting of pragmatic trials: an extension of the CONSORT statement. BMJ 337, a2390 (2008).
Fallowfield, L. J., Leaity, S. K., Howell, A., Benson, S. & Cella, D. Assessment of quality of life in women undergoing hormonal therapy for breast cancer: validation of an endocrine symptom subscale for the FACT-B. Breast Cancer Res. Treat. 55, 187–197 (1999).
Cella, D. et al. Quality of life of postmenopausal women in the ATAC (“Arimidex”, Tamoxifen, Alone or in Combination) trial after completion of 5 years’ adjuvant treatment for early breast cancer. Breast Cancer Res Treat. 100, 273–284 (2006).
Brady, M. J. et al. Reliability and validity of the functional assessment of cancer therapy-breast quality-of-life instrument. J. Clin. Oncol. 15, 974–986 (1997).
Herdman, M. et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual. Life Res. 20, 1727–1736 (2011).
Basulaiman, B. et al. Creating a pragmatic trials program for breast cancer patients: Rethinking Clinical Trials (REaCT). Breast Cancer Res. Treat. 177, 93–101 (2019).
Acknowledgements
This study was funded by a Northern Ontario Academic Medicine Association (NOAMA) Innovation Grant (2020, to MFS and MI) and by the REthinking Clinical Trials Program at the Ottawa Hospital Research Institute, the Ottawa Hospital Foundation, and its generous donors. We are thankful to Ian Tannock for reviewing the finalized manuscript and providing mentorship. We are grateful to patients and their families for their assistance with this study, as well as to physicians for approaching patients.
Author information
Authors and Affiliations
Contributions
Design/conception and preparation of the protocol: M.F.S., M.C., G.P., L.F., A.B., L.V. Principal investigators: M.F.S. and M.I. Study coordinators: D.S. and L.V. Patient recruitment: M.F.S., M.C., M.I., T.N., A.A.A., S.S., F.M., M.J.A., D.S. Data entry: D.S. Statistical analysis: G.P. Manuscript writing: M.F.S., M.C., D.S. Review of the manuscript: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Savard, M.F., Ibrahim, M., Saunders, D. et al. A pragmatic, multicenter, randomized trial comparing morning versus evening dosing of adjuvant endocrine therapy (REaCT-CHRONO Study). npj Breast Cancer 11, 49 (2025). https://doi.org/10.1038/s41523-025-00762-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-025-00762-7
