
Abstract
Cyclin-dependent kinase 4/6 inhibitors (CDK4/6i) plus endocrine therapy (ET) are the mainstay of treatment for hormone receptor positive, HER2 negative (HR + /HER2−) metastatic breast cancer (MBC). However, disease progression is inevitable and unveiling resistance mechanisms is crucial to guide post-CDK4/6i therapeutic strategies. In this study, we retrospectively analyzed a real-world, multi-institutional cohort of patients with HR + /HER2- MBC characterized by circulating tumor DNA (ctDNA) through next-generation sequencing (NGS) before starting second-line treatment. Among 93 patients previously treated with CDK4/6i, PIK3CA (37.6%), ESR1 (46.2%) and TP53 (31.2%) were the most altered genes. Comparing with a CDK4/6i plus ET naïve control cohort, ESR1 (p < 0.001) was significantly associated with first-line exposure. In multivariable analyses, PTEN alterations were independently associated with shorter progression free survival (PFS) (p = 0.008) and overall survival (OS) (p = 0.006), while TP53 (p = 0.031), CCDN1 (p = 0.003) and the ET second-line clinician’s choice (p = 0.011) impacted the OS. Moreover, a low-mutant allele frequency was correlated to longer PFS (p = 0.017) and OS (p = 0.038). These findings highlight the prognostic relevance of specific molecular alterations and support the role of genomic profiling in guiding second-line treatment decisions after CDK4/6i therapy. Prospective validation is warranted to confirm the clinical utility of this approach in HR + /HER2 − MBC.
Similar content being viewed by others

Introduction
The therapeutic approach to hormone receptor-positive, human epidermal growth factor receptor 2–negative (HR + /HER2−) metastatic breast cancer (MBC) has been revolutionized by the introduction of cyclin-dependent kinase 4 and 6 inhibitors (CDK4/6i), which are now considered the standard first-line treatment when combined with endocrine therapy (ET), consisting of either an aromatase inhibitor (AI) or Fulvestrant1,2,3. Despite these advances, prognosis after progression on CDK4/6i and ET remains poor with progression-free survival (PFS) of 2nd line and later line therapies ranging around 4–7 months (for standard regimens such as chemotherapy, ET or everolimus combined therapy)4,5,6. The limited efficacy of treatment used after progression on CDK4/6i could stem from multigene acquired resistance and often empiric treatment selection, due to a lack of objective knowledge of individual molecular resistance factors. In the era of precision medicine, the identification of resistance mechanisms and the optimal therapeutic approach after progression on CDK4/6i and ET still represent an unmet clinical need. This is particularly important considering that patients will be increasingly exposed to prior CDK4/6i, due to the recent transition of CDK4/6i in earlier disease setting as adjuvant therapy for high and moderate-risk disease7,8.
Liquid biopsy has emerged as a promising tool to identify molecular circulating biomarkers to guide treatment decisions. Previous studies revealed genomic alterations in circulating tumor DNA (ctDNA) such as ESR1, TP53, PIK3CA, FGFR1, and RB1 as the most frequently detected alterations following CDK4/6i exposure9,10,11,12. Beyond the detection of single genes mutations, ctDNA analysis allows for estimation of the overall mutational burden through the variant/mutant allele frequency (VAF/MAF). Martinez-Saez and colleagues longitudinally analyzed the changes in mean VAF during CDK4/6i treatment, establishing its prognostic value for the mean of all time points and not for the single values, although no correlations were done with subsequent line outcomes12. Based on the latest guidelines, liquid biopsy is recommended for identification of targetable alterations (somatic alterations in ESR1 and PIK3CA/AKT/mTOR pathway) in the clinical management of patients with MBC progressing on CDK4/6i + ET13. To further evaluate the potential of liquid biopsy to direct the management of HR + /HER2− MBC patients progressing on first line CDK4/6i + ET, we performed this retrospective analysis aimed at investigating the post-CDK4/6i ctDNA genomic landscape and evaluating the prognostic significance of identified circulating biomarkers in the second-line setting.
Results
Cohort characteristics and detected gene alterations
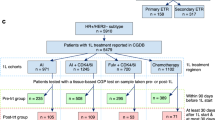
The study cohort included 93 patients with disease progression after first line CDK4/6i in combination with either AI or fulvestrant. The main histology was ductal carcinoma (N = 59; 70.2%) and 30.1% (N = 28) had de novo metastatic disease. The main sites of metastases were bone (N = 75; 80.6%), liver (N = 34; 36.6%), and lung (N = 20; 21.5%) (Table 1). Most of patients, 78.5% (N = 73), were treated with an ET-based second-line therapy (named as ET subgroup), versus 21.5% (N = 20) with a non-ET-based therapy (named no-ET subgroup). Among the 73 patients in the ET subgroup, the main endocrine backbone was a selective estrogen receptor modulators (SERM)/selective estrogen receptor degrader (SERD) (N = 47; 64.4%), followed by AI (N = 24; 32.9%). In the ET subgroup, 68.5% (N = 50) of patients received a targeted therapy (TT) combined with ET, in detail 37% (N = 27) a CDK4/6i beyond progression (BP), 16.4% (N = 12) a mTOR inhibitor and 15.1% (N = 11) a PI3K inhibitor (Table 1). In the no-ET subgroup the main class of drug was chemotherapy except for 2 patients who received a TT alone. The genomic landscape of our population was characterized by ESR1 single nucleotide variants (SNVs) (N=43; 46.2%), PIK3CA SNVs (N=35; 37.6%), TP53 SNVs (N=29; 31.2%), FGFR1 copy number variations (CNVs) (N=12; 12.9%), and CCND1 CNVs (N=10; 10.8%) as the most single gene alterations detected (Fig. 1a, Table S1–S3). Moreover, the most frequently altered genes were involved in pathways such as ER (N = 46; 49.5%), PI3K (N = 39; 41.9%), and p53 (N = 32; 34.4%) (Table S4). To further investigate potential molecular correlates of treatment selection and response, we explored the prevalence of mutations across the different TT classes. As expected, all the patients receiving a PI3K inhibitor harbored a PIK3CA SNV (N = 11; 100%). Moreover, a higher incidence of CDH1 SNVs (N = 2; 18.2%) and EGFR SNVs (N = 2; 18.2%) was also noted in this group. Among patients treated with CDK4/6i-based therapies, KRAS SNVs were more frequently detected (N = 4; 14.9%). No statistically significant enrichment of specific mutations was observed in the subgroup that received mTOR inhibitors in combination with ET as second-line therapy.

a Post-CDK4/6i cohort. b CDK4/6i-naive cohort. The incidence of single alterations (CNV, Fus, Del, Ins, FS, Spl, PTC, and SNV) is represented on the left, with frequency ordered by the sum of all variants in a particular gene. The mutant allele frequency (MAF) of each mutation is shown in the middle. Effect (GoF, LoF, SoF) and pathogenicity (Yes, No, Ukn, Inc) of all detected alterations are displayed on the right. CDK4/6i cyclin-dependent kinase 4/6 inhibitors, CNV copy-number variation, ctDNA circulating tumor DNA, Del deletion, FS frameshift, Fus fusion, GoF gain of function, Inc inconclusive, Ins insertion, LoF loss of function, MAF mutant allele frequency, mut mutated, PTC premature termination codon, SNV single-nucleotide variant, SoF switch of function, Spl splicing variant, Ukn unknown, wt wild-type.
Comparison between post-CDK4/6i and CDK4/6i naïve cohorts’ genomic landscape
Analyzing the control cohort of 181 HR + /HER2- patients naïve for CDK4/6i and ET in the metastatic setting, the top five detected alterations were PIK3CA SNVs (N = 54; 29.8%), TP53 SNVs (N = 52; 28.7%), FGFR1 CNVs (N = 22; 12.2%), AKT1 SNVs (N = 14; 7.7%), and EGFR CNVs (N = 14; 7.7%) (Fig. 1b). The clinical and pathological characteristics of this cohort are summarized in Table S5. The distribution of gene alterations in the latter cohort was compared with the post-CDK4/6i cohort and showed a significantly higher incidence of ESR1 SNVs (46.2% vs. 6.6%, p < 0.001), and ER SNVs pathway mutations (49.5% vs. 15.5%, p < 0.001) in the post CDK4/6i cohort. The low detection rate (2.2%) of BRCA1 SNVs and CHEK2 SNVs in both the cohorts limited any meaningful inference, despite statistical significance. Additionally, our study confirmed a doubled incidence of RB1 SNVs (3.2% vs. 1.1%) following CDK4/6i exposure, although this increasing was not statistically significant (Table S6–8).After multivariable logistic regression—excluding 18 patients known to have received endocrine therapy in the (neo)adjuvant setting in the CDK4/6i naïve cohorts to avoid bias—and after adjustment for sites of metastases and de novo disease, ESR1 SNVs (Odds ratio [OR], 14.46; 95% confidence interval [CI]: 6.35–32.91; p < 0.001) and ER SNV pathway alterations (OR, 5.86; 95% CI: 3.10–11.07; p < 0.001) remained significantly associated with first line exposure to CDK4/6i plus ET combination therapy (Table S9).
Impact of the genomic landscape on post-CDK4/6i cohort survival
The median PFS (mPFS) of the study cohort was 5.6 months (95% CI:4.1–7.1) while the median overall surival (mOS) was 23.6 months (95% CI: 20.2–32.3). According to the type of treatment, the mPFS was 5.7 months (95% CI: 4.3–7.2) and 4.0 months (95% CI: 2.3–5.7) in the ET-combination and no-ET subgroups, respectively. We evaluated the prognostic relationships between clinical, therapeutic, and ctDNA characteristics. With multivariable analysis, SNVs annotated in PTEN (N = 5; 5.4%) (Fig. 2a) showed a negative prognostic impact in terms of PFS (Hazard ratio [HR], 3.75; 95% CI: 1.42–9.95; p = 0.008) together with the presence of liver metastasis (HR, 2.06; 95% CI: 1.23–3.44; p = 0.006) (Table 2, Fig. 3a, c). Meanwhile, the OS multivariable model confirmed a prognostic role of the SNVs annotated in TP53 (N = 29; 31.2%) (Fig. 2b) (HR,1.92; 95% CI: 1.06–3.47; p = 0.031) and PTEN (HR, 6.99; 95% CI: 2.32–21.14; p = 0.001), CCDN1 CNVs (N = 10; 10.8%) (HR, 3.30; 95% CI: 1.48–7.35; p = 0.003), and of the ET second-line clinician’s choice (HR, 0.42; 95% CI: 0.21–0.82; p = 0.011) (Table 2, Fig. 3b, d-f).Cell-cycle and PI3K pathways, together with PIK3CA SNVs, were independently associated with OS only at univariable analysis. In the ET subgroup, mPFS according to the type of second-line treatment was 6.7 months (95% CI: 0–13.4) for CDK4/6i BP, 5.7 months (95% CI: 2.1–9.3) for ET single agent, 5.4 months (95% CI: 4.0–6.9) for PI3K inhibitors, and 4.1 months (95% CI: 1.4–6.7) for mTOR inhibitors. No statistically significant differences in PFS were observed among these treatment groups. Exploring the prognostic impact of ctDNA alterations within the entire ET subgroup (N = 73), PTEN mutations were confirmed as a negative prognostic factor for both PFS (HR 3.01; 95% CI: 1.16–7.83; log-rank test, p = 0.017) and OS (HR 5.83; 95% CI: 1.95–17.41; log-rank test, p = 0.0003). Additionally, alterations in the cell cycle pathway (HR 2.82; 95% CI: 1.08–7.39; log-rank test, p = 0.027) and CCND1 amplification (HR 3.10; 95% CI: 1.42–6.72; log-rank test, p = 0.027) were also significantly associated with shorter OS.

The grey bar represents the entire protein with the different aminoacid positions. The colored boxes are specific functional domains. On top of the lollipops the most frequent variants are annotated as the amino acid change at that specific site. a mutations identified in PTEN gene; b mutations identified in TP53 gene.

a mPFS according to PTEN mutation status (33.0 vs 6.0 months, p = 0.028). b mOS according to PTEN mutation status (6.0 vs 28.6 months, p = 0.0036). c mPFS according to presence of liver metastases (3.1 vs 7.4 months, p = 0.017). d mOS according to TP53 mutation status (17.2 vs 29.8 months, p = 0.052). e mOS according to CCND1 amplification status (13.0 vs 28.8 months, p = 0.0026). f mOS according to second-line endocrine therapy (ET) choice (28.4 vs 15.2 months, p = 0.022). ampl amplification, ET endocrine therapy, mOS median overall survival, mPFS median progression-free survival, mutmutation, mets metastases, OS overall survival, PFS progression-free survival, wt wild-type.
MAF and NOA analyses
In our study cohort, 75 of 93 patients (80.6%) had at least one detectable alteration by Guardant360®, of whom 59 (63.4%) were in the ET subgroup. Considering only patients with detectable alterations, the median MAF was 2.1% (interquartile range [IQR] 0.4%–43.7%) while the median number of alterations (NOA) was 3 (IQR 2–5). The patients were classified according to the median MAF (≤2.1% or >2.1%) and NOA (≤ 3 or >3). We observed that the detection of a low-MAF (i.e. below the median value) was correlated to longer PFS (mPFS 5.7 vs. 4.2 months; HR, 1.92; 95% CI:1.11–3.34; log-rank test, p = 0.017) and OS (mOS 28.6 months vs. 18 months; HR, 1.90; 95%CI: 1.02–3.54; log-rank test, p = 0.038) (Fig. 4a, b). No association of MAF with survival outcomes was observed when narrowing the analysis only to the ET subgroup. Patients with low-NOA (i.e., below the median value) experienced higher OS both in the overall population (mOS 28.8 vs 16.4 months; HR, 2.24; 95%CI: 1.26–3.98; log-rank test, p = 0.033) and in the ET subgroup (mOS 34 vs 22 months; HR, 3.04; 95%CI: 1.52–6.08; log-rank test, p = 0.037) (Fig. 4c, d). No prognostic impact of NOA was observed in terms of PFS.

A low- MAF was correlated to an improved PFS (a) and OS (b) in all post-CDK4/6i cohort. A low-NOA was correlated to an improved OS in the all-study cohort (c) and in ET- subgroup (d). The value of MAF and NOA was categorized in low and high based on the median value. ET endocrine therapy, MAF mutant allele frequency, NOA number of alterations, OS overall survival, PFS progression-free survival.
In the CDK4/6i naïve control cohort, 144 (79.6%) of 181 patients, had at least 1 detectable alteration by Guardant360® test. The median MAF was 2.1% (IQR 0.9%–3%), while the median NOA was 2.0 (IQR 1-4).Comparing the median value of MAF and NOA between the post-CDK4/6i and the CDK4/6i naïve control cohorts, we found, as expected, a statistically significantly higher incidence of NOA after the selective pressure driven by the drugs exposure (p = 0.006), while no difference was observed in terms of MAF (p = 0.23) (Fig S1).
Discussion
To our knowledge, our study represents the first real-world comprehensive genomic analysis after receipt of CDK4/6i as first-line treatment of HR + /HER2- MBC. We aimed to evaluate the prognostic impact of ctDNA alterations with regards to choice of subsequent therapy, mainly represented by FDA-approved targeted therapies. Important to keep in mind that more than half of the population received a TT as second line; in detail, 30.1% a CDK4/6i BP while 12.9% of patients underwent an mTOR and PI3K inhibitors, respectively.
The genomic landscape depicted in our cohort, was characterized by PIK3CA, ESR1 and TP53 as the most frequently mutated genes both in the ET and no-ET subgroups, consistent with the literature14. These genes are historically associated with endocrine resistance and some of them are now targetable with the introduction of new targeted therapies, as seen in trials such as EMERALD, SOLAR-1, and CAPITELLO-29115,16,17.
By comparing the post-CDK4/6i cohort with a CDK4/6i-naïve control group who underwent ctDNA testing prior to the initiation of first-line therapy for metastatic disease, we aimed to elucidate the potential impact of drugs exposure on the accumulation of specific mutations. In line with the recent genomic tissue analysis by Rao and colleagues18, we observed a significantly higher incidence of ESR1 mutations following CDK4/6i + ET exposure, along with an increased NOA. This increased amount of alterations might reflect the higher tumor mutational burden (TMB) reported in their tissue-based study. However, the absence of a matched cohort and the retrospective nature of our data highlight the need for further validation in larger, prospective analyses.
The median PFS ( ~ 6 months) observed in our study cohort is consistent with prospective and retrospective analyses performed in the post-CDK4/6i setting4,5,6. We observed that PTEN alteration was the only significant negative prognostic factor in term of PFS (together with the presence of liver metastases) in both the study cohort and in the ET-subgroup. Preclinical studies support the mechanism of CDK4/6i resistance driven by PTEN loss. These studies showed that PTEN loss leads to the delocalization of p27 from the nucleus, resulting in increased activation of both CDK4 and CDK219,20.
Our results align with previously reported associations between PTEN mutations—evaluated by tissue-based next generation sequencing (NGS) prior to CDK4/6i treatment—and shorter PFS in a similar clinical context, among a small population of HR + /HER2 − MBC patients (6 out of 36 cases)21. Moreover, preclinical and clinical models showed that PTEN-deficient cells are resistant to PIK3CA inhibitors and more sensitive to AKT inhibition19,22,23. These data, combined with the prognostic value of PTEN showed in our analysis, not only support the combination of capivasertib plus fulvestrant as post-CDK4/6i strategy22 but also suggest that its efficacy may be independent from AKT mutations due to the interplay with other altered mechanisms such as PTEN loss. Recently a large real word study showed that PTEN loss was detected at higher rates in tissue than liquid biopsy and this should also justify the low incidence detected in our population24. Since only 5 patients had these mutations, prospective and larger studies are needed to further investigate the potential predictive role of PTEN.
The detection of TP53 and PTEN mutations and CCND1 amplification impacted the OS both in univariable and multivariable analyses. Although it has been reported that 30–50% of HR + /HER2 − CDK4/6i-resistant breast tumors carry TP53 mutations20, the aggressive biology of TP53-mutant tumors—as demonstrated in both arms of the PALOMA-3 trial25—highlights the need to explore alternative therapeutic strategies. The influence of TP53 mutations on CDK4/6i resistance is still under investigation, although recent data have associated TP53 loss-of-function variants with poorer clinical outcomes across multiple treatment settings and CDK4/6i agents. Preclinical analysis suggests that TP53 loss may lead to CDK4/6i resistance by allowing CDK2 to phosphorylate p130, which disrupts the DREAM complex and enables cell-cycle re-entry. Dual inhibition of CDK4/6 and CDK2 may therefore be required to overcome this resistance mechanism in HR+ breast cancer models26. In hematologic malignancies, a functional interaction between CDK6 and TP53 has also been reported27, supporting the hypothesis that CDK4/6 inhibitors may lose efficacy in TP53-deficient tumors.
As known, CCND1 has been implicated in the hyperactivity of the cyclin-D-CDK4/6 pathway28,29. Recently, preclinical data supported the targetability of CCND1 in CDK4/6i resistant model by Polo-like kinase 1(PLK1) inhibitor30 underlining the need to discover new targets, changing the negative prognostic significance of these alterations into positive predictive factors.
Going beyond the single genomic mutations, MAF may be a surrogate of the mutation clonality and a tool for assessing tumor heterogeneity31. In our analysis we confirmed the MAF as a robust prognostic factor for both PFS and OS. This finding could potentially be explained by its correlation with tumor burden, as previously demonstrated32,33.
In our study cohort, 78% of patients received an ET-based second line treatment with a significantly positive impact on the OS. In 68.5% of cases, ET was administered in combination with a TT and in 37% of cases patients received a CDK4/6i BP. This data supports the hypothesis that ctDNA profiling could guide therapeutic decisions by uncovering endocrine sensitivity and identifying the most effective targeted agents to overcome resistance mechanisms. Additionally, ctDNA profiling may help identify more aggressive disease phenotypes that could benefit from a chemotherapeutic approach.
The large clinical use of CDK4/6i BP might have impacted the ET prognostic value, as demonstrated in a previous analysis conducted in the same consortium population in terms of PFS34. The phase II MANTAIN trial tested ribociclib BP + ET resulting in a PFS but not OS advantage. Intriguingly, a subgroup analysis from the MAINTAIN study showed a lack of benefit in the ESR1 mutated population, and 50% of these patients had a concomitant CCDN1 and/or FGFR1 amplification (in our study among the most prevalent alterations)35. The PACE trial failed to demonstrate a survival benefit from the palbociclib BP + fulvestrant combination, although the preliminary ctDNA analysis highlighted an impact of the mutational status on outcome36. Recently, the postMONARCH trial was the first phase III to show the benefit of continuing CDK4/6i BP switching the ET backbone, for HR + /HER2 patients not selected for biomarkers status. At 6 months, the rate of PFS was 50% for the association of Abemaciclib plus Fulvestrant, with the advantage maintained across major genomic subgroups, including patients with ESR1 or PIK3CA mutations at baseline37.
Our study has several limitations. Firstly, the retrospective design introduces potential selection bias. Additionally, the lack of data on exposure to first line CDK4/6i prevented stratification of the population based on this factor. The relatively small sample size of the study cohort, along with the heterogeneity in genomic alterations detected and types of treatments received, limits our ability to establish a robust prognostic interplay among these factors. Finally, the broad and dated period of enrollment limited any hypothesis generation regarding the role of new agents such as antibody-drug conjugates.
In conclusion, our study depicted the genomic landscape of a cohort of MBC patients who had disease progression after receiving CDK4/6i as first-line treatment. The detected alterations were significantly different from those detected in treatment naïve patients. This may impact future treatment strategies most of all with the upcoming of CDK4/6i in the adjuvant setting7,8. The introduction of Abemaciclib in the adjuvant therapeutic algorithm for node positive - high risk patients and the extension of Ribociclib also for high risk node negative population, open the important question of how to treat in case of early relapse these patients, and our finding supports the needing of a genomic reassessment of the disease coupled with a personalized therapeutic genomic-based approach.
Our findings, which confirm the prognostic significance of ctDNA alterations, suggest that the post-CDK4/6i treatment choices could potentially be guided by identifying alterations through ctDNA. Nonetheless, further prospective studies are warranted to validate the clinical utility of this approach in the management of patients with HR + /HER2- MBC.
Methods
Study population and design
We retrospectively analyzed a multi-institutional cohort of 909 patients with HR + /HER2- MBC who underwent NGS-based plasma ctDNA testing from January 2015 to May 2023, in real-world clinical practice, at four academic centers in United States: Weill Cornell Medicine (New York, NY), Northwestern University (Chicago, IL), Massachusetts General Hospital (Boston, MA), and Washington University in St Louis (St Louis, MO). The data sharing and analysis were approved by the institutional review boards (IRBs) of the three sites: Washington University School of Medicine (St. Louis, MO; IRB#202101147), Northwestern University (Chicago, IL; IRB#STU00214133), and Massachusetts General Hospital (Boston, MA; IRB#2013P000848). Weill Cornell Medicine data were collected under an independent IRB- approved protocol (New York, NY; IRB#1812019858). The requirement for informed consent was waived by the IRBs for this de-identified analysis. The study was conducted in accordance with the statutes set by the Health Insurance Portability and Accountability Act and the Declaration of Helsinki.
We limited the study cohort to 93 HR + /HER2- metastatic patients who received CDK4/6i in the first line setting and were candidate for a second-line therapy as determined by the treating physician’s choice and had an available baseline ctDNA analysis before starting a second line treatment. We also included a control cohort of 181 HR + /HER2− patients who were treatment-naïve for metastatic disease (referred to as the CDK4/6i-naïve cohort), with baseline ctDNA analysis performed prior to the initiation of first-line therapy as part of routine clinical practice, in order to compare ctDNA features between the two groups.
ctDNA sample collection and analysis
Ten milliliters (mL) of peripheral blood were collected into two Streck Cell-Free DNA blood collection tubes (BCTs) (Streck, Inc., La Vista, NE), at the timepoints previously described: before starting second-line therapy for the study cohort and before starting a first line of therapy for metastatic disease, for the control cohort. ctDNA profiling was performed using the commercially available Guardant360® (Guardant Health, Inc. Redwood City, CA,USA), a qualitative NGS-based in vitro diagnostic device that uses targeted high throughput hybridization-based capture technology for detection of SNVs, insertions and deletions (indels), CNVs and fusions of both synonymous and non-synonymous alteration in up to 74 genes38,39,40. Based on OncoKB database, mutations were annotated according to their effect (loss of function, gain of function) and pathogenicity39. Only mutations categorized as pathogenic were considered for the analysis. Pathway classification was based on previously defined profiles generated on the Cancer Genome Atlas database (i.e., RTK, RAS, RAF, MEK, NRF2, ER, WNT, MYC, P53, cell cycle, Notch, PI3K)41. The MAF was reported and analyzed based on the highest frequency across all mutated gene detected in the blood samples (overall MAF).
Statistical analysis
Clinical and pathologic variables were reported using descriptive analyses. Categorical variables were reported as frequency distributions, whereas continuous variables were described through median and interquartile ranges (IQRs). Chi-square or Fisher’s exact test were employed to analyze differences in distribution across subgroup, according to sample size. Associations between clinical characteristics and genomics alteration were explored through multivariable logistic regression, inclusive of OR and 95% CI computation.
Regarding the survival outcomes, PFS was defined as the time from the baseline (before starting 2nd line) ctDNA blood draw to progression or death from any cause, whichever came first, while OS was defined as the time from the baseline ctDNA blood draw to death from any cause. Patients without an end-point event at the last follow-up visit were censored. Differences in survival were tested using log-rank test and univariable and multivariable Cox regression with 95% CI and represented by Kaplan-Meier estimator curves. Correction for ctDNA features, main clinical characteristics, and type of treatment was applied to the multivariable model after univariable testing; only variables with a p ≤ 0.10 at the univariable analysis were considered. Only SNVs and CNVs with at least a 5% prevalence were included in the prognostic analyses. Statistical analysis was conducted using IBM-SPSS v. 27.0 and R v.4.1.2 software.
Data availability
De-identified data of the 358 patients will be available upon reasonable request of the authors as the data are currently shared under a data use agreement among the principal investigators of the four institutions with multiple ongoing analyses and manuscripts.
References
Gradishar, W. J. et al. NCCN Guidelines® Insights: Breast Cancer, Version 4. 2023. J. Natl Compr. Cancer Netw. JNCCN 21, 594–608 (2023).
Gennari, A. et al. ESMO Clinical Practice Guideline for the diagnosis, staging and treatment of patients with metastatic breast cancer. Ann. Oncol. J. Eur. Soc. Med. Oncol. 32, 1475–1495 (2021).
Burstein, H. J. et al. Endocrine Treatment and Targeted Therapy for Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer: ASCO Guideline Update. J. Clin. Oncol. J. Am. Soc. Clin. Oncol. 39, 3959–3977 (2021).
Sledge, G. W. et al. The Effect of Abemaciclib Plus Fulvestrant on Overall Survival in Hormone Receptor-Positive, ERBB2-Negative Breast Cancer That Progressed on Endocrine Therapy-MONARCH 2: A Randomized Clinical Trial. JAMA Oncol. 6, 116–124 (2020).
Rossi, L. et al. Clinical outcomes after palbociclib with or without endocrine therapy in postmenopausal women with hormone receptor positive and HER2-negative metastatic breast cancer enrolled in the TREnd trial. Breast Cancer Res. BCR 21, 71 (2019).
Turner, N. C. et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 379, 1926–1936 (2018).
Slamon, D. et al. Ribociclib plus Endocrine Therapy in Early Breast Cancer. N. Engl. J. Med. 390, 1080–1091 (2024).
Johnston, S. R. D. et al. Abemaciclib plus endocrine therapy for hormone receptor-positive, HER2-negative, node-positive, high-risk early breast cancer (monarchE): results from a preplanned interim analysis of a randomised, open-label, phase 3 trial. Lancet Oncol. 24, 77–90 (2023).
Davis, A. A. et al. Genomic Complexity Predicts Resistance to Endocrine Therapy and CDK4/6 Inhibition in Hormone Receptor-Positive (HR+)/HER2-Negative Metastatic Breast Cancer. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 29, 1719–1729 (2023).
Bertucci, F. et al. Genomic characterization of metastatic breast cancers. Nature 569, 560–564 (2019).
Formisano, L. et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun. 10, 1373 (2019).
Martínez-Sáez, O. et al. Circulating tumor DNA dynamics in advanced breast cancer treated with CDK4/6 inhibition and endocrine therapy. NPJ Breast Cancer 7, 8 (2021).
Burstein, H. J. et al. Testing for ESR1 Mutations to Guide Therapy for Hormone Receptor–Positive, Human Epidermal Growth Factor Receptor 2–Negative Metastatic Breast Cancer: ASCO Guideline Rapid Recommendation Update. J. Clin. Oncol. https://doi.org/10.1200/JCO.23.00638 (2023).
Roberto, M. et al. CDK4/6 Inhibitor Treatments in Patients with Hormone Receptor Positive, Her2 Negative Advanced Breast Cancer: Potential Molecular Mechanisms, Clinical Implications and Future Perspectives. Cancers 13, 332 (2021).
Bidard, F.-C. et al. Elacestrant (oral selective estrogen receptor degrader) Versus Standard Endocrine Therapy for Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results From the Randomized Phase III EMERALD Trial. J. Clin. Oncol. J. Am. Soc. Clin. Oncol. 40, 3246–3256 (2022).
Turner, N. C. et al. Capivasertib in Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 388, 2058–2070 (2023).
André, F. et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 380, 1929–1940 (2019).
Rao, X. et al. Clinical and Genomic Characteristics of Patients with Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer Following Progression on Cyclin-Dependent Kinase 4 and 6 Inhibitors. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 29, 3372–3383 (2023).
Costa, C. et al. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kα Inhibitors in Breast Cancer. Cancer Discov. 10, 72–85 (2020).
Wander, S. A. et al. The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor-Positive Metastatic Breast Cancer. Cancer Discov. 10, 1174–1193 (2020).
West, M. T. et al. Real-World Evaluation of Disease Progression After CDK 4/6 Inhibitor Therapy in Patients With Hormone Receptor-Positive Metastatic Breast Cancer. Oncologist 28, 682–690 (2023).
Razavi, P. et al. Alterations in PTEN and ESR1 promote clinical resistance to alpelisib plus aromatase inhibitors. Nat. Cancer 1, 382–393 (2020).
Juric, D. et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 518, 240–244 (2015).
Bhave, M. A. et al. Comprehensive genomic profiling of ESR1, PIK3CA, AKT1, and PTEN in HR(+)HER2(−) metastatic breast cancer: prevalence along treatment course and predictive value for endocrine therapy resistance in real-world practice. Breast Cancer Res. Treat. https://doi.org/10.1007/s10549-024-07376-w (2024).
O’Leary, B. et al. Circulating Tumor DNA Markers for Early Progression on Fulvestrant With or Without Palbociclib in ER+ Advanced Breast Cancer. J. Natl Cancer Inst. 113, 309–317 (2021).
Kudo, R. et al. Long-term breast cancer response to CDK4/6 inhibition defined by TP53-mediated geroconversion. Cancer Cell 42, 1919–1935.e9 (2024).
Bellutti, F. et al. CDK6 Antagonizes p53-Induced Responses during Tumorigenesis. Cancer Discov. 8, 884–897 (2018).
Otto, T. & Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17, 93–115 (2017).
Arnold, A. & Papanikolaou, A. Cyclin D1 in breast cancer pathogenesis. J. Clin. Oncol. J. Am. Soc. Clin. Oncol. 23, 4215–4224 (2005).
Montaudon, E. et al. PLK1 inhibition exhibits strong anti-tumoral activity in CCND1-driven breast cancer metastases with acquired palbociclib resistance. Nat. Commun. 11, 4053 (2020).
Boscolo Bielo, L. et al. Variant of allele frequency: a decision-making tool in precision oncology?. Trends Cancer S2405-8033, 00171–00171 (2023).
Zhang, Q. et al. Prognostic and Predictive Impact of Circulating Tumor DNA in Patients with Advanced Cancers Treated with Immune Checkpoint Blockade. Cancer Discov. 10, 1842–1853 (2020).
Manca, P. et al. Variant allele frequency in baseline circulating tumour DNA to measure tumour burden and to stratify outcomes in patients with RAS wild-type metastatic colorectal cancer: a translational objective of the Valentino study. Br. J. Cancer 126, 449–455 (2022).
Gerratana, L. et al. Cyclin-Dependent Kinase 4/6 Inhibitors Beyond Progression in Metastatic Breast Cancer: A Retrospective Real-World Biomarker Analysis. JCO Precis. Oncol. 7, e2200531 (2023).
A randomized, phase II trial of fulvestrant or exemestane with or without ribociclib after progression on anti-estrogen therapy plus cyclin-dependent kinase 4/6 inhibition (CDK 4/6i) in patients (pts) with unresectable or hormone receptor–positive (HR+), HER2-negative metastatic breast cancer (MBC): MAINTAIN trial. Journal of Clinical Oncology.
Abstract GS3-06: GS3-06 Palbociclib After CDK4/6i and Endocrine Therapy (PACE): A Randomized Phase II Study of Fulvestrant, Palbociclib, and Avelumab for Endocrine Pre-treated ER+/HER2- Metastatic Breast Cancer | Cancer Research | American Association for Cancer Research. https://aacrjournals.org/cancerres/article/83/5_Supplement/GS3-06/717530/Abstract-GS3-06-GS3-06-Palbociclib-After-CDK4-6i.
Kalinsky, K. et al. Abemaciclib plus fulvestrant vs fulvestrant alone for HR+, HER2- advanced breast cancer following progression on a prior CDK4/6 inhibitor plus endocrine therapy: Primary outcome of the phase 3 postMONARCH trial. J. Clin. Oncol. 42, LBA1001–LBA1001 (2024).
Lanman, R. B. et al. Analytical and Clinical Validation of a Digital Sequencing Panel for Quantitative, Highly Accurate Evaluation of Cell-Free Circulating Tumor DNA. PLOS ONE 10, e0140712 (2015).
Zill, O. A. et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 24, 3528–3538 (2018).
Forbes, S. A. et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, D777–D783 (2017).
Sanchez-Vega, F. et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10 (2018).
Acknowledgements
E. Bria was supported by Catholic University of Sacred Heart, Fondazione AIRC per la Ricerca sul Cancro and the Italian Ministry of Health “Ricerca Corrente” 2023 and 2024. No other financial assistance was received in support of the study.
Author information
Authors and Affiliations
Contributions
Study design: L.P., C.R., E.B., C.X.M., A.B., M.C.; Data collection: L.P., A.A.D., E.P., L.G., A.J.M., M.V, S.W.; Data analysis: L.P., D.G., L.G.; Data interpretation: L.P., E.B., C.R., L.G., M.C.; Manuscript Writing and editing: All authors Final approval of manuscript: All authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare the following competing interests: L. Pontolillo reports travel grants from Pifzer and Eli Lilly, all outside the submitted work. A.A. Davis reports grants funding from Breast Cancer Alliance; participating on a Data Safety Monitoring Board or Advisory Board from Pfizer and Biotheranostics; personal fee from Onclive and travel support from DAVA Oncology, all outside the submitted work. L. Gerratana reports consulting fees or payment or honoraria for lectures, presentations, speaker bureaus, manuscript writing or educational events from AstraZeneca, Daiichi Sankyo, Eli Lilly, GlaxoSmithKline, Incyte, Novartis, Pfizer, Merck Sharp & Dohme, Menarini Stemline and Abbvie; travel support form Menarini Stemline; participating on a Data Safety Monitoring Board or Advisory Board from Incyte, Eli Lilly, Menarini Stemline, all outside the submitted work. A.J.Medford reports consulting fees from AstraZeneca, Guardant Health, Illumina, Myriad Genetics, Science for America and payment or honoraria for lectures, presentations, speaker bureaus, manuscript writing or educational events from Natera, all outside the submitted work. K. Clifton reports participating on Advisory Board from Novartis, outside the submitted work. M. Lipsyc-Sharf reports payment or honoraria for lectures, presentations, speaker bureaus, manuscript writing or educational events from Exact Sciences and participating on a Data Safety Monitoring Board or Advisory Board from Natera, all outside the submitted work. E.Bria reports institutional grants funding from AstraZeneca and Roche; payment or honoraria for lectures, presentations, speaker bureaus, manuscript writing or educational events from MSD, AstraZeneca, Roche, Pfizer, Novartis and Takeda; travel support from AstraZeneca, MSD and Roche; participation on a Data Safety Monitoring Board or Advisory Board from MSD, Eli-Lilly, AstraZeneca, Janseen, BMS, Celltrion, all outside the submitted work. C.X. Ma reports research grants from Pfizer, Puma, AstraZeneca, Genentech; consulting fees and participating on advisory board from Puma, Pfizer, Novartis, AstraZeneca, all outside the submitted work. A. Bardia reports grant fundings and consulting fees from Pfizer, Novartis, Genentech, Merck, Menarini, Gilead, Sanofi, AstraZeneca/Daiichi Sankyo, Eli Lilly, travel grants from AstraZeneca, Daiichi Sankyo and Gilead, all outside the submitted work. C. Reduzzi reports grants funding from American Association for Cancer Research, outside the submitted work. M. Cristofanilli reports payment or honoraria for lectures, presentations, speaker bureaus, manuscript writing or educational events from Pfizer, participating on a Data Safety Monitoring Board or Advisory Board from Merck and AstraZeneca, leadership role as President for Inflammatory Breast Cancer International Consortium, all outside the submitted work. J. Wang, E. Nicolò, M. Velimirovic; S. Warrior, E. Podany, M.S. Serafini, L. Munoz Arcos, E. Molteni, C. Gianni, C. Dai, D. Giannarelli declare no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pontolillo, L., Davis, A.A., Gerratana, L. et al. Circulating genomic landscape following cyclin-dependent kinase 4/6 inhibitors exposure in HR + /HER2− metastatic breast cancer: a retrospective multi-institutional Consortium analysis. npj Breast Cancer 11, 93 (2025). https://doi.org/10.1038/s41523-025-00802-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-025-00802-2
