
Abstract
Accurate identification of HER2-low breast cancers remains challenging using standard IHC. We analyzed 3182 breast tumors using transcriptomics, revealing detectable ERBB2 mRNA in 86% of IHC 0 cases. Pathological complete response was most prevalent in anti-HER2-treated patients with highest ERBB2 expression. Our results demonstrate that transcriptomics can sensitively detect HER2 expression and stratify patients beyond IHC classification, supporting its use as a complementary biomarker for guiding anti-HER2 therapy decisions.
Similar content being viewed by others

Precision medicine solutions for breast cancer include gene expression-based molecular subtyping1 and quantification of the expression of biomarkers such as the estrogen receptor, progesterone receptor, and the human epidermal growth factor receptor 2 (HER2), encoded by the ERBB2 gene, to direct targeted treatment. HER2 inhibitors have been a hallmark success in oncology therapies2 since HER2 gene amplification, occurring in around 15% of invasive breast cancers3, was found to be associated with poor prognosis. Standard methods for determining a tumor’s HER2 status include immunohistochemistry (IHC) and in situ hybridization methods such as FISH, which measure ERBB2 gene copy numbers4.
An important extension has recently been made with the demonstration of improved outcome in metastatic HER2-low patients receiving antibody–drug conjugates (ADCs)5,6. The ASCO/CAP 2018 HER2 testing guidelines7 have been updated8 to acknowledge the new indication for ADC therapy, providing best practice recommendations to identify the often subtle differences between HER2-zero (IHC 0) and HER2-low (IHC 1+ or IHC 2+ with negative ISH) in metastatic breast cancer. Meanwhile, recent evidence from the DESTINY-Breast06 trial suggests the existence of a distinct HER2-ultralow category, defined as IHC 0 with faint/incomplete membranous staining in ≤10% of tumor cells—reinforcing the growing recognition of heterogeneity within the HER2-low subgroup. While still formally HER2-negative, such cases may still respond to HER2-directed ADCs due to minimal antigen expression. Notably, Huang et al.9 have emphasized the lack of robust clinical evidence for efficacy in true IHC 0 tumors without any membranous staining, underscoring the need for more precise discrimination among these categories. The updated testing guidelines, however, ultimately affirmed the existing HER2 reporting recommendations, while admitting that it remains challenging to differentiate between HER2-low and HER2-zero based on standard IHC staining, which could result in erroneous treatment recommendations and potentially impact ongoing clinical trials8.
This shortcoming could be rectified by including quantitative ERBB2 mRNA measurements by nCounter-based platforms10,11 or high-throughput transcriptomics such as DNA microarrays12,13 or next generation sequencing (NGS)14,15, as a robust biomarker complementary to conventional IHC in predicting therapeutic benefit. Notably, these methods are, by design, free of well-described pitfalls4 of IHC and FISH such as variations in fixation, choice of antibody (IHC only), and scoring interpretation.
In this study, we sought to explore the relevance of transcriptomics-derived measurement of HER2 in distinguishing between HER2-zero and HER2-low cancers, compared with the histopathology-derived assessment of HER2 status. We collected transcriptomic profiles of 3564 primary breast cancer tumors from 3512 patients treated at Copenhagen University Hospital, Rigshospitalet generated between the years 2014 and 2022. After discarding samples not assigned IHC-derived scores for HER2 (Supplementary Fig. 1), this led to a collection of 3182 samples from 3137 patients, 830 of which had been processed with RNA-Seq and 2352 with tissue microarrays. These two technologies have been shown to generate results with high concordance16, and we indeed observed a similar distribution of expression values between the two sets of sample populations (Fig. 1).

a Histogram of samples profiled with RNA-Seq; b Histogram of samples profiled with microarrays. Distribution of HER2 IHC score is indicated in each bin by a different color; blue: 0; yellow: 1+; green: 2+/normal; orange:2+/amplified; pink: 3+. x-axis: normalized log2-transformed ERBB2 values measured with transcriptomics; y-axis: the number of samples in each bin.
As expression measurements from the two transcriptomic platforms come in different scales and could not be directly compared, we divided the expression value distributions into five expression classes (“very low”, “low”, “intermediate”, “high” and “very high”) delineated by four quantiles (0.05, 0.25, 0.75, 0.95) (Fig. 1). Interestingly, we noticed that for 86% of all samples that had been assigned IHC 0 by standard classification, their ERBB2 expression fell under “low” (41%), “intermediate” (42%), and even “high” (4%) classes. This observation highlights the sensitivity of quantitative transcriptomics in detecting HER2 signal in samples with low levels of the protein, thus adequately distinguishing between HER2-zero and HER2-low.
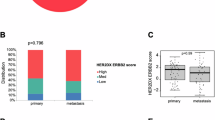
Furthermore, we performed ROC analysis using ERBB2 expression as the predictor and IHC-based HER2 classification as the reference (HER2-low being samples with IHC score of 1+ or 2+ with negative in situ hybridization and HER2-zero being samples with IHC score of 017) (Supplementary Fig. 2). The AUC values were ≥0.75 for both RNA-Seq and microarray datasets, indicating strong performance. Nonetheless, our study aims to establish an expression-based approach to classifying breast tumors, aligning more closely with the definitions of HER2-low and HER2-zero in Tarantino et al.18. In this context, while our “very low” class is not exclusive to IHC = 0 (Fig. 1B) and includes samples with minimal HER2 expression, we consider it equivalent to HER2-zero. Similarly, our ‘low’ class corresponds to HER2-low. Given this distinction, despite previous IHC-based studies reporting higher proportions of HER2-low status among ER-positive breast cancers19, our analysis finds transcriptomics-defined HER2-low to be more prevalent in ER-negative disease (Supplementary Figure 3).
For a subset of 324 samples that received neoadjuvant chemotherapy (NACT), we obtained scores for response to NACT in the form of residual cancer burden (RCB), as well as treatment information. 44% of samples were from patients that received chemotherapy in combination with targeted anti-HER2 therapy (Supplementary Tables 2, 3). To investigate whether ERBB2 expression classes modulate response to NACT in samples of patients either treated or untreated with anti-HER2, we compared the pathological complete response (pCR) rates across ERBB2 mRNA and HER2 IHC classes, stratifying by the receipt of anti-HER2 therapy (Fig. 2). As only patients with an IHC score of 2+ (plus positive ISH) or 3+ are offered targeted treatment, there is an inherent selection bias in treatment assignment. As such, there are no samples with “very high” expression class in the untreated group and no samples of “very low” expression class in the treated group. As expected, among both treated and untreated patients, pCR is more widely distributed across ERBB2 expression classes compared to IHC scores, with the highest proportions of pCR observed in treated patients' tumors classified as “very high”.

The proportion of NACT samples in each treatment response category (RCB), split by ERBB2-expression class (a) and HER2 IHC score (b). The left-hand side displays samples from patients that received chemotherapy and the right-hand side displays samples from patients that received combined therapy. Gray color represents pCR, and blue indicates residual disease (non-pCR). Labels denote the absolute number of samples in each bar segment. RCB: residual cancer burden; class: ERBB2-expression class; HER2 IHC: HER2 status determined by immunohistochemistry.
Using Fisher’s Exact Test, both treated and untreated groups showed significant (p < 0.05) association between ERBB2 expression classes and treatment response in the form of pCR versus residual disease. On the contrary, when examining the association between HER2 IHC scores and treatment response, it was only found significant in the treated group. This is a further indication that NACT could be beneficial for more patients with tumors lacking IHC 3+ score. Notably, although our results link ERBB2 mRNA levels to NACT response, other studies have reported conflicting evidence. In a cohort of patients with HR + /HER2-negative breast cancer20, IHC HER2 status did not correlate with RCB score. Similarly, Schettini et al.21 found no association between baseline ERBB2 mRNA and pathological outcomes after NACT. Thus, more research is needed to determine whether response to neoadjuvant chemotherapy is ERBB2 expression-dependent.
Taken together, our findings underscore the importance of quantifying ERBB2 mRNA in cases where HER2 expression is too low for standard IHC to provide reliable results. Here, transcriptomic measurements better capture the continuum of HER2 expression compared to IHC-based classification (Supplementary Fig. 4). This study has certain limitations, including the lack of external validation and limited RCB data, which may restrict broader applicability of the findings.
Moreover, the DESTINY trial results have spurred renewed interest in developing more sensitive HER2 assays to better capture this heterogeneity and inform treatment decisions22,23,24. Despite its cost and relative complexity, RNA-Seq remains indispensable when comprehensive transcriptome data are required as it enables the identification and quantification of both known and novel transcripts—including non-coding RNAs, single nucleotide variants, fusion genes, and splice variants13. Moreover, RNA-Seq is ideally suited for high-throughput clinical diagnostics25 as it allows pooling and simultaneous processing of dozens of samples. Our combined dataset of 3564 cross-platform, whole-transcriptome ERBB2 mRNA measurements from breast tumors thus provides a reference set for future studies employing this method.
Refinement of HER2-low tumor classification and integration of mRNA profiling into diagnostics could improve patient selection for anti-HER2 therapies and optimize treatment outcomes. Ongoing Phase 2 clinical trials systematically enrolling HER2-low patients6 have the potential to suggest refinements of the targeted treatment strategies.
Methods
Study population and sample selection
The Copenhagen Breast Cancer Genomics Study (CBCGS) population is described in refs. 26,27, while the sample selection is illustrated in Supplementary Fig. 1. This register-based study was conducted with approval of the Danish Data Protection Agency (jr. no.: 2012-58-0004) and Danish Breast Cancer Group (jr. no.: DBCG-2015-14). The study did not include any contact with patients nor use of biological material, and thus ethical approval, including the need to obtain informed consent, was explicitly waived by the Ethical Committee of the Capital Region of Denmark. The NACT cohort comprises two distinct treatment groups. A subset of patients received chemotherapy alone—encompassing both single-agent and multiagent regimens—while another subset, corresponding to HER2-positive cases, received chemotherapy in combination with targeted anti-HER2 therapy (Supplementary Table 2).
NGS library preparation and sequencing
Total RNA was isolated from fresh tumor biopsies as previously described27. For 2,610 tumor samples, gene expression was quantified using Human Genome U133 Plus 2.0 Array (Afymetrix, Santa Clara, CA, USA) while for 954 samples it was quantified using paired-ended RNA-Sequencing on an Illumina NovaSeq (2 × 150 bp) or HiSeq2500 (2 × 126 bp) platform.
Computational data preprocessing
Microarray and NGS data were processed by the computational pipelines described in ref. 27. Briefly, for microarray files, the raw intensity.CEL file was preprocessed by quantile normalization, and probe summaries were extracted via robust multi-array average (RMA) using the affy package28 in R v. 4.0.041. For RNA-seq, fastq files were mapped to the human reference genome (GRCh37.p13 for samples analyzed before 2021, GRCh38 for subsequent samples) using STAR aligner29 v. 2.7.2b and reads overlapping features were quantified using Subread package’s featureCounts v. 1.6.230. Subsequently, values for each sample were normalized by size factors determined by the median ratio of gene counts relative to geometric mean per gene using the DeSeq2 R package31.
Histopathology data
Clinical and sample metadata were retrieved from DCBG, including metadata on histopathology assessments. We used the guidelines of the ASCO/CAP 20187, same as all Danish hospitals for histopathology assessment of HER2. For the non-NACT samples, only the earliest registered genomic sample for each patient was retained. Supplementary Fig. 1. Samples flagged for issues such as low quality, containing a large amount of normal tissue or classified as CIT256 molecular subtype outliers were removed.
Computational data analysis
Histopathology metadata such as HER2 immunohistochemistry scoring, FISH ERBB2 scoring, HER2 final status, and residual cancer burden were matched with transcriptomics measured ERBB2 expression by patient ID. Only HER2 status data that could be uniquely assigned to a transcriptomic sample were retained. R was used to curate histopathology metadata, while statistical analysis and visualization were carried out using Python libraries pandas32, numpy33 and seaborn34. The ROC curve was created using Python library scikit-learn35, whereby the IHC-based definition of HER2-low (IHC = 1+ or 2 + /normal) and HER2-zero (IHC = 0)36 were used as ground truth labels, with transcriptomics-measured HER2 expression as the continuous predictor.
Data availability
Transcriptomic profiles used in this study can be made available from the corresponding author on request.
Code availability
The code supporting this study is not publicly available but can be provided to qualified researchers upon reasonable request to the corresponding author.
References
Parker, J. S. et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 27, 1160–1167 (2009).
Smith, A. E. et al. HER2 + breast cancers evade anti-HER2 therapy via a switch in driver pathway. Nat. Commun. 12, 6667 (2021).
Slamon, D. J. Human breast cancer: correlation relapse survival amplification HER-2/ neu oncogene. Science 235, 177–182 (1979).
Ahn, S., Woo, J. W., Lee, K. & Park, S. Y. HER2 status in breast cancer: changes in guidelines and complicating factors for interpretation. J. Pathol. Transl. Med. 54, 34–44 (2020).
Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N. Engl. J. Med. 387, 9–20 (2022).
Schlam, I., Tolaney, S. M. & Tarantino, P. How I treat HER2-low advanced breast cancer. Breast 67, 116–123 (2023).
Wolff, A. C. et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline focused update. J. Clin. Oncol. 36, 2105–2122 (2018).
Wolff, A. C. et al. Human epidermal growth factor receptor 2 testing in breast cancer. Arch. Pathol. Lab. Med. 147, 993–1000 (2023).
Huang, R., Chen, M. & Shi, Y. Trastuzumab deruxtecan in low or ultralow HER2 metastatic breast cancer. N. Engl. J. Med. 392, 829–831 (2025).
Brasó-Maristany, F. et al. HER2DX ERBB2 mRNA expression in advanced HER2-positive breast cancer treated with T-DM1. J. Natl. Cancer Inst. 115, 332–336 (2023).
Griguolo, G. et al. ERBB2 mRNA expression and response to ado-trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Cancers12, 1902 (2020).
Fakhri, G. B. et al. Concordance between immunohistochemistry and microarray gene expression profiling for estrogen receptor, progesterone receptor, and HER2 receptor statuses in breast cancer patients in Lebanon. Int. J. Breast Cancer 2018, 1–6 (2018).
Mouttet, D. Estrogen-receptor, progesterone-receptor HER2 status determination invasive breast cancer. Concordance between immuno-histochemistry MapQuantTM microarray based assay. PLoS One 11, e0146474 (2016).
Fumagalli, D. et al. RNA sequencing to predict response to neoadjuvant anti-HER2 therapy: A secondary analysis of the NeoALTTO randomized clinical trial. JAMA Oncol. 3, 227–234 (2017).
Wojtaszewska, M. et al. Validation of HER2 status in whole genome sequencing data of breast cancers with the ploidy-corrected copy number approach. Mol. Diagn. Ther. 26, 105–116 (2022).
Pedersen, C. B., Nielsen, F. C., Rossing, M. & Olsen, L. R. Using microarray-based subtyping methods for breast cancer in the era of high-throughput RNA sequencing. Mol. Oncol. 12, 2136–2146 (2018).
Bardia, A. et al. Trastuzumab deruxtecan after endocrine therapy in metastatic breast cancer. N. Engl. J. Med. 391, 2110–2122 (2024).
Tarantino, P. et al. Comprehensive genomic characterization of HER2-low and HER2-0 breast cancer. Nat. Commun. 14, 7496 (2023).
Horisawa, N. et al. The frequency of low HER2 expression in breast cancer and a comparison of prognosis between patients with HER2-low and HER2-negative breast cancer by HR status. Breast Cancer 29, 234–241 (2021).
Schettini, F. et al. Unraveling the clinicopathological and molecular changes induced by neoadjuvant chemotherapy and endocrine therapy in hormone receptor-positive/HER2-low and HER2-0 breast cancer. ESMO Open 9, 103619 (2024).
Schettini, F. et al. Gene expression before and after neoadjuvant chemotherapy or endocrine therapy and survival outcomes in hormone receptor-positive, HER2-negative breast cancer: the NEOENDO study. Res. Sq. https://doi.org/10.21203/rs.3.rs-3937385/v1 (2024).
Prat, A. et al. Development and validation of the new HER2DX assay for predicting pathological response and survival outcome in early-stage HER2-positive breast cancer. EBioMedicine 75, 103801 (2022).
Moutafi, M. et al. Quantitative measurement of HER2 expression to subclassify ERBB2 unamplified breast cancer. Lab. Investig. 102, 1101–1108 (2022).
Torres, E. S. et al. 7P Independent validation of HER2DX ERBB2 mRNA score to predict HER2-positive (HER2+), HER2-low and HER2-0 status in breast cancer. ESMO Open 8, 101231 (2023).
Brueffer, C. et al. Clinical value of RNA sequencing-based classifiers for prediction of the five conventional breast cancer biomarkers: a report from the population-based multicenter Sweden Cancerome Analysis Network-breast initiative. JCO Precis. Oncol. 2, PO.17.00135 (2018).
Berg, T. et al. Molecular subtyping improves breast cancer diagnosis in the Copenhagen Breast Cancer Genomics Study. JCI Insight 9, e178114 (2024).
Jensen, M.-B. et al. Multigene profiles to guide the use of neoadjuvant chemotherapy for breast cancer: a Copenhagen Breast Cancer Genomics Study. NPJ Breast Cancer 9, 47 (2023).
Gautier, L., Cope, L., Bolstad, B. M. & Irizarry, R. affy-analysis Affymetrix GeneChip data probe level. Bioinformatics 20, 307–315 (2004).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
The pandas development team. pandas-dev/pandas: Pandas. Zenodo (2020). https://doi.org/10.5281/zenodo.3509134.
Harris, C. R. et al. Array programming with NumPy. Nature 585, 357–362 (2020).
Waskom, M. seaborn: statistical data visualization. J. Open Source Softw. 6, 3021 (2021).
Pedregosa, F. et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Kook, Y. et al. Differentiating HER2-low and HER2-zero tumors with 21-gene multigene assay in 2,295 h + HER2- breast cancer: a retrospective analysis. Breast Cancer Res. 26, 154 (2024).
Author information
Authors and Affiliations
Contributions
Study conceptualization: M.R.; Data acquisition: M.R., M.R.J.; Methodology: B.E., M.R., M.R.J., M.L.T.; Data curation: M.R., M.B.J., M.L.T., M.A.M.; Data analysis and visualization: M.A.M.; Manuscript writing: M.R., M.A.M.; Critical review of the manuscript: All authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Misiakou, MA., Jensen, MB., Talman, ML. et al. Enhancing HER2-low breast cancer detection with quantitative transcriptomics. npj Breast Cancer 11, 98 (2025). https://doi.org/10.1038/s41523-025-00817-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41523-025-00817-9
