
Abstract
The development and understanding of antifreezing hydrogels are crucial both in principle and practice for the design and delivery of new materials. The current antifreezing mechanisms in hydrogels are almost exclusively derived from their incorporation of antifreezing additives, rather than from the inherent properties of the polymers themselves. Moreover, developing a computational model for the independent yet interconnected double-network (DN) structures in hydrogels has proven to be an exceptionally difficult task. Here, we develop a multiscale simulation platform, integrating ‘random walk reactive polymerization’ (RWRP) with molecular dynamics (MD) simulations, to computationally construct a physically-chemically linked PVA/PHEAA DN hydrogels from monomers that mimic a radical polymerization and to investigate water structures, dynamics, and interactions confined in PVA/PHEAA hydrogels with various water contents and temperatures, aiming to uncover antifreezing mechanism at atomic levels. Collective simulation results indicate that the antifreezing property of PVA/PHEAA hydrogels arises from a combination of intrinsic, strong water-binding networks and crosslinkers and tightly crosslinked and interpenetrating double-network structures, both of which enhance polymer-water interactions for competitively inhibiting ice nucleation and growth. These computational findings provide atomic-level insights into the interplay between polymers and water molecules in hydrogels, which may determine their resistance to freezing.
Similar content being viewed by others

Introduction
Icing is a natural phenomenon that plays a crucial role in sustaining life on Earth, but unwanted icing can cause severe economic, environmental, and life-threatening consequences1,2,3. Considerable efforts have been made to tackle the grand challenge of developing antifreezing (anti-icing) materials, which has significant implications for materials design concepts, antifreezing mechanisms, and many emerging applications for wearable devices, energy storage electronics, artificial tissues, and human-machine interfaces. Conventional antifreezing materials, such as icephobic water-free organics or hydrophilic hydrogels containing antifreezing additives, often exhibit weak mechanical properties when exposed to subzero temperatures, thereby restricting their practical applications. Antifreezing hydrogels possess a distinct advantage over icephobic materials in practical applications, primarily owing to their soft-wet-dynamic similarity to biological tissues4. This characteristic allows them to achieve or maintain desirable functions in cold conditions for bio-related applications.
Current fabrication strategies for antifreezing hydrogels often involve the incorporation of various antifreezing additives into polymer networks, and these antifreezing additives include (1) ionic compounds such as NaCl, ZnCl2, CaCl2, and ionic liquids5,6,7,8, (2) natural biomolecules such as antifreeze proteins, ice-binding proteins, glycoproteins, and silk proteins9,10,11,12,13; (3) carbon/nano materials such as carbon nanotubes, nanofillers, and oleophilic materials14,15,16,17,18,19; and (4) organic/water solvents such as DMSO, benzyltrimethyl ammonium hydroxide, betaine, proline, and glycerol20,21,22,23,24,25. These antifreezing additives function as cryoprotectants by altering the water-ice phase equilibrium at different stages of ice condensation, nucleation, and growth. However, the incorporation of these additives often leads to the formation of heterogeneous network structures, which can compromise the mechanical properties of antifreezing hydrogels during the deformation or freezing processes26.
It is worth noting that, until very recently, there were only two types of fully polymeric antifreezing hydrogels being developed, which did not require the addition of any antifreezing additive. Zhi et al.27 synthesized ethylene glycol-based waterborne anionic polyurethane acrylates/polyacrylamide (EG-waPUA/PAM) hydrogels using a step-growth copolymerization approach by incorporating isophorone diisocyanate monomers, dimethylol propionic acid chain extenders, and ethylene glycol as a crosslinker. While EG-waPUA/PAM hydrogels were reported as fully polymeric antifreezing hydrogels, ethylene glycol used in their synthesis is widely recognized as a water freezing inhibitor. Different from this single antifreezing system, we recently demonstrated a general crosslinking strategy that uses the 4, 9-dioxo-5, 8-dioxa-3, 10-diazadodecane-1, 12-diyl diacrylate (EGINA) to fabricate a family of EGINA-crosslinked double-network hydrogels with intrinsic antifreezing properties, but without any antifreezing additive28. The EGINA crosslinker was designed and synthesized to enable crosslinking of a wide range of hydrogels made of hydrophilic, electrolyte, zwitterionic, and macromolecular polymers with single- or double-network structures. At −20 oC and optimal conditions, the EGINA-crosslinked hydrogels achieved impressive mechanical performance and freeze-tolerance at −20 oC (i.e., high stretchability of 1800%, high tensile stress of 1.1 MPa), and high toughness of 9400 J/m3), with long-term stability for up to 24 h28.
From a mechanistic perspective, the rational design of antifreezing hydrogels is still far from ideal and is currently hindered by a lack of fundamental understanding of how water-polymer interactions affect the structure, dynamics, and behavior of water molecules confined within the hydrogel network, in relation to antifreezing property of hydrogels. Generally speaking, antifreezing mechanisms of hydrogels depend on the structural and dynamic properties of polymer networks, water molecules, and other components included within hydrogels. For hydrogels containing antifreezing additives (i.e., cryoprotectant), frozen-resistance ability primarily stems from the strong interactions between antifreezing additives and water molecules, because these interactions reduce the amount of available water to form ice nuclei. Different from this cryoprotectant-induced antifreezing mechanism, the crosslinked polymer network of hydrogels plays a multifaceted role in controlling their antifreezing property. Chemical, physical, and structural properties of polymer networks, including the degree of hydrophilicity, the chemistry of pendant groups, mesh size, polymer conformation, and network crosslinking density, greatly affect the complex interplay between water-polymer interactions and the confinement effects on water dynamics and structures. Numerous experimental studies from dielectric relaxation29, nuclear magnetic resonance (NMR)30, neutron scattering spectroscopy31,32, and infrared spectroscopy33 have been conducted to characterize the behavior of water confined in the hydrogel networks, in relation to antifreezing properties. Although some of these studies presented different results, a growing body of evidence supports the general observation that some water molecules confined in the hydrogel network exhibit strong interactions with polymer chains, forming a hydration layer around the polymers through multiple non-covalent interactions (e.g., hydrogen bonding, van der Waals interaction, and polar interactions). This hydration layer allows to prevent water molecules themselves from forming ice-like clusters and nuclei, thereby enhancing the frozen resistance of hydrogels under subzero temperatures.
Despite a large number of experimental studies on antifreezing hydrogels34 and computational studies on hydrogels35,36, the application of molecular dynamics (MD) simulations to investigate antifreezing hydrogels has been relatively limited. Ge et al.37 conducted MD simulations of double-network hydrogels made of ion-incorporated polyacrylamide/cellulose nanofibrils. Simulations showed that the addition of LiCl effectively increased the binding affinity of water molecules to polymer networks, leading to the emergence of a non-freezing bound water state, without any observable solid-liquid phase transition. Li et al.38 utilized MD simulations to study the deicing property of electrolyte hydrogels (EHs). They found that electrostatic and van der Waals interactions between the ions and ice crystals facilitated the destruction of the ice crystal structure and the formation of a liquid-like lubricating layer on the EH hydrogels. While these MD simulations offer advantages in revealing the atomic-level molecular interactions between polymers and water molecules, all of these computationally studied hydrogel systems typically incorporate various salts into their hydrogel networks. The antifreezing mechanisms observed in these systems are likely attributed to the strong interactions between these ions and the surrounding water. Such ionic solvation can effectively disrupt the hydrogen bond networks and structural arrangement between water molecules, ultimately hindering the ice nucleation processes.
Evidently, there is a significant knowledge gap in computationally studying fully polymeric antifreezing hydrogels without additives and in providing a mechanistic understanding of how water-polymer interactions influence the structure, dynamics, and interactions of water confined in the hydrogel, particularly in relation to antifreezing mechanisms that depend solely on the properties of the polymer themselves. While advanced molecular system design packages like Moltemplate39 and RdKit (https://www.rdkit.org), in conjunction with subsequent equilibration through soft potentials and techniques such as Monte Carlo runs or distance-based crosslinking, facilitate the creation of extensive polymeric systems, it remains crucial to develop efficient computational algorithms for the construction of polymer networks from individual monomers. As a result, these computationally constructed hydrogel models35,36,37, despite their potential, have contained certain undesirable artifacts, particularly pertaining to initial configuration, topology structures, and simulation time. The initiation of the polymer chains within these models often entails duplicating monomers to achieve a specific length while introducing conformational diversity through randomized torsional angles along the polymer backbone. However, these polymer chains frequently contain a limited number of monomeric units, leading to polymer configurations with improbable entanglements or voids that may deviate from accurate polymer interactions and behaviors. Moreover, the MD simulations executed for periods shorter than a hundred nanoseconds are often insufficient for comprehensive characterization of structural transitions, relaxation phenomena, and true equilibration dynamics. To bridge these gaps, we developed a ‘random walk reactive polymerization’ (RWRP), which imitates a radical polymerization process, to construct a physically-chemically linked polyvinyl alcohol (PVA)/poly(N-(2-Hydroxyethyl)acrylamide) PHEAA double-network (DN) hydrogels from monomers with varying water contents, followed by MD simulations to study the water molecules confined in the polymer networks for better understanding the antifreezing property of PVA/PHEAA hydrogels. The RWRP-constructed PVA/PHEAA DN hydrogels resembled the experimental properties of network structures. MD simulations, complemented by supporting experiments, were then performed to investigate the structure and dynamics of water molecules around both PVA and PHEAA chains in response to different water contents. The sustainable antifreezing property of PVA/PHEAA DN hydrogels is likely stemmed from (i) the formation of tightly bound waters with networks via hydrogen bonds and (ii) the confinement of these waters in highly crosslinked double-network structures, both of which prevent ice nucleation from free water molecules in hydrogel network so as to retain at an unfrozen state. It is worth highlighting that numerous kinetic algorithms have been specifically developed for the purpose of simulating polymer formation and properties through Dissipative Particle Dynamics (DPD) simulations employing coarse-grained (CG) resolutions40,41,42,43,44. The intricacies inherent in DPD simulations with CG resolutions are primarily centered around the treatment of non-bonded interactions and the incorporation of bonded parameters. These simulations are largely confined to modeling linear polymers (rather than hydrogels), often with constraints on conformational variability. Importantly, none of these algorithms have been applied to model atomic-level polymer systems, especially those featuring more intricate double-network structures and explicit water environment. This RWRP algorithm can be extended to investigate a broad range of polymer systems, including elastomers, hydrogels, and polymers synthesized through radical polymerization methods. This opens up opportunities for the computational design of customizable materials with precise control from the molecular scale and beyond.
Methods
Force field parameters
We utilized the ParaChem tool (https://cgenff.paramchem.org/)45 to develop force field parameters for vinyl alcohol (VA) and N-(2-Hydroxyethyl)acrylamide (HEAA) monomers and EGINA crosslinker in the CHARMM27 format46. Figure 1a displays the molecular structures of the VA and HEAA monomers and EGINA crosslinker, while their respective force field parameters in the CHARMM format can be found in Supplementary Table 1. We employed the TIP3P water model in CHARMM27 parameter set with the CMAP correction to describe water molecules.

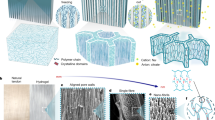
a A step-by-step computational process for constructing EGINA crosslinked PVA/PHEAA DN hydrogel in the presence of water for MD simulations, involving the sequential generation of single-network PVA, double-network PVA/PHEAA, EGINA crosslinked PVA/PHEAA, and solvated PVA/PHEAA hydrogel. The simulation boxes for each individual system have been structured into a cubic shape, with each edge measuring 56 or 78 Å in length. b Molecular structures of VA and PVA, HEAA and PHEAA, and EGINA crosslinker. c A structural representation of EGINA crosslinked PHEAA (left) and a conformation of EGINA crosslinked PHEAA in the simulation models (right). In (c), two chains (chain 1 and chain 2) of PHEAA are crosslinked by EGINA, and each chain is separated into two segments (segment 1 and segment 2 for chain 1, and segment 3 and segment 4 for chain 2) by the cross-linked position.
Random walk reactive polymerization (RWRP)
We reported a one-pot, stepwise polymerization approach for the synthesis of PVA/PHEAA DN hydrogel by forming the first chemically linked PVA network, followed by photo-polymerizing and crosslinking the second chemically linked PHEAA within the first PVA network (Fig. 1a). Here, we developed a computational algorithm of random walk reactive polymerization (RWRP), which mimics the two-step radical polymerization, for computationally constructing PVA/PHEAA DN hydrogels with adjustable water contents. Specifically, a total of 300 VA monomers and 750 HEAA monomers, equivalent to ~3.4 M in experiments, were added randomly to a study system with random orientations and positions. To initiate the first polymerization of the PVA network, 2 mol% of VA monomers were activated and acted as initiators, reacting with other VA monomers to form PVA chains. During the first polymerization step, three index lists were generated: Mlist for VA monomers, Rlist for initiators, and Plist for PVA chains, and these lists were then updated for every subsequent polymerization step. During each polymerization attempt, an initiator (ri) was randomly selected from Rlist. A distance search was then performed to identify nearby VA monomers within a reaction distance of di, which initially started with 10 Å. If no VA monomers were available for polymerization, the reaction distance was adaptively increased by ~10%. If a nearby VA monomer was found within the reaction distance of di, one VA monomer was randomly selected to react with the initiator (ri), forming a new bond. The connectivity table, which included all bonds, angles, and torsions, was then reconstructed, and the Mlist, Rlist, and Plist lists were updated accordingly. Energy minimization was performed to remove any unfavorable contacts and optimize the system. This was followed by a short MD simulation to allow for the diffusion and dynamics of all remaining monomers and PVA chains within the relaxed DN hydrogel. This process was iteratively preceded and then terminated until all VA monomers in Mlist were consumed. Upon the formation of the first PVA network, the second polymerization process including initiator selection, HEAA monomer search, bond formation, connectivity table reconstruction, energy minimization, and MD simulation was iteratively repeated until all HEAA monomers in Mlist were consumed to form the second PHEAA network. It is important to emphasize an evolution in the random walk reactive polymerization (RWRP) algorithm between our studies. In our previous paper47, the pseudo-RWRP algorithm facilitated the construction of the second PAM network, with the Agar network being pre-constructed using crystal structures from the protein databank. However, in this study, we have made substantial enhancements to the RWRP algorithm. It now enables the sequential or simultaneous construction of both networks from monomers.
To simulate the crosslinking of the second PHEAA chains with EGINA crosslinkers, the EGINA molecules were incorporated into the PVA/PHEAA system through a random selection process that involved choosing a pair of PHEAA chains to be crosslinked. Grand canonical Monte Carlo (GCMC) simulation was used to determine the crosslinking acceptation probability by \({P}_{{add}}^{{accept}}=\min \left[1,\exp \left(-\beta \Delta E\right)/\left({N}_{i}+1\right)\right]\) where ΔE is the change in configuration energy due to the addition of EGINA. The change in configurational energy (ΔE) upon the insertion of an EGINA molecule was calculated using an energy evaluation procedure. First, the initial energy of the system with the selected pair of PHEAA chains was recorded. Then, an EGINA molecule was inserted into the system at a random position in the vicinity of the selected PHEAA chains. The new energy of the system after the insertion was calculated. The change in configurational energy (ΔE) was then determined as the difference between the initial energy and the energy after the EGINA insertion. The GCMC simulation was performed iteratively, with multiple attempts to insert EGINA molecules into the system until the desired crosslinking density was achieved. The successful rate of incorporating of crosslinkers is as low as ~0.1% due to steric repulsion effect. The final equilibrated system after the GCMC simulation was used for subsequent molecular dynamics (MD) simulations to study the behavior of water molecules confined within the PVA/PHEAA DN hydrogel network.
MD simulations of double-network hydrogels
After generating PVA/PHEAA DN networks through the RWRP algorithm, we proceeded to solvate these networks with TIP3P water molecules. To prevent atom collapse, we removed any water molecules within 2.4 Å. The resulting PVA/PHEAA DN hydrogel model allows for variations in water content of 50% and 73%, which aligns with experimental conditions. The resultant solvated system (i.e., PVA/PHEAA DN hydrogel) was structured into a cubic shape, with each edge measuring 56 or 78 Å in length depending on water contents, minimized in energy using a combination of steepest descent and conjugate gradient, and equilibrated by short MD simulations of 10–12 ns at the room temperature of 298 K using NPT ensemble and 3D periodical condition. Each hydrogel system underwent an annealing process involving a gradual heating up process from 0 °C to 25 °C, or conversely, a cooling process from 0 °C to −20 °C. This annealing process was orchestrated by adjusting the temperature by 5 °C at every nanosecond interval until equilibrium conditions were reached. Then, extensive long MD production runs of 1 microsecond each were conducted using the all-atom NAMD 2.12 package48 with the CHARMM force field under an NVT (constant number of atoms, constant volume, and constant temperature) ensemble and 3D periodic boundary conditions. The temperature was maintained at 298 K using Langevin thermostat method with a damping coefficient of 1 ps−1. All covalent bonds, including hydrogen bonds, were constrained by RATTLE method, so that velocity Velert method was performed to integrate Newton’s motion equation with a larger timestep of 2 fs. During MD production runs, long-range electrostatic potentials were calculated by Particle Mesh Ewald (PME) with the grad space of 0.5 Å, while short-range van der Waals (VDW) potentials were estimated by the switching function with a twin-range cutoff at 12 Å and 14 Å. All MD trajectories were saved every 2 ps for further analysis. Furthermore, the similar modeling procedure was applied to PVA single network (SN) hydrogel and PHEAA SN hydrogel for comparison. For each hydrogel system, two or three independent simulations were conducted, depending on the convergence of the systems. The final analysis results were then collated and averaged from these independent simulations.
Results and discussion
RWRP-assisted construction of PVA/PHEAA double network hydrogels
In our previous work, we have synthesized and characterized the antifreezing characteristics of PAV/PHEAA DN hydrogel28, consisting of a PVA network as the first physical network linked by hydrogen bonds, and a PHEAA network as the second chemical network crosslinked by EGINA. To computationally build the PVA/PHEAA DN hydrogel, a general ‘Random Walk Reactive Polymerization’ (RWRP) protocol was developed, as shown in Fig. 1a. The computational RWRP protocol employed in this study closely emulates the two-step, one-pot process of free radical polymerization that we utilized for synthesizing the PVA/PHEAA antifreezing hydrogel in our prior research28. To outline its fundamental procedure briefly, the protocol involves the random mixing of precursors—comprising a specific quantity of VA monomers, HEAA monomers, and EGINA crosslinkers (depicted in Fig. 1b). Each molecule in the mixture assumes a random orientation and position. The RWRP approach has been meticulously tailored to replicate the process of chain-growth polymerization via the radical reaction mechanism. This mechanism entails the presence of a single reactive site (radical) at the terminus of each newly forming chain. Initiating the polymerization to create the initial PVA single-network involves generating 23 reactive sites from VA monomers, mirroring the concentration of initiators used in experimental scenarios (1 mol%). The RWRP process advances by selecting, in a random manner, a reactive site and one of the adjacent VA monomers. Their potential to form a bond within a dynamic reaction radius is then assessed. If successful, a new bond is established, and the resultant topological connectivity, encompassing bond, angle, and torsion parameters, is incorporated into the force field considerations. In cases of unsuccessful attempts, the selection process is repeated until all VA monomers are consumed and polymerized into PVA chains. As new bonds form, a combination of energy minimization and brief molecular dynamics (MD) simulations is carried out to optimize network structures for subsequent polymerization phases. This process also facilitates molecule diffusion in response to concentration gradients arising from ongoing reactions. After the initial PVA network is established, the RWRP protocol is re-employed to fabricate the second PHEAA network through a series of iterative stages. These stages encompass initiator selection, seeking HEAA monomers, forming bonds, updating the connectivity table, executing energy minimization, and conducting MD simulations. The GCMC algorithm is subsequently enlisted to crosslink EGINA with two neighboring PHEAA chains, as illustrated in Fig. 1c. This comprehensive methodology effectively reproduces the intricate process of hydrogel formation and enables a detailed exploration of its properties. Subsequently, the resulting EGINA-crosslinked PVA/PHEAA double networks were hydrated with TIP3P-modeled water molecules to create hydrogel states with varying water contents (50 wt% and 73 wt%) for MD simulations, which permit the study of water dynamics, structure, and interaction within hydrogel networks in relation to the antifreezing property of hydrogels. To provide a comparative analysis, PVA single-network hydrogels and PHEAA single-network hydrogels with varying water contents were also computationally generated and investigated, alongside the EGINA-crosslinked PVA/PHEAA double-network hydrogels (Table 1). A visual representation of the complete RWRP process utilized to computationally produce the PVA/PHEAA double network is shown in Supplementary Movie 1.
Network structure and dynamics of PVA/PHEAA hydrogels at subzero temperature
Given the pure polymeric nature of PVA/PHEAA hydrogels without any antifreezing additives, the polymer network is considered as a primary factor in preventing ice nucleation and growth, thus providing the hydrogels with its antifreezing property49. Here, the presence of a double-network structure in PVA/PHEAA hydrogels results in the formation of highly compacted networks with very small pores, typically measuring <10.0 Å in size. These pores are generated when multiple polymer chains cluster together to form bundles, leading to the creation of numerous small voids within the hydrogel structure. This arrangement gives rise to a high probability of small pore formation, contributing to the unique properties of the hydrogels. By visually inspecting the MD trajectories of PVA/PHEAA hydrogels at various temperatures ranging from −20 °C to 25 °C, it was observed that as the temperature increased, the polymer chains exhibited a tendency to cluster together (Fig. 2a). The clustering of polymer chains in PVA/PHEAA hydrogels with increasing temperatures can be attributed to two primary factors. At −20 °C, intermolecular forces such as hydrogen bonding between polymer chains are relatively strong, resulting in a more extended conformation of the chains. However, as the temperature rises, these intermolecular interactions weaken, allowing the chains to come closer together and form clusters. This is due to the reduced strength of intermolecular forces at higher temperatures. Furthermore, increasing the temperature raises the thermal energy of the system, leading to greater molecular motion. As a result, the polymer chains become more mobile and have a higher likelihood of interacting and forming clusters. The combination of weakened intermolecular forces and increased molecular motion at higher temperatures promotes the clustering phenomenon in PVA/PHEAA hydrogels. As the water content increased from 50 wt% to 73 wt%, the polymer chains experience reduced steric hindrance and can adopt more extended and flexible configurations. Visual inspection of the MD trajectories revealed noticeable trends in the pore formation as the water content increased and the crosslinking density reduced. The observed increase in the formation of larger pores was consistent with the higher flexibility and swelling behavior of the hydrogel at higher water content.

a Representative MD snapshots of the EGINA crosslinked PVA/PHEAA hydrogels with 73 wt% at −20 °C (top) and 25 °C (bottom). PVA, PHEAA, and EGINA are characterized by blue CPK, red sphere, and yellow sphere, respectively. The figures do not display the pedant groups of PHEAA. b Probability distribution of network-based root mean square fluctuations (RMSFs) for the PVA (top) and PHEAA (bottom) networks. RMSF values are calculated based on the carbon atoms in the backbone of the two networks (C4 in PVA and C13 in PHEAA). c The number of hydrogen bonds between polymer networks across different temperatures and water contents. The data in (b) and (c) were obtained from the last 500-ns MD trajectories.
To investigate the impact of temperature on the mobility of polymer chains, Fig. 2b presents a comparison of the root-mean-square fluctuation (RMSF) of atomic positions for each heavy atom in both PVA and PHEAA networks of the hydrogels across a temperature range from −20 °C to 25 °C. Upon initial observation, it appears that the PVA network within the hydrogels, regardless of water content and temperature, exhibited the higher mobility than the PHEAA network, as indicated by RMSF values. The difference in mobility between the PVA and PHEAA networks can be attributed to the presence of three distinct hydrogen bonding groups (OH–, NH–, and O=) in PHEAA, in contrast to only the OH- group in a PVA chain. These additional hydrogen bonding groups in the PHEAA network result in stronger interactions with water molecules and neighboring chains. Consequently, these stronger interactions impose a restraining effect on the local dynamics of the PHEAA network chains, leading to reduced mobility compared to the PVA network. Furthermore, with an increase in water content from 50 wt% to 73 wt% in the hydrogels at the same temperature, the RMSF peaks in the RMSF distribution profiles became broader and exhibited lower probabilities. This observation indicates that the polymer networks in water-rich hydrogels exhibit higher mobility compared to hydrogels with lower water content. The higher mobility can be attributed to the less tightly packed polymer chains in water-rich environments, allowing for greater movement and flexibility.
More importantly, it is apparent that temperature plays a significant role in influencing the mobility of polymer networks within the hydrogels. With a decrease in temperature from 25 °C to −20 °C, notable changes can be observed in the RMSF profiles of both PVA and PHEAA networks. The prominent peaks in the RMSF profiles become narrower and shift towards lower RMSF values, indicating a decrease in mobility. Specifically, for the PVA network, the RMSF values transited from the range of 4.5–6.1 Å to 1.8–2.4 Å with a higher probability. Similarly, for the PHEAA network, the RMSF values shifted from around 2 Å to the range of 1.5–1.8 Å. These findings suggest that lower temperatures induce a thermodynamic damping effect, resulting in a more constrained mobility of the polymer chains within the hydrogels. This thermodynamic damping effect, together with the decreased thermal energy and increased intermolecular interactions, restricts the chain dynamics and limits their ability to move and fluctuate, resulting in a decrease in RMSF values at lower temperatures. Furthermore, as illustrated in the Fig. 2c, we observed that increasing the water content from 50 wt% to 73 wt% did not produce a significant change in the number of hydrogen bonds between polymer networks at both −20 °C and 25 °C. This finding indicates that the polymer network maintains a relatively stable conformation and internal interactions across this range of water content. Conversely, when examining hydrogels with the same water content, we noted a decrease in the number of hydrogen bonds between polymer networks as the temperature decreased from 25 °C to −20 °C. This implies that at lower temperatures, these polymer networks provide an increased number of binding sites for water molecules, resulting in the formation of stronger water-binding polymer networks. These networks play a critical role in preventing ice formation within the polymer structure at lower temperatures.
Water structure and dynamics confined in PVA/PHEAA DN hydrogels at subzero temperature
It is generally accepted that there exist two distinct types of water molecules within hydrogel network, i.e., bound and nonbound water. At a molecular level, bound and nonbound water can be characterized and distinguished by several parameters, including self-diffusion coefficient, residence time, and radial distribution function (RDF) of water molecules around polymer chains. Figure 3a shows the radius distribution function (RDF) of water molecules around polymer chains in four hydrogels with different water contents (50 wt% and 73 wt%) and at different temperatures (253 K to 300 K). At first glance, the g(r) curves for all hydrogel systems appeared to follow similar trends, with the first, dominate peak occurring at a distance of ~2.85 Å and a second, broader peak at a distance of ~5.45 Å. However, closer inspection of these curves also revealed certain differences in the water structure of hydrogel systems in response to changes in water content and temperature. As the higher RDF peak indicates an increased presence of water molecules around the polymer networks, the decrease in temperature from 25 oC to -20 oC for the same hydrogels caused an increase in RDF intensities, indicating that water molecules have the high tendency to cluster and condense around the polymer chains at lower temperatures. Moreover, the increase in water content from 50 wt% to 73 wt% in hydrogels resulted in a decrease in RDF intensities at the same temperature. This suggests that a higher packing density of polymer chains, which occurs at lower water content, is more effective for attracting and trapping the surrounding water molecules. The RDF results indicate that an increase in polymer density and a decrease in temperature lead to a tighter packing of water molecules around the polymer chains, resulting in a corresponding increase in observed RDF intensities.

a RDF, b coordination number, c self-diffusion coefficient, and d residence time of water molecules around the PVA network for the four different double-network PVA/HEAA hydrogels with 50 wt% and 73 wt% at −20 °C and 25 °C.
To further analyze RDF profiles, The coordination number (Nw) of water molecules surrounding the polymer chains in different hydrogels at various temperatures was determined by integrating the first hydration shell of RDFs from Fig. 3a. As shown in Fig. 3b, Nw of the PVA network exhibited a decreasing order: 1.68 in 50 wt% hydrogels at −20 oC > 1.38 in 50 wt% hydrogels at 25 oC and 1.69 in 73 wt% hydrogels at −20 oC > 1.54 in 73 wt% hydrogels at 25 oC. A similar phenomenon also can be observed for Nw of the PHEAA network (Supplementary Fig. 1). These differences in Nw provide further evidence that more water molecules are favorable to stay in close proximity to densely packed polymer chains at lower temperatures, as compared to more loosely packed polymer chains at higher temperatures.
From a dynamic perspective of water molecules around polymer networks in hydrogels, we measured the self-diffusion coefficients (SDCs) of water molecules within the first and the second hydration shells of polymer chains in hydrogels. In Fig. 3c, SDCs of water molecules in all hydrogels (0.67–2.07 × 10−5 cm2 s−1) were much smaller than that of bulk water (2.54 × 10−5 cm2 s−1)50, clearly indicating the existence of polymer-water interactions that substantially impede the movement of water within the polymer network. Moreover, decreasing the temperature from 25 °C to −20 °C resulted in a reduction in the SDCs of water molecules in both 50 wt% hydrogels from 1.28 × 10−5 cm2 s−1 to 0.67 × 10−5 cm2 s−1 and 73 wt% hydrogels from 2.07 × 10−5 cm2 s−1 to 1.06 × 10−5 cm2 s−1. This finding is consistent with our intuition that water molecules tend to move more rapidly at elevated temperatures. In line with our findings regarding the impact of temperature on water molecule mobility, increasing the water content of hydrogels at a constant temperature resulted in a corresponding increase in the SDC of water molecules within hydrogel networks. This provides additional evidence that water molecules exhibit greater mobility within hydrogels containing higher water content.
In addition to SDC, another key dynamic property of water molecules is their residence time around polymer chains. A longer residence time implies a more stable association between polymer chains and water molecules. The residence time is determined by fitting the function of \({C}_{R}\left(t\right)=\frac{1}{{N}_{w}}\mathop{\sum }\limits_{j=1}^{{N}_{{water}}}\frac{ < {P}_{{Rj}}(0){P}_{{Rj}}(t) > }{ < {P}_{{Rj}}{(0)}^{2} > }\) with \({C}_{R}\left(t\right)=A\exp (-\frac{t}{{\tau }_{s}})\), where PRj is a binary function, value of 1 (0) indicates the jth water stays (leaves) in (from) a layer with a thickness of R at a time of t. In Fig. 3d, the rate of decay of \({C}_{R}\left(t\right)\) curves in hydrogels was slower at low temperatures and with low water content compared to hydrogels with high water content and at higher temperatures, showing a decreased order of 50 wt% hydrogels at −20 oC (126.7 ps) > 73 wt% hydrogels at −20 oC (49.7 ps) > 50 wt% hydrogels at 25 oC (37.8 ps) > 73 wt% hydrogels at 25 oC (25.2 ps), consistent with SDC results. When considering different computational properties together, it is evident that interaction strength between polymer networks and water molecules varies, with more water molecules being inclined to form a stronger and more stable association with densely packed polymer chains at lower temperatures. This, in turn, leads molecules to the formation of a strongly bound state for water molecules, which are resistant to freezing.
Polymer-water interactions in PVA/PHEAA DN hydrogels at subzero temperature
To gain deeper insights into the antifreezing mechanism of EGINA crosslinked PVA/PHEAA hydrogels, it is crucial to investigate their spatial conformation and the intermolecular interactions between water molecules and polymer networks. In Fig. 4a, given that both PVA and PHEAA networks possess robust hydrogen bonding groups such as OH–, NH–, and O=, they exhibited a strong propensity to interact with water molecules through hydrogen bonds. Such hydrogen-bonding interactions occurred between the oxygen and nitrogen atoms in these functional groups and the hydrogen atoms in water. Consequently, bound water molecules orient themselves in a way to establish a hydrogen-bond network within the hydrogels. To quantitatively analyze the hydration energy responsible for the antifreezing property of hydrogels, the total number of hydrogen bonds and the intermolecular interactions between water molecules and polymer chains were calculated and averaged from the last 500-ns simulations. As shown in Fig. 4b, both hydrogels with distinct water contents of 50 wt% and 73 wt% at −20 oC contained more hydrogen bonds than the same hydrogels at 25 oC. As the temperature increases, the hydrogen-bonded water molecules within the hydrogels were progressively replaced by free water. Specifically, the elevated temperature acted to destabilize the hydrogen bonds formed between water molecules and hydrophilic polymers. This computational phenomenon results in a shift from a more structured hydrogen-bonded network to a state where water molecules are no longer bound in the same manner to the hydrophilic polymers, consistent with recent spectroscopy measurements51, where hydrogen bonds become disrupted at higher temperatures. In addition, the PHEAA network exhibited a higher number of hydrogen bonds with water molecules compared to the PVA network. This can be attributed to the presence of additional hydrophilic NH- and O= groups in PHEAA. The higher number of hydrogen bonds restricts the mobility of PHEAA network more, as a result of the stronger intermolecular interactions between water molecules and PHEAA chains, consistent with the lower RMSF values (indicating less mobility) of PHEAA as shown in Fig. 2b.

a MD snapshots depicting five distinct hydrogen bonds formed between water molecules and PVA or PHEAA in PVA/PHEAA DN hydrogels, including water-PVA(O8), water-PHEAA(O11), water-PHEAA(O8), PHEAA(O11)-water, and PHEAA(N9)-water, where O8, O11, and N9 represent oxygen and nitrogen atoms, respectively. Hydrogen bonds are indicated by yellow dashes. b Average polymer-water hydrogen-bond number and c average polymer-water interaction energies in the PVA/PHEAA DN hydrogel with the water content of 50 wt% and 73 wt% at temperatures of −20 °C and 25 °C. Average values in (b) and (c) are calculated by extracting data from the last 500-ns MD trajectories.
In Fig. 4c, all four hydrogels, each with different water contents and subjected to varying temperatures, displayed significant polymer-water interactions. This was indicated by the average solvation energy (intermolecular interaction energy) in the following decreasing order: ~−24,522 kcal/mol for 73 wt% hydrogel at −20 °C < ~−21,655 kcal/mol for 50 wt% hydrogel at −20 °C < −20,785 kcal/mol for 73 wt% hydrogel at 25 °C < ~−18,761 kcal/mol for 50 wt% hydrogel at 25 °C. Among the four hydrogel systems, it was observed that the hydrogels at −20 °C exhibited much lower solvation energies than the same hydrogels at 25 °C. This suggests that at lower temperatures, the polymer chains have a stronger affinity for and preferential binding to water molecules through hydrogen bonds, thus presenting strongly bound water molecules (nonfreezing water molecules) around polymer chains. Hypothetically, the strengthening of water-polymer bonds results in a tightening of the hydrogel networks. This tightening of the network structure leads to a reduction in volume, creating a significant negative pressure within the hydrogels. Consequently, this negative pressure further decreases the surface vapor pressure, enhancing the hydrogels’ resistance to freezing. At high temperatures, it is anticipated that the hydrogen bonds between polymers will be disrupted, resulting in an enhanced mobility of the side chains. However, the number of hydrogen bonds between polymers is not significantly affected by temperature, as indicated by small global conformational transitions of polymer chains. Regarding the dependence on water content, the increase in water content in hydrogels, ranging from 50 wt% to 73 wt%, resulted in a proportional increase in polymer-water interactions and polymer-water hydrogen bonds. Specifically, at −20 oC, the polymer-water interactions increased from −21,655 kcal mol−1 to −24,522 kcal mol−1, while at 25 oC, they increased from −18,761 kcal mol−1 to −20,785 kcal mol−1. Similarly, the number of hydrogen bonds formed between polymers and water molecules also showed a notable increase. At −20 oC, the number of hydrogen bonds rose from 6487 to 7269, while at 25 oC, it increased from 5800 to 6379.
Therefore, the increase in water content in hydrogels provides more water molecules for interaction, facilitates polymer hydration, and disrupts polymer–polymer hydrogen bonds, all of which contribute to the enhanced polymer-water interactions and increased number of hydrogen bonds between polymers and water molecules. Subsequently, the strong polymer–water interactions and hydrogen bonds between polymers and water molecules hinder the organization and alignment of water molecules required for ice crystal formation (Fig. 5). This interference prevents the nucleation and growth of ice crystals within the hydrogel matrix. Moreover, the increased polymer–water interactions and hydrogen bonding help maintain the mobility of water molecules within the hydrogel, even at low temperatures. This retained mobility prevents water molecules from freezing into a solid state and allows them to remain in a more fluid-like state, further inhibiting ice crystal formation. Overall, the increased polymer–water interactions and hydrogen bonding in hydrogels contribute to the antifreezing mechanism by impeding ice crystal formation, creating a water-rich environment, and preserving the mobility of water molecules. These factors collectively help the hydrogel maintain its integrity and functionality at low temperatures, making it suitable for applications requiring antifreezing properties, such as in biomedical and environmental applications.

The hydrogel with the excellent antifreezing property exhibits the strong polymer–water and weak water–water interactions, while the hydrogel with a weak antifreezing efficiency has the weak polymer-water and strong water–water interactions.
In summary, while antifreezing hydrogels have primarily relied on the use of antifreezing additives such as salts, organics, and biomolecules, there has been a lack of practical strategies for fabricating fully polymeric antifreezing hydrogels and a fundamental understanding of the polymer-induced antifreezing mechanism. We recently presented a simple and universal crosslinking strategy to fabricate a family of EGINA-crosslinked hydrogels with intrinsic, built-in antifreezing and mechanical properties without any antifreezing additive. To better understand the associated antifreezing property of this new type of hydrogels, we have developed a ‘random walk reactive polymerization’ (RWRP) algorithm that mimics the process of radical polymerization, enabling to construct physically-chemically linked PVA/PHEAA double-network (DN) hydrogels with varying water contents from monomers. PVA/PHEAA DN hydrogels constructed by the RWRP closely resemble the experimental properties of network structures. This RWRP method goes beyond the conventional timescales of MD simulations (typical hundreds of nanoseconds), thereby enabling the utilization of these advanced algorithms and methodologies to explore a diverse range of hydrogel systems and their associated properties. Next, we performed molecular dynamics (MD) simulations to study the behavior of water molecules confined within the polymer networks. MD results show that the antifreezing property of hydrogels is heavily reliant on the water-binding state, which is determined by a complex interplay between water-polymer interactions and the confinement effect of the polymer network on the water molecules. Specifically, the ability of the hydrogel to resist freezing is affected by a balance between enthalpy-driven tightly bound water molecules and the entropy-driven formation of a quasi-liquid water phase within polymer networks. More specifically, at lower temperatures, the polymer chains become highly packed and attract water molecules closely, resulting in the formation of extensive hydrogen bonds with the water molecules. This creates a strongly bound state, which reduces the mobility of the water molecules and provides a physical network barrier that inhibits ice growth. To further validate our computational findings, it is necessary to additional experimental studies, such as quasi-elastic neutron scattering to examine water dynamics, structures, and interactions in the hydrogels.
Data availability
All data, particularly force field parameters, used in this work are publicly available at GitHub.com/zhenglab-Akron/RWRP, and can also be found in main text and Supplementary Information.
Code availability
The codes developed for this work are available at GitHub.com/zhenglab-Akron/RWRP.
References
Lv, J., Song, Y., Jiang, L. & Wang, J. Bio-inspired strategies for anti-icing. ACS Nano 8, 3152–3169 (2014).
He, Z. et al. Bioinspired multifunctional anti-icing hydrogel. Matter 2, 723–734 (2020).
He, Z., Liu, K. & Wang, J. Bioinspired materials for controlling ice nucleation, growth, and recrystallization. Acc. Chem. Res. 51, 1082–1091 (2018).
Liu, Z. et al. Highly compressible and superior low temperature tolerant supercapacitors based on dual chemically crosslinked PVA hydrogel electrolytes. J. Mater. Chem. A 8, 6219–6228 (2020).
Morelle, X. P. et al. Highly stretchable and tough hydrogels below water freezing temperature. Adv. Mater. 30, 1801541 (2018).
Liu, Z. et al. Poly(ionic liquid) hydrogel-based anti-freezing ionic skin for a soft robotic gripper. Mater. Horiz. 7, 919–927 (2020).
Rao, P. et al. Ductile “ice”: frozen hydrogels with high ductility and compressive yielding strength. Extrem. Mech. Lett. 28, 43–49 (2019).
Zhang, X.-F. et al. Inorganic salts induce thermally reversible and anti-freezing cellulose hydrogels. Angew. Chem., Int. Ed. 58, 7366–7370 (2019).
Biggs, C. I. et al. Polymer mimics of biomacromolecular antifreezes. Nat. Commun. 8, 1546 (2017).
Zhao, X. et al. Bioinspired ultra-stretchable and anti-freezing conductive hydrogel fibers with ordered and reversible polymer chain alignment. Nat. Commun. 9, 3579 (2018).
Meister, K. et al. Observation of ice-like water layers at an aqueous protein surface. Proc. Natl Acad. Sci. USA 111, 17732 (2014).
Wu, L., Li, L., Qu, M., Wang, H. & Bin, Y. Mussel-inspired self-adhesive, antidrying, and antifreezing poly(acrylic acid)/bentonite/polydopamine hybrid glycerol-hydrogel and the sensing application. ACS Appl. Polym. Mater. 2, 3094–3106 (2020).
Wang, W. et al. Physically cross-linked silk fibroin-based tough hydrogel electrolyte with exceptional water retention and freezing tolerance. ACS Appl. Mater. Interfaces 12, 25353–25362 (2020).
Gao, H. et al. Adaptive and freeze-tolerant heteronetwork organohydrogels with enhanced mechanical stability over a wide temperature range. Nat. Commun. 8, 15911 (2017).
Lu, N. et al. Rational design of antifreezing organohydrogel electrolytes for flexible supercapacitors. ACS Appl. Energy Mater. 3, 1944–1951 (2020).
Ma, D. et al. Wearable, antifreezing, and healable epidermal sensor assembled from long-lasting moist conductive nanocomposite organohydrogel. ACS Appl. Mater. Interfaces 11, 41701–41709 (2019).
Han, L. et al. Mussel-inspired adhesive and conductive hydrogel with long-lasting moisture and extreme temperature tolerance. Adv. Funct. Mater. 28, 1704195 (2018).
Bai, G., Gao, D., Liu, Z., Zhou, X. & Wang, J. Probing the critical nucleus size for ice formation with graphene oxide nanosheets. Nature 576, 437–441 (2019).
Wei, P. et al. Conductive self-healing nanocomposite hydrogel skin sensors with antifreezing and thermoresponsive properties. ACS Appl. Mater. Interfaces 12, 3068–3079 (2020).
Rong, Q., Lei, W., Huang, J. & Liu, M. Low temperature tolerant organohydrogel electrolytes for flexible solid-state supercapacitors. Adv. Energy Mater. 8, 1801967 (2018).
Chen, H., Ren, X. & Gao, G. Skin-inspired gels with toughness, antifreezing, conductivity, and remoldability. ACS Appl. Mater. Interfaces 11, 28336–28344 (2019).
Wu, J. et al. Ultrastretchable and stable strain sensors based on antifreezing and self-healing ionic organohydrogels for human motion monitoring. ACS Appl. Mater. Interfaces 11, 9405–9414 (2019).
Wang, Y., Zhang, L. & Lu, A. Transparent, antifreezing, ionic conductive cellulose hydrogel with stable sensitivity at subzero temperature. ACS Appl. Mater. Interfaces 11, 41710–41716 (2019).
Sui, X. et al. Zwitterionic osmolyte-based hydrogels with antifreezing property, high conductivity, and stable flexibility at subzero temperature. Adv. Funct. Mater. 30, 1907986 (2020).
Su, X. et al. A solvent co-cross-linked organogel with fast self-healing capability and reversible adhesiveness at extreme temperatures. ACS Appl. Mater. Interfaces 12, 29757–29766 (2020).
Liu, X., Inda, M. E., Lai, Y., Lu, T. K. & Zhao, X. Engineered living hydrogels. Adv. Mater. 34, 2201326 (2022).
Mo, F. et al. A flexible rechargeable aqueous zinc manganese-dioxide battery working at −20 °C. Energy Environ. Sci. 12, 706–715 (2019).
Zhang, D. et al. A general crosslinker strategy to realize intrinsic frozen resistance of hydrogels. Adv. Mater. 33, 2104006 (2021).
Nandi, N., Bhattacharyya, K. & Bagchi, B. Dielectric relaxation and solvation dynamics of water in complex chemical and biological systems. Chem. Rev. 100, 2013–2046 (2000).
Alam, T. M., Childress, K. K., Pastoor, K. & Rice, C. V. Characterization of free, restricted, and entrapped water environments in poly(N-isopropyl acrylamide) hydrogels via 1H HRMAS PFG NMR spectroscopy. J. Polym. Sci. Part B: Polym. Phys. 52, 1521–1527 (2014).
Deriu, A., Cavatorta, F., Cabrini, D., Carlile, C. J. & Middendorf, H. D. Water dynamics in biopolymer gels by quasi-elastic neutron scattering. Europhys. Lett. 24, 351 (1993).
Noferini, D. et al. Disentangling polymer network and hydration water dynamics in polyhydroxyethyl methacrylate physical and chemical hydrogels. J. Phys. Chem. C 123, 19183–19194 (2019).
Yan, C., Kramer, P. L., Yuan, R. & Fayer, M. D. Water dynamics in polyacrylamide hydrogels. J. Am. Chem. Soc. 140, 9466–9477 (2018).
Jian, Y. et al. Biomimetic anti-freezing polymeric hydrogels: keeping soft-wet materials active in cold environments. Mater. Horiz. 8, 351–369 (2021).
Wang, K. W., Betancourt, T. & Hall, C. K. Computational study of DNA-cross-linked hydrogel formation for drug delivery applications. Macromolecules 51, 9758–9768 (2018).
Tamai, Y., Tanaka, H. & Nakanishi, K. Molecular dynamics study of polymer−water interaction in hydrogels. 1. Hydrogen-bond structure. Macromolecules 29, 6750–6760 (1996).
Ge, W., Cao, S., Yang, Y., Rojas, O. J. & Wang, X. Nanocellulose/LiCl systems enable conductive and stretchable electrolyte hydrogels with tolerance to dehydration and extreme cold conditions. Chem. Eng. J. 408, 127306 (2021).
Li, T. et al. Self-deicing electrolyte hydrogel surfaces with Pa-level ice adhesion and durable antifreezing/antifrost performance. ACS Appl. Mater. Interfaces 12, 35572–35578 (2020).
Jewett, A. I. et al. Moltemplate: a tool for coarse-grained modeling of complex biological matter and soft condensed matter physics. J. Mol. Biol. 433, 166841 (2021).
Lenzi, V., Ramos, M. M. D. & Marques, L. S. A. Dissipative particle dynamics simulations of end-cross-linked nanogels. Mol. Simul. 47, 27–36 (2021).
Nikolov, S., Fernandez-Nieves, A. & Alexeev, A. Mesoscale modeling of microgel mechanics and kinetics through the swelling transition. Appl. Math. Mech. 39, 47–62 (2018).
Moreno, A. J. & Lo Verso, F. Computational investigation of microgels: synthesis and effect of the microstructure on the deswelling behavior. Soft Matter 14, 7083–7096 (2018).
Grest, G. S. & Kremer, K. Statistical properties of random cross-linked rubbers. Macromolecules 23, 4994–5000 (1990).
Kacar, G., Peters, E. A. J. F. & de With, G. Mesoscopic simulations for the molecular and network structure of a thermoset polymer. Soft Matter 9, 5785–5793 (2013).
Vanommeslaeghe, K., Raman, E. P. & MacKerell, A. D. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Modeling 52, 3155–3168 (2012).
Brooks, B. R. et al. CHARMM - a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4, 187–217 (1983).
Zhang, M. et al. A multiscale polymerization framework towards network structure and fracture of double-network hydrogels. npj Comput. Mater. 7, 39 (2021).
Phillips, J. C. et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 (2005).
Qiu, M. et al. Tailoring water structure with high-tetrahedral-entropy for antifreezing electrolytes and energy storage at −80 °C. Nat. Commun. 14, 601 (2023).
Hafner, R., Guevara-Carrion, G., Vrabec, J. & Klein, P. Sampling the bulk viscosity of water with molecular dynamics simulation in the canonical ensemble. J. Phys. Chem. B 126, 10172–10184 (2022).
Liu, Y. et al. Polymer–water interaction enabled intelligent moisture regulation in hydrogels. J. Phys. Chem. Lett. 12, 2587–2592 (2021).
Acknowledgements
We thank financial support from NSF-DMR-2311985 and ACS-ND-65277. We also trained three high school students - Bowen Zheng from Copley High School, Alice Xu from Hudson High School, and Keven Gong from Western Reserve Academy – by this project.
Author information
Authors and Affiliations
Contributions
Y.L. and J.Z. initiated the idea of this project. Y.L. and D.Z. designed and conducted simulations. Y.L., Y.T. and D.Z. analyzed the results. J.Z. supervised this project. The manuscript was written through the inputs of all the authors. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
Yes.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Y., Zhang, D., Tang, Y. et al. Development of a radical polymerization algorithm for molecular dynamics simulations of antifreezing hydrogels with double-network structures. npj Comput Mater 9, 209 (2023). https://doi.org/10.1038/s41524-023-01161-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-023-01161-x
This article is cited by
-
Investigating the role of temperature and moisture on the degradation of 3D-printed polymethyl methacrylate dental materials through molecular dynamics simulations
Scientific Reports (2024)
-
Virtual screening of acetylcholinesterase inhibitors through pharmacophore-based 3D-QSAR modeling, ADMET, molecular docking, and MD simulation studies
In Silico Pharmacology (2024)
