
Abstract
We demonstrate a machine learning-based approach which predicts the properties of crystal structures following relaxation based on the unrelaxed structure. Use of crystal graph singular values reduces the number of features required to describe a crystal by more than an order of magnitude compared to the full crystal graph representation. We construct machine learning models using the crystal graph singular value representations in order to predict the volume, enthalpy per atom, and metal versus semiconductor/insulator phase of DFT-relaxed organic salt crystals based on randomly generated unrelaxed crystal structures. Initial base models are trained to relate 89,949 randomly generated structures of salts formed by varying ratios of 1,3,5-triazine and HCl with the corresponding volumes, enthalpies per atom, and phase of the DFT-relaxed structures. We further demonstrate that the base model is able to be extended to related chemical systems (isomers, pyridine, thiophene and piperidine) with the inclusion of 2000 to 10,000 crystal structures from the additional system. After training a single model with a large number of data points, extension can be done at significantly lower cost. The constructed machine learning models can be used to rapidly screen large sets of randomly generated organic salt crystal structures and efficiently downselect the structures most likely to be experimentally realizable. The models can be used as a stand-alone crystal structure predictor, but may serve CSP efforts best as a filtering step in more sophisticated workflows.
Similar content being viewed by others

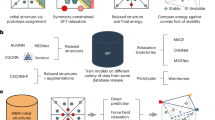

Introduction
Organic crystals form the basis of many common goods, including pharmaceuticals1, pesticides2, and pigments3, and have applications in emerging technologies such as thin film semiconductors4, catalysts5, and optoelectronics6. An involved problem in materials design is the engineering of molecular crystals with targeted features (see e.g. refs. 7,8,9,10,11, among many others). This task involves two major challenges: identifying a molecular target with promising solid-state properties, and the prediction and control of its crystal structure12,13,14,15. Crystal structure predictions (CSP) of organic solids are nowadays pursued using effective algorithms and great computational resources. Yet, they have been shown to be very complex as unlike the constituents of inorganic crystals, organic molecules are generally conformationally flexible, resulting in numerous polymorphs with local energy minima within only a few kJ mol−1 of the global minimum energy structure9,16,17,18. As algorithms become more efficient, crystal structures of increasingly complex organic systems become accessible to investigation. The most recent Seventh CSP Blind Test has tasked participants with predicting crystal structures for: metal-organic compounds, organic molecules containing combinations of Si, I, S, and F, and multicomponent crystals19.
Contemporary CSP approaches typically require 10,000’s to 100,000’s of force field potentials, molecular dynamics, or density functional theory (DFT) calculations to relax and calculate crystal energies and properties20,21,22, with the goal of identifying as many experimentally feasible crystal forms (e.g. polymorphs) as possible8,23,24,25,26,27. The consideration of such a large number of putative crystal structures and the marginal differences in their energies render the identification of plausible structures exceedingly difficult. While approaches to improve the prediction of organic crystal structures have recently been developed28, further steps need to be taken to make CSP more versatile29, accurate, affordable, and routine. With this in mind, we have developed a machine learning approach that could reduce the number of required energy calculations, thus lowering the computational cost of CSP8,24,25,26, which otherwise can take up more than 100,000 CPU hours10.
Machine learning is a rapidly developing tool for predicting properties of organic crystal structures that can address this issue. The predictive power of machine learning models depends on the data used to train the model, the method for constructing representations of crystals, and the choice of machine learning algorithm. Numerous studies have found success predicting properties of organic crystals30,31,32,33,34,35 by employing a wide range of machine learning approaches. Common limitations are that either training data will contain crystals not in energetic local minima, which may not be representative of the relaxed structures, or generated structures need to be relaxed into an energetic minimum using ab initio methods, which again increases the computational cost.
In this work, we demonstrate an approach to accelerate CSP by constructing machine learning (ML) models which can predict properties of DFT-relaxed organic crystals based on the structures and chemical compositions of randomly generated unrelaxed crystal structures. By constructing ML models for enthalpy and volume of the corresponding relaxed structures, our approach allows downselection of structures which can then be explicitly evaluated using more expensive DFT calculations. The downselection process removes randomly generated structures which are likely to relax into high energy configurations, leaving only the most relevant initial configurations. Work with similar goals has been carried out by Honrao et al. 36 and Gibson et al. 37. Honrao et al. constructed support vector regression models for binary Al-Ni and Cd-Te systems featurized using radial and angular distribution functions. Gibson et al. trained crystal graph convolutional neural networks to predict formation energies using inorganic structures from the Materials Project Database.
Presented here are two further developments on machine learning modeling for CSP. First, we describe and validate a crystal graph singular value representation of crystal structures, which reduces the required number of descriptors by several orders of magnitude. Crystal graph descriptors were developed by Xie et al.38 as an effective method for representing crystal structures which can be used to train high accuracy convolutional neural network (CGCNN) models. The approach has been used in numerous studies, including refs. 37,39,40,41,42, yet requires a large number of values to describe each structure, causing data files to become quite large. As an example, in the case of a triazine hydrochloride salt (C3H4N3Cl) crystal with one formula unit in a cell, each atom is represented by a 12 × 41 matrix, for a total of 5412 descriptors required to represent the structure. By using the singular values, the representation is reduced to fewer than 300 values. Second, we employ random forest models which require fewer hyperparameters and are fit without the iterative backpropagation process required for neural network algorithms43. The machine learning approach developed in this work provides a method for predicting properties of relaxed organic crystals knowing only the structure of the randomly generated unrelaxed structures, while requiring only a small number of DFT relaxations to form a training set. We apply our machine learning methods to salts formed from HCl (and HBr in Extension A) and six small organic ring molecules: 1,2,3-triazine, 1,2,4-triazine, 1,3,5-triazine, pyridine, thiophene, and piperidine, shown in Fig. 1. The nitrogen-based heterocyles were selected as model compounds based on their ability to form salts, while thiophene (a compound known to polymerize in acidic conditions44) was used to evaluate the ability to predict the chemical behavior of thiophene in the presence of hydrochloric acid. The type of acid and the stoichiometric ratios of the reactants were varied to explore a larger structural landscape. All crystal structures were initialized assuming salt formation45.

From left to right: 1,2,3-triazine, 1,2,4-triazine, 1,3,5-triazine, pyridine, thiophene, and piperidine.
The machine learning approach we develop here does not make any assumptions about the chemical composition of the system, the range of structures, the method used to generate trial structures, or the approach used to optimize structures after generation. This makes the approach broadly applicable to both organic and inorganic crystals. Our work will demonstrate a machine learning approach which is broadly applicable to organic crystals and may be incorporated into sophisticated CSP workflows as a filter to remove thermodynamically unfavorable structures. We especially encourage attempts to integrate our approach into organic CSP based on machine learning-trained interatomic potentials.
Methods
Overview
Complete descriptions of the choices of molecular crystals, DFT and CSP methods, model construction, and model evaluation are included in the Supplementary Information Sections 1–4.
Random crystal structures were generated using the AIRSS46,47 software package. The input structure contained specified numbers of both protonated and unprotonated organic molecules along with the number of Cl− ions (or other anion) equal to the number of protonated organic molecules. Unprotonated molecules would be present only if the unit cell contained more organic molecules than units of HCl. Organic molecules were generated with fixed 2D structures and arranged in approximately close-packed structures without any imposed symmetry. The unit cells were initialized with volumes less than the close-packed volumes so that structures would expand during relaxation. During relaxation, the unit cell shape, relative positions and orientations between molecules, and geometries of individual molecules were optimized simultaneously using the conjugate gradient algorithm implemented in the Vienna Ab-Initio Software Package (VASP) version 5.4.448,49. While the machine learning method demonstrated in this work is broadly applicable, we emphasize that any individual model is strongly dependent on the approaches used to generate and relax structures.
Base models are constructed to relate descriptors of unrelaxed structures to DFT-computed unit cell volume (V), enthalpy per atom (h) (from which the relative enthalpic stability, ΔH, of different crystal structures can be readily calculated), and metal or semiconductor/insulator phase (phase). The base ML models are fit using data from four crystal structure prediction runs for 1,3,5-triazine. In order to test the ability of the learned model to predict properties of crystals of different organic molecule salts, we consider three levels of extension,
-
Extension A: 1,3,5-triazine HBr, 1,2,3-triazine HCl, and 1,2,4-triazine HCl
-
Extension B: pyridine (C5H5N) HCl
-
Extension C: thiophene (C4H4S) HCl and piperidine (C5H11N) HCl
Extension A includes two sets of structures in which the organic molecule is an isomer of 1,3,5-triazine and an organic salt of 1,3,5-triazine with HBr instead of HCl. Extension B considers salts of pyridine, another small nucleophillic molecule based on a six member ring. CSP for pyridine has been previously shown to be a challenging problem due to having numerous computed structures in the energy gap between the observed and most stable structure as well as a complex, asymmetric unit cell50. Extension C contains thiophene HCl and piperidine HCl. Extension C is the largest extension away from 1,3,5-triazine HCl with thiophene being a five-membered, sulfur containing heterocyclic compound that does not protonate under conventionally used crystallisation conditions. These structures have therefore not been thoroughly examined. Instead, the thiophene HCl structures are included to test model performance on high energy structures. (A survey of the Cambridge Structural Database51 (version 5.45, March 2024 update) revealed a lack of entries involving thiophene derivatives with protonated sulfur atoms.) Piperidine HCl is computationally more demanding because of its conformational flexibility. While the relaxation process does not explicitly prevent molecules from relaxing into different conformations, starting structures were generated using only a single conformer. A thorough CSP study of piperidine HCl would require generating and examining structures which result from all piperidine conformations.
Constructed models are tested for overfitting using a nested cross validation scheme. The accuracy of each regressor model is quantified using the mean absolute error (MAE), mean absolute fractional error (MAFE), and Spearman coefficient (ρ). Classifier models are evaluated using the average precisions (AP) for each class and mean average precision (mAP) based on the precision-recall curves. Further details of all quantities are listed in the Supplementary Information Section 4.
Descriptor choice
For every crystal structure, three sets of descriptors are compiled: crystal graph singular values, Coulomb matrix eigenvalues, and crystal structure parameters.
Crystal graph representations of every structure are generated using code provided by Xie et al.38 on the authors’ Github repository52. The approach describes local chemical environments for each atom as graphs characterizing the bonding arrangements between atoms. An n × m matrix, ci, is constructed to represent each of the a atoms in the unit cell. The crystal graph matrix for each atom in the unit cell is used to construct an an × am block diagonal matrix B,
The singular values of B are used as descriptors for our ML models, and are referred to as Crystal Graph Singular Value (CGSV) descriptors. Performance of models including CGSV descriptors is discussed in Supplementary Information Section 5. The code by Xie et al. was used solely to generate the crystal graph representations; no results were generated from the authors’ pretrained models.
The Coulomb matrix Cij of a structure is defined as:
where Zi is the atomic number of atom i and rij is the minimum distance between atom i and all periodic images of atom j53,54. Indices i and j include only the atoms in a single unit cell. The Coulomb matrix descriptors are then found as the sorted eigenvalues of Cij. Further descriptors are formed from: number of positive eigenvalues, number of negative eigenvalues, \({\rm{Tr}}({C}_{ij})\), and \(\det ({C}_{ij})\).
We note that \({\rm{Tr}}({C}_{ij})={\sum }_{k}{\lambda }_{k}=\frac{1}{2}{\sum }_{k}{Z}_{k}^{2.4}\) where {λi} are the eigenvalues of the Coulomb matrix. The trace of the Coulomb matrix, then, provides a unique irrational number which characterizes the atomic composition of the unit cell. This allows the trace to be used as a categorical descriptor to identify the atomic contents of the cell. However, this trace descriptor only identifies the chemical contents of the cell, not the bonding arrangement. The descriptor formed from \({\rm{Tr}}({C}_{ij})\) distinguishes, e.g. 1,3,5-triazine (C3H3N3) from pyridine (C5H5N), but would not distinguish 1,3,5-triazine from 1,2,3-triazine.
The crystal structure descriptors consist of the unit cell edge lengths a, b, and c and unit cell angles α, β, and γ.
While all molecules studied here are rigid and flat, the chosen descriptors are applicable to more complex 3D and conformationally flexible molecules.
Model extension
The ability of models to extend to crystals with different chemical compositions is tested by incrementally incorporating structures from an extension CSP run into the 1,3,5-triazine HCl training sets. Structures in four 1,3,5-triazine HCl CSP runs are randomly divided into fitting, validation, and testing sets as described in Supplementary Information Section 3. N structures from the extension CSP run are randomly added to the fitting set and all remaining extension CSP structures are added to the testing set. The fitting set is weighted such that the total weight of 1,3,5-triazine HCl structures is four, corresponding to four CSP runs, and the total weight of the added CSP run structures in the fitting set is one. Random forest models are fit using the combined fitting set then applied to the validation and testing sets. Performance of the random forest models are evaluated for the 1,3,5-triazine HCl runs and extension run separately. Monitoring separate performances for 1,3,5-triazine HCl and the included extension run checks decreased accuracy for 1,3,5-triazine HCl due to inclusion of added structures and ability of the model to extrapolate outside the 1,3,5-triazine HCl data set.
Results
Descriptor selection
We minimize the random forest regressor model training errors by optimizing the number of descriptors to be used for model construction. An initial decision tree regressor is fit to the full set of 506 descriptors with a maximum tree depth of 20 layers with the model Gini importances for all descriptors tabulated. The Gini importance of a descriptor quantifies the importance of a descriptor in a tree-based model by considering both the number of times each descriptor is used in the fitted model and the number of samples split by the descriptor43,55,56. Features with Gini importances greater than “c × average Gini importance” are retained for fitting subsequent random forest regressors. c is assigned values between 0.01 and 1.0, with the corresponding number of descriptors dependent on the choices of fitting set and target quantity. Figure 2a shows the corresponding fitting, validation, and testing MAEs for the constructed random forest regressors. The Spearman correlation coefficients between VDFT and VML are plotted against the number of included descriptors in Fig. 2b. The fitting MAE decreases monotonically with increasing number of descriptors, which we attribute to the increasing number of possible splitting criteria for fitting random forest regressors. The testing set MAE shows a minimum at 70 included descriptors. For the number of descriptors below 70, the number of splitting criteria is not sufficient to capture the relation between the descriptors and V. However, increasing the number of descriptors to fit the random forest models beyond 70 leads to more severe overfitting, which manifests itself in the decreasing fitting set MAE, while the testing MAE increases. With more than 13 descriptors included, the Spearman coefficients for the fitting, validation, and testing sets are all above 0.95. Similar results are obtained for models constructed for the enthalpy per atom (h) regressor and metal versus semiconductor/insulator (phase) classifier. From these results, we determine an appropriate criterion for downselecting descriptors: include only descriptors with Gini importances greater than 0.1 times the average Gini importance.

a MAE for volume model trained on 1,3,5-triazine HCl crystal structures versus number of descriptors included in model construction. b Spearman correlation coefficient for volume model trained on 1,3,5-triazine HCl crystal structures versus number of descriptors included in model construction.
The number of descriptors selected for each model in this paper are listed in Table 1. The number of descriptors used for each target quantity shows little change based on the CSP run added to the fitting set. For example, in constructing models for V, the number of descriptors included varies between 63 and 70. The only large difference is observed in the model of h for thiophene HCl, which takes 56 descriptors. For the other five h models, 110 to 122 descriptors are selected.
The required number of descriptors can be rationalized by consideration of the Spearman correlation coefficients (ρ) between the descriptors and target quantities, shown in Fig. 3. The strongest correlations between descriptors and target values are found for VDFT. Out of 506 possible descriptors, 380 descriptors have ∣ρ∣≥0.8 with VDFT and only 6 descriptors have ∣ρ∣≤0.2. This indicates that the chosen descriptors are strongly correlated with VDFT. For h and phase, no descriptors with ∣ρ∣≥0.8 are present. Strong correlations between descriptor and target values allows the construction of low error models with fewer fitting parameters. ML models constructed for VDFT would be expected to require fewer descriptors than for hDFT and phase. Of the three target quantities, V has the most direct connection to the chosen descriptors. To take the Coulomb matrix as an example, the off-diagonal elements have a 1/rij dependence. Increasing the volume of the unit cell will increase the typical distances between atoms in the cell, thereby decreasing the off-diagonal elements of the Coulomb matrix and the magnitudes of the eigenvalues. Similarly, the off-diagonal elements of the Coulomb matrix are the potential energies due to the Coulomb interactions between pairs of nuclei, thereby providing a measure of one source of potential energy in the crystal. However, the Coulomb matrix neglects all other contributions to the enthalpy, such as electron-electron and electron-nucleus interactions. There is no direct connection between the chosen descriptors and the phase.

Spearman correlation coefficients between target quantities and Coulomb matrix and crystal graph singular value descriptors for the four datasets of 1,3,5-triazine HCl CSP. The orange curve plots ρ for VDFT, blue curve plots ρ for hDFT and the green curve plots ρ for metal versus insulator from DFT. Descriptors 0 through 44 to the left of the vertical black line corresponds to Coulomb matrix descriptors while descriptors 45 through 500 correspond to the crystal graph singular value descriptors.
1,3,5-triazine crystal base model construction
All results in this subsection refer to models trained with the four datasets of 1,3,5-triazine with HCl listed in Supplementary Table 1.
Fitting set size
A persistent consideration in constructing ML models is the number of datapoints required to train the model. If too few datapoints are used in training, the ML algorithm may be unable to find patterns relating the descriptors and target values, resulting in either overfit or underfit models. Using arbitrarily large training sets is also undesirable because it increases the cost to generate the training set and the cost to fit the model. As an example, we consider performance of random forest regressor models to predict crystal structure volumes which are trained with varying fitting set sizes. Results are plotted in Fig. 4. From the four CSP runs with 1,3,5-triazine and HCl, 9 × Nfitting/8 structures are randomly selected to form the fitting and validation sets. All remaining structures are placed in the testing set. As in the rest of this work, we iterate model construction over 10 random splittings of the fitting and validation sets with Nfitting structures in each fitting set and Nfitting/8 structures in each validation set. For the testing set, values of the MAE, MAFE, and Spearman coefficient are consistent over the range 5000≤Nfitting≤70, 000. Improvement in the model with respect to Nfitting is observed through the increase in MAE and MAFE of the fitting set and decrease in the difference of the Spearman coefficients for the fitting set and testing set. Adding more fitting data increases the model error for the fitting data, while decreasing the overfitting of the model. Moving forward, we use the full data set of 1,3,5-triazine and HCl for the base model. Due to our validation scheme, this corresponds to 71,959 structures in the fitting set for each iteration.

Performance of the base 1,3,5-triazine HCl V model versus number of randomly selected samples in the fitting set, characterized by (a) Spearman coefficient, (b) MAE, and (c) MAFE.
Volume model
One design guideline for determining which crystal polymorphs are experimentally realizable is that the structure should have high density, with a packing density of 60% to 80%11,57. The general principle of preferring high density crystal structures removes highly porous structures with large voids from consideration. Porous structures are often unfavorable due to the ability of constituent atoms and molecules to rearrange into lower energy configurations by filling the voids. Despite the general preference for organic molecules to pack into high density arrangements, there are several technologically relevant classes of organic-based materials for which this principle does not hold. Some organic molecules can form highly porous structures due to the influence of solvents58 or be a component of covalent59 or metal-organic frameworks60 displaying large voids. There are also organic salts61,62 which form porous structures due to effects from covalent bonding and weak intermolecular interactions. In such cases, the preference for low volume structures would prevent identifying synthesizable structures using the approach described in this paper. It would then fall on the user to identify more suitable target quantities.
Random forest regressor trained on DFT calculated volumes and comparison of the machine-learned and DFT-calculated volumes for the 1,3,5-triazine HCl training set is shown in Fig. 5. The model produces MAEs of 45 Å3 for the fitting set, 50 Å3 for the validation set, and 49 Å3 for the testing set. This indicates that with the optimum choice of the maximum tree depth hyperparameter, overfitting can be minimized, but is still observable: the validation and testing MAEs are approximately factors of 1.1 and 1.09 larger than the fitting MAE, respectively. While the validation and testing MAEs are larger than the fitting MAE, the discrepancies are small.

a Heat map showing the distribution of testing data of VDFT obtained via DFT and VML predicted by random forest regressor. Plotted are the results from the testing set average values averaged over all 10 iterations. Dashed red line guides the eye to show VDFT = VML. b Distribution of ML errors of V for 10 fitting-validation iterations. c Distribution of ML fractional errors in V for 10 fitting-validation iterations.
The random forest regressor model produces non-Gaussian distributions of fitting and validation errors in VML, see Fig. 5b. This plot shows the differences between machine-learning predicted VML and DFT-calculated VDFT for fitting and validation sets over all 10 fitting-validation iterations. Error distributions were computed by aggregating results from all 10 fitting-validation iterations into one distribution for the fitting and validation sets. The fitting set errors are centered around 0.018 Å3 with 1-σ standard deviation width of 62 Å3. The validation set error distribution is centered at 0.10 Å3 with 1-σ standard deviation width of 70 Å3. The testing set error distribution (not included in Fig. 5b, c) closely follows the validation set error distribution, with the center at 0.10 Å3 and a 1-σ standard deviation width of 70 Å3. Throughout the results, the validation and testing set error distributions are nearly identical, thus only the fitting and validation set results will be shown and discussed in order to demonstrate the absence of overfitting. We observe a small bias toward underestimating VDFT, with the constructed model predicting smaller values for 51% of both fitting and validation materials. The unimodal distribution of errors indicates that there is no subgroup of initial structures for which the model consistently fails. Further, the Spearman correlation coefficients between the DFT-calculated volumes and the model predicted volumes are calculated as 0.95 for both the fitting and validation sets. By considering the fractional error distributions in Fig. 5c, bias and non-Gaussian behavior in the constructed model is further shown. The fitting set fractional errors are centered around of -1.4% and the validation set fractional errors are centered around -1.6%. While the absolute errors show nearly symmetrical distributions, the fitting and validation fractional error distributions are skewed, with long tails of samples with negative fractional errors and rapidly decaying tails with positive fractional errors.
Volume is an extensive variable which depends strongly on the number of atoms in the unit cell. One consequence is clusters visible in Fig. 5a corresponding to different CSP runs with different numbers of atoms in the cell. If in a CSP run multiple different values for the number of organic molecule units in the cell are included, the volume minimization criterion would select structures with the fewest organic molecules. Taking into account different cell contents requires using the intensive quantity of volume per atom v. Normalizing the volumes plotted in Fig. 5a per atom in each CSP run produces the heat map in Fig. 6. The model trained on total volume retains high Spearman correlation coefficients of 0.96 for the fitting set, 0.95 for the validation set, and 0.88 for the testing set when applied to volume per atom. The model trained on total volume is readily adapted to predict volume per atom. This provides the potential to train on small unit cells and extrapolate to larger cells.

Heat map showing the performance of the volume model on the 'testing set', with results from the testing set averaged over all 10 fitting-validation iterations. Data shown corresponds to Fig. 5a with volumes normalized per atom in the unit cell. Colors toward the red end of the color spectrum indicate regions of higher densities of data points. Dashed red line guides the eye to show vDFT = vML.
For this work, we did not find it necessary to spend excessive resources developing highly accurate ML models of the volume of relaxed crystal structures. However, the model still outperforms selecting generated structures based solely on the unrelaxed volume, as shown in a test case in the Supplementary Information Section 6. Instead we use a less accurate model which reproduces general trends as a coarse filter and can then explicitly check the predictions by performing DFT simulations for downselected randomly generated initial unrelaxed structures. While the volume model does not produce sufficiently accurate predictions to replace explicit DFT calculation, it does show remarkable agreement in error distributions between the fitting, validation, and testing sets.
Enthalpy model
Our second criterion for selecting polymorphs is the enthalpy per atom of the structure. Enthalpy is chosen as a thermodynamic criterion because it includes consideration of pressure and volume effects in the CSP. A random forest regressor trained on DFT calculated enthalpies per atom and comparison of the machine-learned and DFT-calculated enthalpies per atom for the 1,3,5-triazine HCl training set is shown in Fig. 7. The model produces MAEs of 0.044 eV atom−1 for the fitting set, 0.048 eV atom−1 for the validation set, and 0.047 eV atom−1 for the testing set. This indicates that with the optimum choice of the maximum tree depth hyperparameter, overfitting is still present, but minimal: the validation and testing MAEs are approximately factors of 1.09 and 1.07 larger than the fitting MAE, respectively. As in the volume model, the MAE values for the validation and testing sets closely match the MAE of the fitting set.

a Parity plot comparison of hDFT obtained via DFT and hML predicted by random forest regressor. Plotted are the results from 10 fitting-validation iterations as well as testing set average values and error bars from all 10 iterations. b Distribution of ML errors of V for 10 fitting-validation iterations. The distributions are unimodal and symmetric about their means. c Distribution of ML fractional errors in V for 10 fitting-validation iterations.
The model for h has MAFE values of 0.0070 for the fitting set, 0.0077 for the validation set, and 0.0076 for the testing set. The enthalpy per atom model produces MAFE values which are an order of magnitude smaller than the MAFE values of the volume model.
For the purpose of downselecting structures to relax, one would not be interested in crystal structures over the entire range of hML, only low hML structures. Considering only the testing set structures with the 1000 lowest hML values, the MAE drops to 0.026 eV atom−1 and the MAFE to 0.0040. Thus, the h model is considerably more accurate in the region of interest.
The random forest regressor model produces non-Gaussian distributions of fitting and validation errors in hML, see Fig. 7b. This plot shows the differences between machine-learning predicted hML and DFT-calculated hDFT for fitting and validation sets over all 10 fitting-validation iterations. The fitting set errors are centered around 1.0 × 10−4 eV atom−1 with 1-σ standard deviation width of 0.066 eV. The validation set error distribution is centered around 1.6 × 10−5 eV atom−1 with a 1-σ standard deviation width of 0.080 eV. Errors between the DFT-computed and the ML predicted enthalpies per atom show unimodal distributions, indicating that there is no subgroup on which the model performs particularly poorly. We observe a small bias toward overestimating hDFT, with the constructed model predicting larger values for 57% of samples in the fitting set and 58% of samples in the validation set. Further, the Spearman correlation coefficients between the DFT-calculated enthalpies per atom and the model predicted enthalpies per atom are calculated as 0.88 for the fitting set and 0.87 for the validation set.
By considering the fractional error distributions in Fig. 7c bias and non-Gaussian distribution in the constructed model is further shown. The distributions of the fractional errors are centered at -0.016% for the fitting set and –0.019% for the validation set. While the absolute errors show nearly symmetrical distributions, the fitting and validation fractional errors are skewed, appearing as long tails in the region of negative fractional error but rapidly decaying in positive fractional error.
The constructed random forest regressor models are able to predict volumes and enthalpies of relaxed structures of 1,3,5-triazine HCl over a range of number of constituent components in the unit cell. MAE values, MAFE values and Spearman coefficients for the constructed models are similar between the fitting, validation, and testing sets. These indicate that the models have not been overfit and therefore could be applied to unseen data. The utility of the present machine learning approach comes from the ability to rank unrelaxed structures by the predicted relaxed volume or enthalpy then show further consideration only for the structures most likely to relax into high density, low enthalpy final configurations. There is the potential to combine our machine learning property prediction with trained force-field approaches, see e.g. refs. 63,64,65. In such a hypothetical workflow, one would simultaneously train both the force-fields and the property prediction models using the same data. Once both are trained, many trial structures would be generated and the property prediction model used as a filter to remove those which are likely to become thermodynamically unfavorable after optimization.
Metal vs insulator model
Material properties can be drastically and discontinuously altered by small changes which result in a metal-semiconductor/insulator transition66. A material may have both metallic and semiconductor/insulator phases, corresponding to different crystal structures. Based on the composition, it may be possible to anticipate which phase is more probable. The phase model demonstrated here can be used to retain only structures in the more probable phase. Removing structures likely to relax into the undesired metal or semiconductor/insulator phase can further speed up crystal structure prediction. A grid search was performed over random forest classifiers with allowed maximum depths between 8 and 20 layers and minimum samples for splitting between 5 and 20 as classifiers for metallic versus semiconducting/insulating relaxed structures. Using the mAP of the precision-recall curve as the optimization criterion, a maximum tree depth of 10 layers with a minimum splitting criterion of 10 samples was selected. Figure 8 plots the precision-recall curve for prediction of the semiconductor/insulator phase in crystal structures of 1,3,5-triazine HCl. The model performs well in the case of the semiconductor/insulator phase with average precisions of 0.94 for the fitting and validation sets and 0.97 for the testing set. The model struggles on the minority metal phase, with average precisions of 0.59 for the fitting set, 0.24 for the validation set, and 0.26 for the testing set. These results give mAP values of 0.77 for the fitting set, 0.59 for the validation set, and 0.61 for the testing set. The imbalances between the average precisions for semiconductors/insulators and metals indicates that the phase model prediction of a structure as a semiconductor/insulator is more reliable than the prediction as a metal. This may be a consequence of the strongly imbalanced classes which could be improved by considering a chemical composition which has a more equitable division of semiconductor/insulator and metallic structures.

Precision-recall curve for prediction of semiconductor/insulator in structures of 1,3,5-triazine HCl crystals.
Comparison with CGCNN
We benchmark the performance of the random forest regressor algorithm used here against the CGCNN approach by Xie et al. as implemented in their publicly accessible code38,52. The two algorithms are compared by training models on the base model of 1,3,5-triazine HCl with varying numbers of structures in the fitting sets. The tests also consider the number of epochs used to fit the CGCNN model, with 30 epochs sufficient to converge the MAE values for Nfitting = 70, 000 and 200 epochs sufficient to converge the MAE values for Nfitting = 900. The results are plotted in Fig. 9, with the models’ fitting and testing MAEs compared in Fig. 9a. The random forest regressor displays lower MAE values over the entire range of Nfitting compared to the CGCNN trained with either 30 epochs or 200 epochs. The CGCNN reaches the accuracy of the random forest regressor only once the fitting set includes over 70,000 structures. Figure 9b plots the computational cost of fitting each model. The time required to fit both models scales as \(t\propto {N}_{{\rm{fitting}}}^{1}\), with scaling of the CGCNN fitting time consistent between 30 and 200 epochs. However, while the scaling with number of fitting samples is identical, the CGCNN algorithm requires two orders of magnitude more time to fit. We also note that in the random forest approach 10 random forests are fit while the CGCNN approach fits only one neural network. The random forest approach detailed in this work produces more accurate model with fewer structures needed for fitting and is fit significantly faster than the CGCNN. These factors make the random forest approach preferred for scaling up to larger, chemically diverse training data.

Comparison of performance of random forest regressor V model and CGCNN V model measured by (a) MAE and (b) fitting time.
Extending the models
In Section III B, three ML models predicting properties of 1,3,5-triazine HCl crystals were constructed and tested within an interpolative regime. In order for a ML model to make reliable predictions for new chemical compositions, it must first see some samples with the new chemical composition. Results in this subsection clarify the number of structures which are required to extend the use of the models and characterize their performance on the new chemical compositions.
We examine a representative example of model performance as structures from a different chemical system are gradually added to a model. In Fig. 10, structures of 1,2,3-triazine HCl are incorporated into the model training set. The Spearman coefficient plotted in Fig. 10a requires ~10,000 added structures in order to converge. With 10,000 structures of 1,2,3-triazine HCl added to the fitting set, the Spearman coefficient for the testing set is 0.73. Adding up to 18,000 structures of 1,2,3-triazine HCl only increases the Spearman coefficient to 0.77. The MAE and MAFE shown in Fig. 10b, c converge with fewer added structures, requiring as few as 2000 samples of 1,2,3-triazine HCl added to the fitting set. These results demonstrate two important factors for training the models. First, the number of structures from a new chemical composition which must be added to the base model for training is dependent on the measure used to evaluate the model. Second, the models can be extended by adding as few as 2000 to 10,000 structures with different chemical compositions.

a Spearman coefficient, (b) MAE, and (c) MAFE as 1,2,3-triazine HCl structures are added to the training set. Values consider only structures of 1,2,3-triazine HCl.
Full results for extension tests are summarized in Tables 2, 3, 4. For both the volume and enthalpy per atom models, the MAFE values of the added structure testing sets are close to the base model MAFE values. The effect of extension more significantly influences the values of the added structure testing set Spearman values. The volume base model has a testing set Spearman coefficient of 0.95. The testing set Spearman coefficients for the added structures decreases, to 0.72 - 0.73 for the A and B extension cases and to 0.62 and 0.40 for the C extension case. Similarly for the enthalpy per atom models, the fitting MAE and MAFE values display limited variation between the base model and the models with added structures. In the case of the enthalpy per atom models, the base model has a fitting set Spearman coefficient of 0.87. Adding structures from new CSP runs approximately halves the fitting set Spearman coefficient for the added structures to 0.41 - 0.49 for the A and B extension cases and to 0.32 and 0.38 for the C extension case. Similarly in the phase model, mAP values for both fitting and testing sets show substantial drops relative to the base model when new crystal structures are added to the training data.
The model struggles with extending directly from the 1,3,5-triazine HCl to piperidine HCl. The testing Spearman coefficient indicates a weak correlation between the values of VDFT and VML. The model is able to achieve a relatively low MAFE compared to other extensions by “guessing” the average value rather than learning a reliable relation between the initial structures and the final relaxed volume. Future work will investigate if extension can be improved by training on a broader initial chemical space.
The decreases in testing set Spearman coefficients for the added structures fundamentally limits the accuracy of the ML approach. The low Spearman coefficient causes the constructed models to have difficulty ranking the volumes and enthalpies per atom of new structures. While the model approach cannot be used alone to identify experimentally obtainable structures, it can be used as a tool for downselecting structures for further computational study.
Discussion
In this work we have trained ML models to predict the properties of DFT-relaxed crystal structures of molecular salts based on only the unrelaxed structures. The goal is to produce a machine learning method which filters out molecular crystal structures in CSP workflows by identifying which structures are likely to relax into physically unfavorable crystals. We considered three key quantities: volume, enthalpy per atom, and metal versus semiconductor/insulator phase. The chemical systems included small ring molecules of 1,2,3-triazine, 1,2,4-triazine, 1,3,5-triazine, pyridine, thiophene, and piperidine combined with varying concentrations of HCl. Our approach has two key components to speed up model construction: we use crystal graph singular values instead of the full crystal graph representations, and random forests instead of neural networks. Use of crystal graph singular values reduces the total number of descriptors by at least two orders of magnitude. Random forests are fit more rapidly than neural networks and require tuning of fewer hyperparameters. Each model is fit at low computational cost, each one requiring on the order of minutes to train on an individual workstation. The structure evaluation and machine learning approach demonstrated in this work is not intended as a stand-alone CSP algorithm. As presented, the ability to identify rare polymorphs would be slowed by the reliance on DFT for geometric optimization and the region of the structural space explored by the randomly generated structures. Instead, the utility of the machine learning approach is as a filtering step in other CSP efforts involving groups of related chemical compounds. Integrating into other CSP efforts is beyond the scope of the presented work, but is the focus of ongoing studies.
The models performed consistently well in the interpolative regime, with the testing and validation error distributions closely matching the fitting error distributions. Performance of the models was inconsistent between target quantities in the extrapolative regime. Testing volume and enthalpy per atom MAE and MAFE values for materials added to the base model were comparable to the testing MAE and MAFE values found for the base model. Instead, difficulty in the extrapolative regime appeared as marked decreases in the Spearman coefficients between the DFT calculated and ML predicted values. In the case of predicting semiconductor/insulator versus metallic phases, the models showed additional difficulty consistently identifying the minority metallic phase when new organic salts were added to the training data. While this work has demonstrated that there is some ability to construct ML models by training on a large base data set then incorporating data from 2000-10,000 structures of a new chemical system, this approach still requires development and refinement. Difficulty in extrapolating to new chemical spaces is typical of machine learning models. Within our approach, extrapolation could be improved by broadening the chemical space included in the initial training set and using more sophisticated approaches from transfer learning. Our choices of organic molecules were largely limited to small, rigid molecules. Numerous applications and challenges for organic CSP require such considerations, yet it would also be worthwhile to test the ability of the presented machine learning approach to predict properties of crystals based on more flexible molecules. Future work will generate and relax structures of salts of flexible molecules starting from multiple conformers in order to test the reliability of our machine learning approach on current CSP challenges.
It is also important to note our model building method shows several advantages compared to the widely used CGCNN approach. While the time complexity for both neural networks and random forests is linear in the dimensionality of the material representation43,56, the computational cost of fitting the CGCNN is at least two orders of magnitude larger than fitting the random forest. Further, the random forest regressors produce lower error models for smaller fitting sets. Our set of crystal graph singular value descriptors accelerates model construction compared to the full crystal graph representation by reducing the required number of descriptors needed to characterize each material, while improving the accuracy of models fit with multiple chemical compositions. While both neural networks (e.g. Refs. 38,67,68,69) and random forests (e.g. refs. 70,71,72,73) have shown success in predicting materials’ properties, random forests tend to be easier to train due to requiring tuning of fewer hyperparameters.
The limitations of incorporating the machine learning method developed here into CSP workflows are that it assumes the experimentally observable polymorphs can be determined from only thermodynamic considerations and that sufficient training data covering the appropriate regions of configuration space could be generated to construct usable models. There are many cases among pharmaceutical molecules in which the thermodynamically most stable structure is kinetically hindered, and therefore not observed74. Large, flexible molecules pose unique challenges to current organic CSP efforts. Beyond introducing additional degrees of freedom which must be considered, small changes in bond and torsion angles can drastically change the energetic stability of a crystal structure9,75. The challenge for machine learning methods becomes both sampling the configuration space and learning rapidly varying functions. Only limited work has been performed on developing machine learning approaches to discontinuous functions76.
The model building approach taken in this work is general and can be extended in multiple directions. A wider range of organic molecule components can be tested and incorporated into the models’ training sets. Target values and optimization criteria can be refined to better search for experimentally realizable polymorphs. With the model training set sufficiently expanded, it can rank proposed polymorph structures to downselect which structures should receive further computational examination. Our approach could be extended to include more complex systems: larger organic molecules, cocrystals, intercalated systems, organometallic complexes, and diasteroemeric salts. Finally, the machine learning approach here is not limited to using quantities predicted with DFT. It could instead be combined with data generated using, as an example, force-field methods65,77,78.
Current developments in organic CSP look beyond predicting crystal structures and toward the rational design of materials across numerous applications, including pharmaceuticals, organic semiconductors, and porous organic materials. The challenge in rational design requires considering the interplay between crystal structure and organic molecule while accounting for real-world influences from effects including: temperature, solvents, and crystallization kinetics79. Solving such problems will require novel computational approaches toward accelerating CSP. One of the currently best performing workflows for organic CSP, developed by Firaha et al.27, utilizes the GRACE software package65,80. This workflow requires performing multiple force field and ab initio calculations for numerous trial structures to obtain highly accurate optimized crystal structures. The approach demonstrated in our work may assist in two ways: either providing a coarse initial screening to narrow the configuration space in which the CSP approach by Firaha et al. should search or allowing the CSP method by Firaha et al. to generate initial configurations then using machine learning models to downselect which configurations should be considered for the most expensive theromodynamic calculations.
Code availability
Machine learning code is available at ref. 84.
References
Datta, S. & Grant, D. J. W. Crystal structures of drugs: advances in determination, prediction and engineering. Nat. Rev. Drug. Discov. 3, 42–57 (2004).
Yang, J. et al. Ddt polymorphism and the lethality of crystal forms. Angew. Chem. 129, 10299–10303 (2017).
Hao, Z. & Iqbal, A. Some aspects of organic pigments. Chem. Soc. Rev. 26, 203–213 (1997).
Kumar, B., Kaushik, B. K. & Negi, Y. S. Perspectives and challenges for organic thin film transistors: Materials, devices, processes and applications. J. Mater. Sci. Mater. Electron. 25, 1–30 (2014).
Corma, A., Garcia, H. I. & Llabres i Xamena, F. X. Engineering metal organic frameworks for heterogeneous catalysis. Chem. Rev. 110, 4606–4655 (2010).
Bai, F. et al. Organic optoelectronics (John Wiley & Sons, 2012).
Motherwell, W. D. S. et al. Crystal structure prediction of small organic molecules: A second blind test. Acta. Crystall. B-Stru. 58, 647–661 (2002).
Oganov, A. R., Lyakhov, A. O. & Valle, M. How evolutionary crystal structure prediction works and why. Accounts. Chem. Res. 44, 227–237 (2011).
Price, S. L. Predicting crystal structures of organic compounds. Chem. Soc. Rev. 43, 2098–2111 (2014).
Reilly, A. M. et al. Report on the sixth blind test of organic crystal structure prediction methods. Acta. Crystall. B-Stru. 72, 439–459 (2016).
Corpinot, M. K. & Bucar, D.-K. A practical guide to the design of molecular crystals. Cryst. Growth Des. 19, 1426–1453 (2018).
Maddox, J. Crystals from first principles. Nature 335, 201–201 (1988).
Cruz-Cabeza, A. J. Crystal structure prediction: are we there yet? Acta. Crystall. B-Stru. 72, 437–438 (2016).
Price, S. L. Control and prediction of the organic solid state: a challenge to theory and experiment. P. Roy. Soc. A-Math. Phy. 474, 20180351 (2018).
Cheng, C. Y., Campbell, J. E. & Day, G. M. Evolutionary chemical space exploration for functional materials: computational organic semiconductor discovery. Chem. Sci. 11, 4922–4933 (2020).
Lommerse, J. P. M. et al. A test of crystal structure prediction of small organic molecules. Acta. Crystall. B-Stru. 56, 697–714 (2000).
Nyman, J. & Day, G. M. Static and lattice vibrational energy differences between polymorphs. CrystEngComm 17, 5154–5165 (2015).
Greenwell, C. & Beran, G. J. O. Inaccurate conformational energies still hinder crystal structure prediction in flexible organic molecules. Cryst. Growth Des. 20, 4875–4881 (2020).
Hunnisett, L. M. et al. The seventh blind test of crystal structure prediction: Structure generation methods. J. Acta. Cryst. submitted.
Sontising, W. & Beran, G. J. O. Combining crystal structure prediction and simulated spectroscopy in pursuit of the unknown nitrogen phase ζ crystal structure. Phys. Rev. Mater. 4, 063601 (2020).
Conway, L. J., Pickard, C. J. & Hermann, A. Rules of formation of h–c–n–o compounds at high pressure and the fates of planetary ices. Proc. Natl. Acad. Sci. USA 118, e2026360118 (2021).
Nelson, J. R., Needs, R. J. & Pickard, C. J. Navigating the ti-co and al-co ternary systems through theory-driven discovery. Phys. Rev. Mater. 5, 123801 (2021).
Day, G. M. Current approaches to predicting molecular organic crystal structures. Crystallogr. Rev. 17, 3–52 (2011).
Atahan-Evrenk, S. & Aspuru-Guzik, A. Prediction and calculation of crystal structures. Top. Curr. Chem. 345, 95–138 (2014).
Yang, J. et al. Large-scale computational screening of molecular organic semiconductors using crystal structure prediction. Chem. Mater. 30, 4361–4371 (2018).
Curtis, F. et al. Gator: a first-principles genetic algorithm for molecular crystal structure prediction. J. Chem. Theory Comput. 14, 2246–2264 (2018).
Firaha, D. et al. Predicting crystal form stability under real-world conditions. Nature 623, 324–328 (2023).
Butler, P. W. V. & Day, G. M. Reducing overprediction of molecular crystal structures via threshold clustering. Proc. Natl. Acad. Sci. USA 120, e2300516120 (2023).
Villeneuve, N. M., Dickman, J., Maris, T., Day, G. M. & Wuest, J. D. Seeking rules governing mixed molecular crystallization. Cryst. Growth Des. 23, 273–288 (2022).
Musil, F. et al. Machine learning for the structure–energy–property landscapes of molecular crystals. Chem. Sci. 9, 1289–1300 (2018).
McDonagh, D., Skylaris, C.-K. & Day, G. M. Machine-learned fragment-based energies for crystal structure prediction. J. Chem. Theory Comput. 15, 2743–2758 (2019).
Egorova, O., Hafizi, R., Woods, D. C. & Day, G. M. Multifidelity statistical machine learning for molecular crystal structure prediction. J. Phys. Chem. A 124, 8065–8078 (2020).
Wengert, S., Csanyi, G., Reuter, K. & Margraf, J. T. Data-efficient machine learning for molecular crystal structure prediction. Chem. Sci. 12, 4536–4546 (2021).
Balodis, M., Cordova, M., Hofstetter, A., Day, G. M. & Emsley, L. De novo crystal structure determination from machine learned chemical shifts. J. Am. Chem. Soc. 144, 7215–7223 (2022).
Kilgour, M., Rogal, J. & Tuckerman, M. Geometric deep learning for molecular crystal structure prediction. J. Chem. Theory Comput. 19, 4743–4756 (2023).
Honrao, S. J., Xie, S. R. & Hennig, R. G. Augmenting machine learning of energy landscapes with local structural information. J. Appl. Phys. 128, 085101 (2020).
Gibson, J., Hire, A. & Hennig, R. G. Data-augmentation for graph neural network learning of the relaxed energies of unrelaxed structures. Npj Comput. Mater. 8, 1–7 (2022).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Chen, C., Ye, W., Zuo, Y., Zheng, C. & Ong, S. P. Graph networks as a universal machine learning framework for molecules and crystals. Chem. Mater. 31, 3564–3572 (2019).
Park, C. W. & Wolverton, C. Developing an improved crystal graph convolutional neural network framework for accelerated materials discovery. Phys. Rev. Mater. 4, 063801 (2020).
Karamad, M. et al. Orbital graph convolutional neural network for material property prediction. Phys. Rev. Mater. 4, 093801 (2020).
Lee, J. & Asahi, R. Transfer learning for materials informatics using crystal graph convolutional neural network. Comput. Mater. Sci. 190, 110314 (2021).
Breiman, L. Random forests. Mach. Learn. 45, 5–32 (2001).
Valencia, D., Whiting, G. T., Bulo, R. E. & Weckhuysen, B. M. Protonated thiophene-based oligomers as formed within zeolites: understanding their electron delocalization and aromaticity. Phys. Chem. Chem. Phys. 18, 2080–2086 (2016).
Cruz-Cabeza, A. J. Acid–base crystalline complexes and the p k a rule. CrystEngComm 14, 6362–6365 (2012).
Pickard, C. J. & Needs, R. J. High-pressure phases of silane. Phys. Rev. Lett. 97, 045504 (2006).
Pickard, C. J. & Needs, R. J. Ab initio random structure searching. J. Condens. Matter Phys. 23, 053201 (2011).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Anghel, A. T., Day, G. M. & Price, S. L. A study of the known and hypothetical crystal structures of pyridine: why are there four molecules in the asymmetric unit cell? CrystEngComm 4, 348–355 (2002).
Groom, C. R., Bruno, I. J., Lightfoot, M. P. & Ward, S. C. The cambridge structural database. Acta. Crystall. B-Stru. 72, 171–179 (2016).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties https://github.com/txie-93/cgcnn (2018).
Rupp, M., Tkatchenko, A., Mueller, K.-R. & Von Lilienfeld, O. A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 108, 058301 (2012).
Montavon, G. et al. Learning invariant representations of molecules for atomization energy prediction. Adv. Neur. Int. 25, 449–457 (2012).
Menze, B. H. et al. A comparison of random forest and its gini importance with standard chemometric methods for the feature selection and classification of spectral data. BMC bioinformatics 10, 1–16 (2009).
Pedregosa, F. et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Kitaigorodskii, A. I. Theory of close packing of molecules. In Organic Chemical Crystallography, 65–112 (Consultants Bureau, New York, 1961).
Little, M. A. & Cooper, A. I. The chemistry of porous organic molecular materials. Adv. Funct. Mater. 30, 1909842 (2020).
Cote, A. P. et al. Porous, crystalline, covalent organic frameworks. Science 310, 1166–1170 (2005).
Cai, G., Yan, P., Zhang, L., Zhou, H.-C. & Jiang, H.-L. Metal–organic framework-based hierarchically porous materials: synthesis and applications. Chem. Rev. 121, 12278–12326 (2021).
Yu, S., Xing, G.-L., Chen, L.-H., Ben, T. & Su, B.-L. Crystalline porous organic salts: from micropore to hierarchical pores. Adv. Mater. 32, 2003270 (2020).
Xing, G., Peng, D. & Ben, T. Crystalline porous organic salts. Chem. Soc. Rev. 53, 1495–1513 (2024).
Nikhar, R. & Szalewicz, K. Reliable crystal structure predictions from first principles. Nature Commun. 13, 3095 (2022).
Metz, M. P. et al. Crystal structure predictions for 4-amino-2, 3, 6-trinitrophenol using a tailor-made first-principles-based force field. Cryst. Growth Des. 22, 1182–1195 (2022).
Mattei, A. et al. Efficient crystal structure prediction for structurally related molecules with accurate and transferable tailor-made force fields. J. Chem. Theory Comput. 18, 5725–5738 (2022).
Gebhard, F. Metal—insulator transitions. In The Mott Metal-Insulator Transition, 1–48 (Springer, 1997).
Ye, S., Li, B., Li, Q., Zhao, H.-P. & Feng, X.-Q. Deep neural network method for predicting the mechanical properties of composites. Appl. Phys. Lett. 115, 161901 (2019).
Feng, S., Zhou, H. & Dong, H. Using deep neural network with small dataset to predict material defects. Mater. Design 162, 300–310 (2019).
Kim, B., Lee, S. & Kim, J. Inverse design of porous materials using artificial neural networks. Sci. Adv. 6, eaax9324 (2020).
Nagasawa, S., Al-Naamani, E. & Saeki, A. Computer-aided screening of conjugated polymers for organic solar cell: classification by random forest. J. Phys. Chem. Lett. 9, 2639–2646 (2018).
Takahashi, K., Takahashi, L., Miyazato, I. & Tanaka, Y. Searching for hidden perovskite materials for photovoltaic systems by combining data science and first principle calculations. ACS Photonics 5, 771–775 (2018).
Wang, T., Zhang, C., Snoussi, H. & Zhang, G. Machine learning approaches for thermoelectric materials research. Adv. Funct. Mater. 30, 1906041 (2020).
Goodall, R. E. A. & Lee, A. A. Predicting materials properties without crystal structure: Deep representation learning from stoichiometry. Nat. Commun. 11, 1–9 (2020).
Neumann, M. A. & van de Streek, J. How many ritonavir cases are there still out there? Faraday Discuss. 211, 441–458 (2018).
Iuzzolino, L., McCabe, P., Price, S. L. & Brandenburg, J. G. Crystal structure prediction of flexible pharmaceutical-like molecules: density functional tight-binding as an intermediate optimisation method and for free energy estimation. Faraday Discuss. 211, 275–296 (2018).
Moustapha, M. & Sudret, B. Learning non-stationary and discontinuous functions using clustering, classification and gaussian process modelling. Comput. Struct. 281, 107035 (2023).
Neumann, M. A., Leusen, F. J. J. & Kendrick, J. A major advance in crystal structure prediction. Angew. Chem. Int. Ed. 47, 2427–2430 (2008).
Nyman, J., Pundyke, O. S. & Day, G. M. Accurate force fields and methods for modelling organic molecular crystals at finite temperatures. Phys. Chem. Chem. Phys. 18, 15828–15837 (2016).
Beran, G. J. O. Frontiers of molecular crystal structure prediction for pharmaceuticals and functional organic materials. Chem. Sci. 14, 13290–13312 (2023).
Neumann, M. A. & Perrin, M.-A. Energy ranking of molecular crystals using density functional theory calculations and an empirical van der waals correction. J. Phys. Chem. B 109, 15531–15541 (2005).
Blaiszik, B. et al. The materials data facility: Data services to advance materials science research. JOM 68, 2045–2052 (2016).
Blaiszik, B. et al. A data ecosystem to support machine learning in materials science. MRS Commun. 9, 1125–1133 (2019).
Shapera, E. P., Bucar, D.-K., Prasankumar, R. P. & Heil, C. Dataset for the paper “accelerating crystal structure prediction of organic salts via machine learning” https://acdc.alcf.anl.gov/mdf/detail/organic_crystal_prediction_v1.4/ (2023).
Machine-learning-assisted-prediction-of-organic-salt-structure-properties. https://github.com/EthanPShapera/Machine-learning-assisted-prediction-of-organic-salt-structure-properties.
Acknowledgements
We thank C. J. Pickard for assistance with AIRSS. We thank V. Hegde and B. Meredig for helpful discussions. Computational time was provided by the dCluster of the Graz University of Technology. Funding was provided by Enterprise Science Fund, Intellectual Ventures. Supported by TU Graz Open Access Publishing Fund.
Author information
Authors and Affiliations
Contributions
E.P.S. and C.H ran high throughput crystal structure prediction calculations. E.P.S. developed the machine learning approach. D.K.B. determined the chemical systems to study and design criteria. E.P.S. and C.H. contributed in writing the manuscript. All authors contributed to final editing of the manuscript. R.P.P. and C.H. procured funding and supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shapera, E.P., Bučar, DK., Prasankumar, R.P. et al. Machine learning assisted prediction of organic salt structure properties. npj Comput Mater 10, 176 (2024). https://doi.org/10.1038/s41524-024-01355-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41524-024-01355-x
