
Abstract
Graphene foam (GF), synthesized via Chemical Vapor Deposition (CVD), has been proven to be the ideal bulk porous material. The addition of poly(dimethylsiloxane) (PDMS) within the porous structure enables enhancement of mechanical strength and alteration of heat transfer behavior. This study focuses on the thermodynamic behavior of GF/PDMS composites during deformation, and employs stochastic modeling and neuroevolution potential (NEP) for complex material modeling with precise prediction of microscopic mechanisms governing thermal property variations. The results demonstrate that the composite with a 5% doping rate of PDMS achieves the optimal mechanical performance and shows a 7.13-fold modulation in thermal resistance during the deformation from 40% stretching to 50% compression. Findings indicate PDMS fortifies structural stability while enabling dynamic thermal conductivity modulation in GF. This research provides critical insights into the micro-mechanisms of GF/PDMS composites and offers a theoretical foundation for applications in dynamic thermal management and self-powered sensor networks.
Similar content being viewed by others

Introduction
Three-dimensional graphene foam (GF) fabricated via chemical vapor deposition (CVD) possesses a large specific surface area and inherits the high-quality characteristics of two-dimensional graphene, making it highly promising for robust applications in flexible electronics, dynamic thermal management, electromagnetic shielding and sensing technologies1,2,3,4,5. While this synthetic approach yields GF with excellent thermal and electrical conductivity, the self-supporting structure of free-standing GF is inherently fragile and prone to fracture under external mechanical loading6. To address this limitation, PDMS, a versatile, elastomeric polymer renowned for its mechanical flexibility, chemical stability, and processability, has emerged as a key material for reinforcing GF. By integrating PDMS, researchers can substantially enhance the mechanical resilience of GF, such as increasing its Young’s modulus and fracture toughness7,8, while simultaneously tailoring its functional properties9,10,11. Xu et al.12 demonstrated that a bilayer GF/PDMS/poly (ethylene terephthalate) (PET) composite exhibits a sixfold higher relative resistance change under bending compared to a single-layer GF/PDMS composite, highlighting PDMS’s role in modulating electro-mechanical behavior. Du et al.13 further revealed that PDMS-doped GF composites show an 8-fold tunability in thermal conductance during compression, enabling the design of continuously adjustable thermal switches. These findings underscore how PDMS integration not only strengthens GF mechanically but also endows it with dynamically tunable electrical, thermal, and electromagnetic properties, making GF/PDMS composites indispensable for advanced applications requiring multifunctionality and durability.
Experimental methods effectively characterize the adjustable macroscopic properties of the GF/PDMS system. However, it is particularly challenging to explore the influence of microscale deformation, notably on thermal transport, owing to the system’s heterogeneous nature as a composite involving interactions between inorganic and organic components7,14,15.
Current studies often utilize simplified theoretical frameworks and traditional molecular dynamics (MD) simulations, such as volume fractions and representative volume element (RVE), to investigate the mechanism underlying its mechanical and thermodynamic behaviors16,17,18. Analyses based on these simplified equivalent models may exhibit limitations under extreme deformation conditions. While traditional MD approaches have provided many significant insights, their limitations in modeling system-wide deformations, in combination with high computational complexity, often restricted their application to small-scale local models19. The development of more efficient and precise quantitative predictive tools remains critical to inform rational material optimization and accelerate translational applications.
In this study, we introduced a stochastic modeling framework that integrates Density Functional Theory (DFT) data with machine learning to derive an enhanced interatomic potential to explore the thermodynamic properties of the GF/PDMS composites20,21,22. The accuracy and efficiency of this method have been validated through comparisons with inter-molecular spacing energies in C60 systems23, the Tersoff potential for pristine graphene24, and reactive force field simulations of magnesium ion dissolution25. The behaviors of GF with different PDMS doping rates were disclosed during the process of stretching and compression. The microscopic mechanism underlying the relationship between the regulation of thermal properties and the deformation was revealed. The decoding of thermal tuning in GF composites provides theoretical support for the design and preparation of pressure-sensitive functional materials for superior-performance thermal regulation systems.
Results
Physically constrained random contact-based model construction
In the CVD method, the skeleton of GF is constructed by stacked graphene layers. As the composite is fabricated by immersing GF in PDMS solution, the connection between GF and PDMS mainly relies on Van der Waals’ force4.
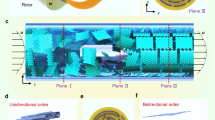
The physical model utilized in this simulation is based on the experimental preparation of the GF/PDMS composite. Limited by the computational capability of the Vienna Ab initio Simulation Package (VASP), PDMS models are constructed by repeating units ranging from 3 to 8. The molecular structures of PDMS with different units are presented in Fig. 1a. The GF model mimics the layer-by-layer structure. Additionally, a supercell is employed to guarantee structural uniformity and the non-randomness of cross-plane heat transfer.

a Chain structure of PDMS molecules with 4, 6 and 8 repeating units; b Point, edge and face contacts between PDMS with graphene; c GF with varying PDMS doping rates in MD simulations.
To comprehensively explore all possible integration of PDMS with GF without compromising computational efficiency, a stochastic contact algorithm was implemented through Python code. The primary interaction sites between PDMS and graphene are CH–π interactions, where methyl hydrogen atoms in PDMS engage with the π-electron cloud of graphene. These interactions, supplemented by potential crosslinking between the CH3 side groups of PDMS and graphene, form the basis of the interfacial modeling strategy26,27.
Given the localized nature of these interactions, our method strategically places PDMS chains via three distinct contact modes-point, edge, and face contacts- anchored at randomly selected carbon atoms on the graphene surface, as shown in Fig. 1b. Each attachment is geometrically optimized to prevent atomic overlaps, with molecular orientations sampled from a uniform distribution. This approach effectively captures the distribution of interfacial configurations observed in experimental composites, as validated by the dynamic behavior of contact points during MD equilibration.
Following stochastic placement, energy minimization is performed to relax the system into stable adhesion structures. The resulting supercell models (60Å × 60Å × 300Å) systematically vary PDMS doping from 0% to 10%, with all PDMS molecules bound to the GF framework through non-covalent interactions, as depicted in Fig. 1c. Detailed parameters for the five representative models are provided in Table 1.
Neuroevolution potential training
Traditional empirical potentials for MD simulations exhibit limited transferability to the continuous compression behavior of GF/PDMS composites. This limitation arises from the heterogeneous organic-inorganic interfaces and multiscale structural evolution inherent to such hybrid systems. To address this gap, we developed a machine-learning potential (MLP) trained on quantum mechanical datasets derived from the DFT calculations, as shown in Fig. 2. This approach leverages the Neuroevolution Potential (NEP-4), which integrates the unique advanced Seperable Natural Evolution Strategy (SNES) for the global optimization of atomic interactions. The NEP-4 architecture achieves sub-meV/atom accuracy in energy predictions while showing higher computational efficiency comparable to classical MD potentials28,29. Hence, the NEP-4 model was adopted for potential training in our research.

The process involves: (i) obtaining configurations through AIMD or MD; (ii) screening configurations that cannot be adequately described by the potential; (iii) training the potential function through active learning to achieve a comprehensive description of nearly all structural features of the system; (iv) conducting thermodynamic and mechanical property analyses based on the optimized potential function.
The interatomic potential was trained on a multiscale dataset encompassing defective and pristine CVD-fabricated graphene, pure PDMS chains with tunable crosslinking densities, and GF/PDMS nanocomposite interfaces. Figure 3a presents the scanning electron microscope (SEM) characterization of GF/PDMS. The initial model trained on the dataset accurately reproduces structural features at the microscopic scale. The number of atoms in all configurations ranges roughly from 40 to 150. Ab Initio Molecular Dynamics (AIMD) generated initial configurations for GF/PDMS systems. The VASP software suite (DFT-based) provided reference energies, forces, and virial tensors. Using the NPT ensemble, we perturbed and simulated configurations in the training set to filter out cases inadequately described by the potential. Representative structures were selected via farthest-point sampling30,31, with their energy, force, and virial data fed into training and testing sets to iteratively improve model accuracy. The Bussi-Donadio-Parrinello thermostat and Bernetti-Bussi barostat maintained stable temperature and pressure conditions during simulations, while GPUMD (a GPU-accelerated MD tool) enabled efficient training32,33.

a SEM images of GF/PDMS composite45 (Reproduced with permission of Elsevier, 2016); b Loss functions of training and testing datasets with the number of training steps; Plots comparing NEP predictions against DFT reference values for energy (c), atomic forces (d), and virial stresses (e); f Convergence check results in atomic forces; g The comparison of Reaxff and DFT; h Comparison of computational speeds between AIMD and MLP-NEP; i Size dependence of thermal conductivity in GF/PDMS composites with a 5% doping rate at different sizes of 30 Å, 45 Å, 60 Å, 75 Å, 300 Å and 600 Å.
The fitting results of the loss function are shown in Fig. 3b. Convergence behavior ensures consistency between computed values and fitted data. Comparative analysis of DFT versus NEP predictions, shown in Fig. 3c–e), revealed sub-meV/atom energy root-mean-square errors (RMSEs) and sub-0.1 eV/Å force deviations, consistent with state-of-the-art NEP benchmarks23,33. The diagonal clustering of data points confirms the potential accuracy in modeling GF/PDMS interfacial dynamics.
To validate the robustness of the potential, we performed convergence checks during the final 10 ns of MD simulations, compared the differences in energy, force, and virial obtained by NEP and DFT under extreme deformation conditions, ensuring that the prediction errors in energy, forces and virials remained within the bounds established by the training data. Testing configurations (uniformly sampled) showed that NEP-predicted energy, force, and virial errors remained within training data bounds. As shown in Fig. 3f, the root-mean-square error (RMSE) for forces between NEP and DFT was 291.3 meV/Å, confirming the potential’s accuracy and stability23,24,25.
To further assess the accuracy of our model, we used part of the training dataset to compare the atomic forces predicted by the trained NEP-MLP with those from a reactive force field34 (Reaxff) (Fig. 3g) based on previously reported force field. The Reaxff-predicted atomic forces exhibited a significantly higher RMSE of 7170.9 meV/Å compared to NEP-MLP’s 384.2 meV/Å (Fig. 3g), clearly demonstrating the machine learning potential’s superior accuracy and reliability for capturing complex interactions in GF/PDMS systems.
With comparable accuracy and identical computational resources, NEP-MLP achieves a speedup of ~30,000,000 times over traditional AIMD (Fig. 3h), providing the computational foundation for the discoveries presented in this work. Due to the substantial computational variability observed in small systems (as discussed in Section “Strain-dependent mechanics of GF/PDMS composites”), simulations involving large-scale systems—on the order of tens of thousands of atoms—are essential. In this context, the extremely low time complexity of NEP provides a significant advantage, enabling efficient and accurate calculating of large systems.
Size-convergence validation
In the calculation of thermal conductivity based on MD, it has been commonly observed that the size effect tends to induce an underestimation of the thermal conductivity values of materials. This phenomenon indicates that the size of the material must be greater than the average free path of phonons. To mitigate finite-size effects in non-equilibrium molecular dynamics (NEMD) thermal conductivity calculations, the GF/PDMS composite with a 5% doping rate is systematically evaluated across six system sizes ranging from 30 Å to 600 Å, as shown in Fig. 3f. Sub-75Å model exhibits atomic crossovers between group layers during the relaxation process due to the intergroup displacements less than 6 Å, distorting temperature gradient calculations.
Above 75 Å, the proportion of atoms involved in crossovers decreases exponentially (R2 = 0.97) with system size, stabilizing thermal conductivity values within 2% variance beyond 300 Å, as depicted in Fig. 3i. Limited by computational resource constraints, the 300 Å model is selected in the calculations here.
Strain-dependent mechanics of GF/PDMS composites
To explore the mechanical and thermal response of GF/PDMS composites under uniaxial strain, the compression and stretching are imposed along the z-axis direction on the GF with different PDMS doping rates. The deformation features of the GF/PDMS composite are depicted in Fig. 4a–e. Fixed boundary conditions in x-y planes eliminate lateral expansion artifacts. In the process of compression, the squeezing effect between atoms accumulates continuously until the box cracks irreversibly. Terminating compression at 50% prevents cell fracture. It shows that the graphene layers experience bending and wrinkling during compression. The inter-layer space is squeezed until the graphene layers stack together. In the process of stretching, the strain increases with the stretching length. Percolative fracture networks emerge at 40% tensile strain, which leads to the disruption of continuous thermal pathways. Therefore, the stretching length is confined to 40%.

a Pristine structure; b Structure 25% compression ratio; c Structure with 50% compression ratio; d Structure 20% stretching ratio; e Structure with 40% stretching ratio; f Tensile stress variation with varying PDMS doping rates under stretching; g Maximum stretchability rate with varying PDMS doping rates; h Tensile stress variation with varying PDMS doping rates under compressions; i The relationship between PDMS doping rates and tensile modulus, tensile strength, and fracture strain.
Five models selected from Table 1 serve as base models, within 0.5 ns stretching and compressive deformations applied along the z-axis. Figure 4f–i illustrate their evolution from the initial state to fracture under varying PDMS doping rates. As doping levels increase, the maximum stretching ratio gradually improves, indicating that PDMS effectively enhances the tensile strength of pristine GF. A critical doping threshold emerges between 2.5% and 5%, where the maximum tensile strain increases by 4.5%. In the 2.5% doped model, insufficient PDMS quantity results in inadequate interlayer connections between graphene layers. Consequently, the graphene undergoes fracture at the unbonded regions where PDMS is absent, and thus the stretching curve almost mirrors that of pure GF. At 5% doping, PDMS bridges nearly all graphene layers, enabling load transfer significantly that elevates peak tensile strength to 1.7 GPa at 10% doping. Force curve fluctuations indicate graphene layer fracture: incomplete force drops to 0 GPa signal sustained interlayer connections via PDMS chains. Additionally, the frequency of stress drops also increases with doping levels. Figure 4i quantifies mechanical parameter improvements of 20% rise in tensile modulus and tensile strength in the z-direction with PDMS doping rates. While fracture strain improvements are subtle, consistent enhancements are observable. The behavior during compression follows a trend that is observed during stretching. As illustrated in Fig. 4h, at equivalent strain, higher doping reinforces compressive pressure (negative force), attributed to the rigid PDMS framework restricting the global deformation. Notably, the 5% PDMS compressive stress curve lies significantly below 0% and 2.5% counterparts, underscoring its role in forming a stable rigid structure that stabilizes thermal conduction pathways consistent with its mechanical reinforcement function.
Thermal transport modulation in GF/PDMS composites
While PDMS enhances the mechanical properties of GF, its role as a dopant in modulating phonon-mediated heat transport remains critical for dynamic thermal management applications. Here, we measured the thermal conductivity and thermal conductance of five initial composite models using NEMD simulations. The simulation box is partitioned into 13 axial groups along the heat-flow direction, with a temperature gradient imposed along the z-axis by defining a hot plane (330 K) and a cold plane (270 K). Heat transfer is induced via kinetic energy exchange between particles in these planes, such that energy removed from the hot region is exactly balanced by energy added to the cold region. A linear increase in this energy exchange over time indicates the establishment of steady-state thermal transport.
Figure 5a illustrates the temporal evolution of energy in the hot (blue line) and cold (red line) regions. During the initial 0.5 ns, non-uniform heat distribution prevented steady-state conditions. After this transient period, the thermal transfer of the system gradually stabilizes, and the heat flux between hot and cold regions approaches equilibrium. Figure 5b shows the temperature profile of each axial group from the onset of steady state (0.5 ns) to 2 ns, demonstrating uniform thermal gradient development across the system.

a Temporal evolution of energy gain (red) and loss (blue) in cold and hot regions; b Z-direction temperature profile across axial groups from the establishment of steady-state thermal transport (0.5 ns) to 2 ns; c Variation of thermal conductivity and thermal conductance of GF with different doping rates of PDMS (T = 300 K); d Equivalent thermal circuit of pure GF (without PDMS); e Comparison of heat-flow pathways with and without PDMS; f Equivalent thermal circuit of GF doped with PDMS; g Thermal conductivity variation with the varying deformation; h Thermal conductance variation with the varying deformation.
The thermal properties of GF doped with PDMS are investigated under both compression and stretching. Figure 5c shows the increasing doping rate of PDMS directly results in the decline of the total thermal conductivity of the composite. This is because the thermal conductivity of PDMS (0.14 W/(m·K)) is an order of magnitude lower than that of pure GF at the identical temperature (T = 300 K) and density (ρ = 0.49 g/cm3). Here, thermal resistance analysis is employed to reveal the underlying mechanism of thermal property variation with doping rates. The thermal circuit of pristine GF, composed of stacked graphene layers, is displayed in Fig. 5d. Each of the graphene layers acts as a parallel thermal resistance in the heat-transfer process. When PDMS molecules are introduced, regardless of the contact form, additional branch thermal resistance arises at graphene-PDMS interfaces. Figure 5e compares the heat-flow pathways with and without PDMS, while Fig. 5f depicts the equivalent thermal circuit after PDMS doping. It shows PDMS incorporation increases the total thermal resistance, which is the primary reason for the decline in thermal conductivity. However, the thermal resistance increases moderately for limited PDMS loadings, leading to an insignificant reduction in the overall thermal conductivity.
The thermal conductivity and the thermal conductance of GF under compression are measured, as shown in Fig. 5g, h. Results indicate that across all PDMS doping rates, the thermal conductivity λ decreases as the model height reduces, aligning with increased contact thermal resistance from gradual graphene layer stacking during compression. The simulated thermal conductivity values are in good agreement with experimental results, exhibiting the same order of magnitude as the literature-reported data: 0.94 W/(m·K) at 8vol.% PDMS doping and 0.56 W/(m·K) at 0.7 wt.% PDMS doping. This close correspondence validates the reliability of our simulation framework19,35,36. Within the Tight Binding model framework37, electrons are confined near atomic nuclei with interactions primarily between adjacent atoms. Morphological changes induced by PDMS incorporation and composite compression enhance van der Waals forces between graphene layers, introducing stronger hindrances to atomic thermal motion. It amplifies phonon-phonon scattering during propagation, shortening phonon lifetimes and reducing thermal conductivity.
Thermal conductance variations across models with differing PDMS doping rates exhibit an intriguing trend. Over the deformation spectrum from 50% compression to 40% stretching, the thermal conductivity scales approximately linearly with model length. Since thermal conductance depends on the ratio of thermal conductivity to model length, thermal conductance is governed primarily by the relative shifts in thermal conductivity. Below a 20% stretching ratio, the thermal conductance shows a minor initial decrease followed by an increase, a behavior rationalized by a spring model38 rooted in the effective medium theory.
During the compression from the initial state to 50%, the limited formation of new internal bonds, especially in low-density CVD models, is offset by increased thermal pathways, driving a continuous rise in overall thermal conductance consistent with computational results. When the stretching ratio is below 20%, the gradual alignment of graphene chains equalizes thermal resistances across pathways, enhancing the uniformity of heat flow and thermal efficiency, which are key factors for the concurrent increase in both thermal conductivity and thermal conductance. Once the stretching ratio exceeds 20%, although thermal conductivity continues to rise, the overall thermal conductance declines across all doping rates. The decline stems from the fracture of some graphene layers, which reduces the effective thermal pathways and consequently leads to a drop in thermal conductance. However, if the elongation of the model has a more pronounced effect on thermal conductivity than the reduction in thermal pathways, the overall thermal conductance may still maintain an upward trend.
Notably, the thermal conductance remains within the range of 0.026 GW/(m2·K) and 0.033 GW/(m2·K) across all models, irrespective of doping or deformation mode. This stability arises because the applied strains remain below the critical threshold for effective medium behavior, thereby preventing significant fluctuations of thermal conductance. It is crucial to note that when the stretching ratio exceeds 20%, the dampening effect of PDMS doping on thermal conductivity diminishes. It results from the stochastic nature of thermal pathways fracturing during stretching, which introduces variable impacts on thermal resistance. Consequentially, models with different PDMS doping rates show increased intercomputation variability in thermal conductance, reflected in larger standard deviations across multiple simulations.
Heat-transfer mechanisms in GF/PDMS composite
VDOS provides insights into the vibration modes of a material at different frequencies, typically visualized as the distribution of acoustic frequency. The phonon density and the thermal conductivity are generally positively correlated. In other words, a higher peak in the VDOS spectrum signifies a larger phonon density, driving enhanced thermal transport.
Heat transfer in GF/PDMS composites occurs via phonons across a wide frequency range from 0 THz to 60 THz, where nearly all phonons act as heat carriers. Figure 6a shows the VDOS spectra of the composites with different doping rates under a 40% compression ratio. This deformation ensures robust connectivity between PDMS and graphene layers, facilitating heat transfer through PDMS molecular structures.

a VDOS plots for composites with varying PDMS doping rates under 50% compression; b VDOS plots for the composite with 5% PDMS doping rate under different compression ratios; c VDOS plots for the composite with 5% PDMS doping rate under different stretching ratios; d Relationship between phonon frequency functions and corresponding thermal conductivity of GF composites with different PDMS-doping rates.
In the HNEMD calculations, an uncompressed model was initially employed to conduct an in-depth analysis of the VDOS and phonon contributions in models with doping rates of 0%, 5%, and 10%. For clarity in the analysis, phonons are categorized into three distinct frequency ranges: the relatively low-frequency (0 ~ 13 THz), the relatively mid-frequency (13 ~ 26 THz), and the relatively high-frequency (26 ~ 60 THz). The integral of the phonon frequency function within specified limits quantifies its contribution to the thermal conductance. Figure 6b depicts the contributions of phonons in different frequency ranges at 300 K, where GF/PDMS composites exhibit a wide range of phonon characteristics. Results are discussed collectively as both Fig. 6a, d correspond to models with identical heights (300 Å). For the pristine GF, conspicuous peaks in the spectral thermal conductivity appear across all frequency domains, indicating that phonons across the entire spectrum drive thermal transport. Nevertheless, in the GF doped with 5% PDMS, the contributions of both low- and high-frequency phonons to the overall thermal conductivity exhibit a diminishing trend. The decline intensifies with higher doping, as PDMS introduces interfacial thermal resistance that impedes heat flux and increases composite structural complexity, enhancing phonon scattering and reducing transport efficiency. The synergistic effects lower the composite’s overall thermal conductivity.
The VDOS analysis provides strong evidence to corroborate this trend. As the doping rate increases, the VDOS curve shifts downward overall, with the most pronounced changes in high- and low-frequency regions. Notably, the contribution of low-frequency phonons decreases continuously, whereas the high-frequency phonons initially experience a slight reduction upon PDMS addition but stabilize above 5% doping. The suppression of low-frequency phonons is identified as the dominant factor contributing to the reduction in thermal conductivity of the composite with the increasing PDMS content. This phenomenon can be elucidated by the fact that, although the changes in high-frequency phonons are more significant in the VDOS spectrum, the low-frequency phonons, owing to their higher group velocity, possess stronger thermal conductivity capabilities. Consequently, the low-frequency phonons play a more pivotal role in the variation of thermal conductivity.
Deformation-induced changes in model dimensions profoundly affect phonon contributions to thermal conductivity in GF/PDMS composites. During compression, reduced internal spacing suppresses high-frequency phonon propagation due to dense packing and increased interatomic forces, which enhance scattering. Conversely, new ligament connections formed under compression create additional thermal pathways for low- and mid-frequency phonons, partially offsetting the heat transport losses. However, interfacial thermal resistances from PDMS limit the overall recovery of thermal conductivity.
During stretching, expanded internal space activates high-frequency phonons, while diminishing the contribution of low-frequency phonons. Reduced interatomic forces decrease phonon scattering, favoring high-frequency transport. Considering the effects of group velocity, increased high-frequency phonons slightly outweigh decreased low-frequency phonons, leading to a modest rise in thermal conductivity. These dimensional-dependent phonon dynamics highlight the interplay between structural geometry and thermal transport in deformable composites.
Under cyclic tensile-compressive deformation, the pristine GF exhibits a thermal conductivity decrease from 1.18 W/(m·K) at 40% tensile strain to 0.49 W/(m·K) at 50% compression. In contrast, the thermal conductivity of the GF doped with 10% PDMS shows a smaller decline from 1.02 W/(m·K) to 0.43 W/(m·K). Across all doping concentrations, the changes in thermal conductivity are modest relative to structure deformations, ranging from 2.38 to 2.55 times. Thermal resistance calculations demonstrate that within the elastic range, the 5% PDMS-doped model undergoes the largest change in thermal resistance, increasing by a factor of 7.13. It highlights its promise for thermal tunable materials. While higher PDMS doping could further improve the mechanical robustness, computational limitations restrict the maximum simulated compression to 50%, lower than the real-world scenarios. This implies that a greater thermal resistance modulation could be achieved in practical applications, reinforcing the utility of GF/PDMS composites for adaptive thermal management.
Discussion
The NEP-based machine-learning framework was employed to investigate the deformation-induced property variation, demonstrating outstanding performance in simulating complex systems at the tens-of-nanometers scale, significantly expanding accessible computational size. Compared with traditional empirical potentials, machine learning potentials offer superior predictive capabilities for extreme structural configurations, enabling accurate characterization of the mechanical and thermodynamic properties of GF/PDMS composites under conditions of stretching and compression.
This research also illustrates the mechanical and thermal behaviors of GF/PDMS composites during deformation, revealing the microscale mechanisms governing this complex material system. PDMS doping reduces thermal conductivity through enhanced phonon scattering and interfacial resistance but provides significant mechanical reinforcement. The mechanism of the unique interaction between GF and PDMS is clarified by VDOS and phonon spectrum analyses. It demonstrates that PDMS introduces interfacial thermal resistances and amplifies the low-frequency phonon scattering, significantly suppressing thermal conduction. The stacking of graphene layers during compression and the disruption of thermal pathways during stretching further modulate thermal transport. The GF with a 5% PDMS doping rate achieves an optimal balance, exhibiting the largest thermal resistance variation 7.13-fold increase within the elastic range and robust structural stability across deformations from 50% compression to 40% stretching. These findings highlight the potential of GF/PDMS composites for adaptive thermal devices, such as tunable thermal switches and sensors, by leveraging the deformation-dependent thermal and mechanical properties.
Methods
Machine learning potential function fitting method
In the NEP, the descriptor is an important parameter composed of a set of radial and angular terms to encode local atomic environments. For atom i, the radial component \({q}_{n}^{i}\) quantifies the cumulative influence of all neighboring atoms within a certain cutoff distance, resulting in \(({n}_{\max }^{R}+1)\) radial descriptor components. And the radial component is expressed in Eq. (1)30.
The angular components are described by the integrated four-body descriptors \({q}_{n{l}_{1}{l}_{2}{l}_{3}}^{i}\) (where \(0\le n\le {n}_{max}^{A}\), and \(1\le {l}_{1}={l}_{2}={l}_{3}\le {l}_{max}^{3b}\)) in Eq. (2), incorporating the three-body descriptors \({q}_{nl}^{i}\) (where \(0\le n\le {n}_{max}^{A}\) and \(1\le l\le {l}_{max}^{3b}\)) as defined in Eq. (3). In the equations, spherical harmonics \({Y}_{lm}({{\theta }}_{ij},{{\phi }}_{ij})\) capture angular dependencies, where \({{\theta }}_{ij}\) (polar) and \({{\phi }}_{ij}\) (azimuthal) define the orientation of atom j relative to i in Eq. (4).
The calculation of loss is minimized through SNES to optimize the free parameters in the NEP model. The total loss function is defined as the weighted sum of several individual losses, as shown in Eq. (5).
Here ΔU, ΔF, and ΔW are the root-mean-square errors (RMSE) between the predicted and reference values of energy, force, and virial, respectively. L1 and L2 are two regularization parameters, while \({\lambda }_{e}\), \({\lambda }_{f}\), \({\lambda }_{v}\), \({\lambda }_{1}\), and\({\lambda }_{2}\) are the weights of each term.
NEP-4 training leverages a hybrid strategy balancing system complexity and computational efficiency. Key hyperparameters (Table 2) are initialized following established NEP frameworks, including radial/angular cutoffs (\({r}_{C}^{R}\)=6 Å, \({r}_{C}^{A}\)=4 Å), radial and angular descriptor dimensions (\({n}_{max}^{R}\)=12, \({n}_{max}^{A}\)=8), number of basis functions for the radial and angular descriptors (\({N}_{bas}^{R}\)=12, \({N}_{bas}^{A}\)=8), training batch size(\({N}_{bat}\)=100), and evolutionary algorithm populations (\({N}_{pop}\)=50)33. The network architecture comprises 40 neurons per hidden layer (\({N}_{neu}\)=40), trained over 1,000,000 steps (\({N}_{gen}\)=1,000,000) to ensure energy, force and virial convergence.
Thermodynamic calculation methods of molecular dynamics
To ensure system stability and minimize the energy of the initial model, it is generally necessary to perform energy minimization and relaxation for over 0.2 ns in GPUMD. The process allows the interaction between GF and PDMS to equilibrate to a natural state, avoiding structural fragmentation or abnormal box deformation during subsequent thermodynamic calculations. Here, the calculation of heat flux and thermal conductivity is conducted using the nonequilibrium molecular dynamics (NEMD) and harmonic nonequilibrium molecular dynamics (HNEMD) methods. The NEMD method relies on Fourier’s Law39,40, as shown in Eq. (6).
where \(J\) represents the heat flux along the z-axis, and \(\nabla T\) denotes the temperature gradient. The heat flux is determined by the rate of energy transfer. If \(E\) is the accumulated energy and \(S\) is the cross-sectional area of the supercell, the heat flux \(J\) can be written as Eq. (7).
Here the derivative term describes the temporal change in accumulated energy per unit area, consistent with Fourier’s law formulation for steady-state heat transport. Before the calculation, the model is initialized at 0 Pa and 300 K to ensure thermal and mechanical equilibrium. To maintain the stability of the box during the pressure-temperature adjustment, two different ensembles are employed for relaxation. The model undergoes a 0.2 ns relaxation in the NPT ensemble, followed by another 0.2 ns relaxation in the NVE ensemble to make the atomic states in the system approximate those in reality. Subsequently, the temperatures at both hot and cold ends are set to 330 K and 270 K, respectively, to establish a 60 K temperature gradient for heat transport simulations. For a finite system, the equivalent thermal conductivity is calculated by Eq. (8).
where \(\Delta T\) denotes the temperature difference across each group with the heat transport length L and \(Q/S\) represents the heat flux using the Langevin method, the model is divided into 13 groups along the z-axis, with each group treated as a heat-transfer unit, and the atomic temperature defined by the average temperature of the group. This approach enables the quantification of energy exchange between the heat source and all layers within the transport regions41. A heat-transfer simulation of 2 ns is conducted to guarantee steady-state thermal transmission, during which each group functioned as an independent thermal unit to characterize spatial temperature profiles and flux distribution.
In the HNEMD method, the non-equilibrium state in large GF/PDMS models is induced via external potential fields or driving forces sustained until the heat transfer reaches equilibrium to characterize thermal properties under compression. A linear relationship exists between the driving force parameter \({{\bf{F}}}_{{\bf{e}}}^{{\boldsymbol{\beta }}}\) and heat flux \({{\bf{J}}}^{{\boldsymbol{\alpha }}}\), as shown in Eq. (9). \(T\) represents the system temperature, \(V\) is the system volume, and \({{\boldsymbol{\kappa }}}^{{\boldsymbol{\alpha }}{\boldsymbol{\beta }}}\) denotes the proportionality constant for the thermal conductivity tensor component \({\boldsymbol{\alpha }}{\boldsymbol{\beta }}\). The heat flux is decomposed as:
By decomposing the heat flux in the frequency domain, the spectral thermal conductivity \({{\boldsymbol{\kappa }}}^{{\boldsymbol{\alpha }}}\left({\boldsymbol{\omega }}\right)\) can be derived from Eq. (10)23.
where \({U}_{j}\) represents the potential energy of atom \(j\) and \({{\bf{r}}}_{{\bf{i}}{\bf{j}}}\) is the distance between two atoms, defined as \({{\bf{r}}}_{{\bf{i}}{\bf{j}}}\equiv {{\bf{r}}}_{{\bf{i}}}-{{\bf{r}}}_{{\bf{j}}}\) where \({{\bf{r}}}_{{\bf{i}}}\) is the position vector of atom \(i\) and \({\boldsymbol{\upsilon }}({\bf{t}})\) is the velocity vector of atoms at time t.
In the HNEMD simulation, a time step of 1 fs is chosen, and the simulation is conducted at a temperature of 300 K and a pressure of 0 Pa. After a series of convergence tests, it is found that the system achieves equilibrium and stability most rapidly when the driving force parameter is set to 0.2 μm−1, which is therefore adopted as the final value. To ensure the system is in a natural state, energy minimization is applied to the initial model, followed by the relaxation for 0.25 ns in NVE and NPT ensembles. For spectral decomposition aimed at obtaining the relationship between phonon frequencies and thermal conductivity, the middle 50 Å of the supercell model is chosen as the analysis region.
The VDOS is obtained through the Fourier transform of the atomic velocity autocorrelation function. Firstly, the atomic velocity autocorrelation function is calculated to characterize the dynamic evolution of atoms over time. Subsequently, the function is transformed into the frequency domain via the Fourier transform, yielding the phonon density of states for the system. The calculation follows the equations presented in Eq. (11)42.
where \(\nu\) represents the phonon frequency and \(\gamma\) denotes the velocity autocorrelation function, which can be calculated based on Eq. (12).
where \({{\boldsymbol{\nu }}}_{{\bf{i}}}({\bf{0}})\) and \({{\boldsymbol{\nu }}}_{{\bf{i}}}({\bf{t}})\) are the velocity vectors of the \(i\) th atom at the initial state and at time \(t\), respectively.
Mechanical characterization methods of molecular dynamics
Mechanical properties of GF/PDMS composites with different PDMS doping rates were analyzed by GPUMD, applying periodic boundary conditions in all three dimensions and conducting tensile and compressive tests along the z-axis. The mechanical simulations began with 0.1 ns of energy minimization, followed by 0.2 ns of relaxation in the NPT ensemble and 0.2 ns in the NVE ensemble to ensure system stability. Subsequently, tensile and compressive deformations were applied along the z-axis at a specific strain rate with temperature controlled via the Bussi-Donadio-Parrinello thermostat43, and the lateral stress regulated by a stochastic cell rescaling barostat44. This setup enables systematic investigation of how PDMS doping (from 0% to 10%) influences the mechanical behavior of the GF/PDMS composite system under uniaxial deformation.
Data availability
The data that support the findings of this study are available from: https://github.com/Intronss/GF-PDMS.
Code availability
The data that support the findings of this study are available from: https://github.com/Intronss/GF-PDMS.
References
Li, N. et al. Three-dimensional graphene foam as a biocompatible and conductive scaffold for neural stem cells. Sci. Rep. 3, 1604 (2013).
Wang, X. B. et al. Three-dimensional strutted graphene grown by substrate-free sugar blowing for high-power-density supercapacitors. Nat. Commun. 4, https://doi.org/10.1038/ncomms3905 (2013).
Yang, Z. X., Chen, H. M., Wang, N. N., Xia, Y. D. & Zhu, Y. Q. Permeability studies on 3D Ni foam/graphene composites. J. Phys. D. Appl. Phys. 50, https://doi.org/10.1088/1361-6463/aa8078 (2017).
Chen, Z. P. et al. Three-dimensional flexible and conductive interconnected graphene networks grown by chemical vapour deposition. Nat. Mater. 10, 424–428 (2011).
Lin, L., Deng, B., Sun, J. Y., Peng, H. L. & Liu, Z. F. Bridging the gap between reality and ideal in chemical vapor deposition growth of graphene. Chem. Rev. 118, 9281–9343 (2018).
Pettes, M. T., Ji, H. X., Ruoff, R. S. & Shi, L. Thermal transport in three-dimensional foam architectures of few-layer graphene and ultrathin graphite. Nano. Lett. 12, 2959–2964 (2012).
Reddy, S. K., Ferry, D. B. & Misra, A. Highly compressible behavior of polymer mediated three-dimensional network of graphene foam. RSC. Adv. 4, 50074–50080 (2014).
Zhang, C., Li, A., Zhao, Y. H., Bai, S. L. & Zhang, Y. F. Thermal, electrical and mechanical properties of graphene foam filled poly (methyl methacrylate) composite prepared by polymerization. Compos. Part B Eng. 135, 201–206 (2018).
Kong, K. T. S., Mariatti, M., Rashid, A. A. & Busfield, J. J. C. Enhanced conductivity behavior of polydimethylsiloxane (PDMS) hybrid composites containing exfoliated graphite nanoplatelets and carbon nanotubes. Compos. Part B Eng. 58, 457–462 (2014).
Owuor, P. S. et al. High Toughness in Ultralow Density Graphene Oxide Foam. Adv. Mater. Interfaces 4, https://doi.org/10.1002/admi.201700030 (2017).
Li, T., Du, J., Xu, M., Song, Z. Y. & Ren, M. F. Lightweight and flexible graphene foam composite with improved damping properties. Nanomaterials 12, https://doi.org/10.3390/nano12081260 (2022).
Xu, R. Q. et al. Facile fabrication of three-dimensional graphene foam/poly(dimethylsiloxane) composites and their potential application as strain sensor. ACS Appl. Mater. Inter. 6, 13455–13460 (2014).
Du, T. T. et al. Wide range continuously tunable and fast thermal switching based on compressible graphene composite foams. Nat. Commun. 12, 4915 (2021).
Anand, S. et al. Dual 3D networks of graphene derivatives based polydimethylsiloxane composites for electrical insulating electronic packaging materials with outstanding electromagnetic interference shielding and thermal dissipation performances. Chem. Eng. J. 462, https://doi.org/10.1016/j.cej.2023.142017 (2023).
Samad, Y. A., Li, Y. Q., Alhassan, S. M. & Liao, K. Novel Graphene foam composite with adjustable sensitivity for sensor applications. ACS Appl Mater. Inter 7, 9195–9202 (2015).
Chan, K. C., Tso, C. Y., Hussain, A. & Chao, C. Y. H. A theoretical model for the effective thermal conductivity of graphene coated metal foams. Appl. Therm. Eng. 161, https://doi.org/10.1016/j.applthermaleng.2019.114112 (2019).
Mandavi, S. M., Adibnazari, S., del Monte, F. & Gutiérrez, M. C. Compressive modulus and deformation mechanisms of 3DG foams: experimental investigation and multiscale modeling. Nanotechnology, 32, https://doi.org/10.1088/1361-6528/ac1a3e (2021).
Pedrielli, A., Taioli, S., Garberoglio, G. & Pugno, N. M. Mechanical and thermal properties of graphene random nanofoams via Molecular Dynamics simulations. Carbon 132, 766–775 (2018).
Khosravani, S., Sadr, M. H., Carrera, E., Pagani, A. & Sanchez-Majano, A. R. Multi-scale analysis of thermoelastic properties of graphene foam/PDMS composites. Comp. Mater. Sci. 216, 111842 (2023).
Fan, Z. Y., Chen, W., Vierimaa, V. & Harju, A. Efficient molecular dynamics simulations with many-body potentials on graphics processing units. Comput. Phys. Commun. 218, 10–16 (2017).
Fan, Z. Y. et al. Neuroevolution machine learning potentials: Combining high accuracy and low cost in atomistic simulations and application to heat transport. Phys. Rev. B. 104, https://doi.org/10.1103/PhysRevB.104.104309 (2021).
Fan, Z. Y., Dong, H. K., Harju, A. & Ala-Nissila, T. Homogeneous nonequilibrium molecular dynamics method for heat transport and spectral decomposition with many-body potentials. Phys. Rev. B 99, https://doi.org/10.1103/PhysRevB.99.064308 (2019).
Dong, H. K. et al. Anisotropic and high thermal conductivity in monolayer quasi-hexagonal fullerene: a comparative study against bulk phase fullerene. Int. J. Heat Mass Tran. 206, https://doi.org/10.1016/j.ijheatmasstransfer.2023.123943 (2023).
Zhang, S. D. et al. Theoretical investigation on the dynamic thermal transport properties of graphene foam by machine-learning molecular dynamics simulations. Int. J. Therm. Sci. 210, https://doi.org/10.1016/j.ijthermalsci.2024.109631 (2025).
Liu, Z. L. et al. Understanding magnesium dissolution through machine learning molecular dynamics. Chem. Eng. J. 516, 163578 (2025).
Hong, H. et al. In situ polymer-solution-processed graphene-PDMS nanocomposites for application in intracranial pressure sensors. Nanomaterials 14, https://doi.org/10.3390/nano14050399 (2024).
Li, B. W. et al. Multilayer graphene/PDMS composite gradient materials for high-efficiency photoresponse actuators. Macromol. Mater. Eng. 307, https://doi.org/10.1002/mame.202100868 (2022).
Behler, J. Perspective: machine learning potentials for atomistic simulations. J. Chem. Phys. 145, 170901 (2016).
Fan, Z. Y. et al. GPUMD: A package for constructing accurate machine-learned potentials and performing highly efficient atomistic simulations. J. Chem. Phys. 157, https://doi.org/10.1063/5.0106617 (2022).
Song, K. K. et al. General-purpose machine-learned potential for 16 elemental metals and their alloys. Nat. Commun. 15, https://doi.org/10.1038/s41467-024-54554-x (2024).
Bartók, A. P. et al. Machine learning unifies the modeling of materials and molecules. Sci. Adv. 3, e1701816 (2017).
Aktulga, H. M., Fogarty, J. C., Pandit, S. A. & Grama, A. Y. Parallel reactive molecular dynamics: Numerical methods and algorithmic techniques. Parallel Comput 38, 245–259 (2012).
Fan, Z. Y. Improving the accuracy of the neuroevolution machine learning potential for multi-component systems. J. Phys-Condens. Mat. 34, https://doi.org/10.1088/1361-648X/ac462b (2022).
Ying, P. H. et al. Atomistic insights into the mechanical anisotropy and fragility of monolayer fullerene networks using quantum mechanical calculations and machine-learning molecular dynamics simulations. Extreme. Mech. Lett. 58, https://doi.org/10.1016/j.eml.2022.101929 (2023).
Zhang, Y. F., Zhao, Y. H., Bai, S. L. & Yuan, X. W. Numerical simulation of thermal conductivity of graphene filled polymer composites. Compos. Part B Eng. 106, 324–331 (2016).
Zhao, Y. H., Wu, Z. K. & Bai, S. L. Study on thermal properties of graphene foam/graphene sheets filled polymer composites. Compos. Part A 72, 200–206 (2015).
Morell, E. S., Correa, J. D., Vargas, P., Pacheco, M. & Barticevic, Z. Flat bands in slightly twisted bilayer graphene: tight-binding calculations. Phys. Rev. B 82, https://doi.org/10.1103/PhysRevB.82.121407 (2010).
Xiong, Z. X., Marconnet, A. & Ruan, X. L. Unconventional and dynamically anisotropic thermal conductivity in compressed flexible graphene foams. ACS Appl. Mater. Inter. 14, https://doi.org/10.1021/acsami.2c10880 (2022).
Ikeshoji, T. & Hafskjold, B. Nonequilibrium molecular-dynamics calculation of heat-conduction in liquid and through liquid-gas interface. Mol. Phys. 81, 251–261 (1994).
Jund, P. & Jullien, R. Molecular-dynamics calculation of the thermal conductivity of vitreous silica. Phys. Rev. B 59, 13707–13711 (1999).
Li, Z. et al. Influence of thermostatting on nonequilibrium molecular dynamics simulations of heat conduction in solids. J. Chem. Phys. 151, https://doi.org/10.1063/1.5132543 (2019).
Schmitz, G., Yönder, Ö, Schnieder, B., Schmid, R. & Hättig, C. An automatized workflow from molecular dynamic simulation to quantum chemical methods to identify elementary reactions and compute reaction constants. J. Comput. Chem. 42, 2264–2282 (2021).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, https://doi.org/10.1063/1.2408420 (2007).
Bernetti, M. & Bussi, G. Pressure control using stochastic cell rescaling. J. Chem. Phys. 153, https://doi.org/10.1063/5.0020514 (2020).
Zhao, Y. H., Zhang, Y. F., Bai, S. L. & Yuan, X. W. Carbon fibre/graphene foam/polymer composites with enhanced mechanical and thermal properties. Compos. Part B Eng. 94, 102–108 (2016).
Acknowledgements
This work was supported by the Shenzhen Science and Technology Program (JCYJ20220530141009020); the Taishan Industry Leading Talents Program (tscx202312007); National Natural Science Foundation of China (52372007); Natural Science Foundation of Shandong Province (ZR2023ME125). This work was also supported by AI TENNessee Initiative Seed Funds and Center for Materials Processing University-Industry Partnership Seed Grant at University of Tennessee Knoxville.
Author information
Authors and Affiliations
Contributions
T.D. conceptualized and designed the research. P.Z. and S.Z. conducted the simulations. P.Z., S.Z., Y.Q. and L.W. analyzed the data and interpreted the results. P.Z., S.Z., T.D. and X.L. jointly wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
T.D., P.Z., L.W. and S.Z. are the inventors of a provisional patent application with CN 2024112847845 filed on Sep. 13, 2024.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, P., Zhang, S., Qin, Y. et al. Machine learning-driven molecular dynamics decodes thermal tuning in graphene foam composites. npj Comput Mater 11, 214 (2025). https://doi.org/10.1038/s41524-025-01710-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41524-025-01710-6
