
Abstract
Foundational machine learning interatomic potentials that can accurately and efficiently model a vast range of materials are critical for accelerating atomistic discovery. We introduce universal potentials based on the graph atomic cluster expansion (GRACE) framework, trained on several of the largest available materials datasets. Through comprehensive benchmarks, we demonstrate that the GRACE models establish a new Pareto front for accuracy versus efficiency among foundational interatomic potentials. We further showcase their exceptional versatility by adapting them to specialized tasks and simpler architectures via fine-tuning and knowledge distillation, achieving high accuracy while preventing catastrophic forgetting. This work establishes GRACE as a robust and adaptable foundation for the next generation of atomistic modeling, enabling high-fidelity simulations across the periodic table.
Similar content being viewed by others
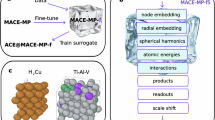

Data availability
Training datasets (MPTrj, sAlex and OMat24) are publicly available. GRACE foundational potentials are available at https://gracemaker.readthedocs.io/en/latest/gracemaker/foundation.
Code availability
Code for GRACE potential is available at github.com/ICAMS/grace-tensorpotential.
References
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Bartók, A. P., Payne, M. C., Kondor, R. & Csányi, G. Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons. Phys. Rev. Lett. 104, 136403 (2010).
Shapeev, A. V. Moment tensory potentials: a class of systematically improvable interatomic potentials. Multiscale Model. Simul. 14, 1153 (2016).
Drautz, R. Atomic cluster expansion for accurate and transferable interatomic potentials. Phys. Rev. B 99, 014104 (2019).
Unke, O. T. et al. Machine learning force fields. Chem. Rev. 121, 10142–10186 (2021).
Musaelian, A. et al. Learning local equivariant representations for large-scale atomistic dynamics. Nat. Commun. 14, 579 (2023).
Jacobs, R. et al. A practical guide to machine learning interatomic potentials–status and future. Curr. Opin. Solid State Mater. Sci. 35, 101214 (2025).
Kulichenko, M. et al. Data generation for machine learning interatomic potentials and beyond. Chem. Rev. 124, 13681–13714 (2024).
Wang, G. et al. Machine learning interatomic potential: bridge the gap between small-scale models and realistic device-scale simulations. iScience 27, 109673 (2024).
Thiemann, F. L., O’Neill, N., Kapil, V., Michaelides, A. & Schran, C. Introduction to machine learning potentials for atomistic simulations. J. Phys. Condens. Matter 37, 073002 (2024).
Yuan, E. C. Y. et al. Foundation models for atomistic simulation of chemistry and materials https://arxiv.org/abs/2503.10538 (2025).
Pettifor, D. G. Theory of the crystal structures of transition metals. J. Phys. C Solid State Phys. 3, 367 (1970).
Pettifor, D. G. The structures of binary compounds. I. Phenomenological structure maps. J. Phys. C Solid State Phys. 19, 285 (1986).
Pettifor, D. G. Bonding and Structure in Molecules and Solids (Oxford University Press, 1995).
Seiser, B., Drautz, R. & Pettifor, D. TCP phase predictions in Ni-based superalloys: structure maps revisited. Acta Materialia 59, 749–763 (2011).
Bialon, A. F., Hammerschmidt, T. & Drautz, R. Three-parameter crystal-structure prediction for sp-d-valent compounds. Chem. Mater. 28, 2550–2556 (2016).
Faber, F., Lindmaa, A., von Lilienfeld, O. A. & Armiento, R. Crystal structure representations for machine learning models of formation energies. Int. J. Quantum Chem. 115, 1094–1101 (2015).
Parsaeifard, B. et al. An assessment of the structural resolution of various fingerprints commonly used in machine learning. Mach. Learn. Sci. Technol. 2, 015018 (2021).
Lopanitsyna, N., Fraux, G., Springer, M. A., De, S. & Ceriotti, M. Modeling high-entropy transition metal alloys with alchemical compression. Phys. Rev. Mater. 7, 045802 (2023).
Cerqueira, T. F. T., Wang, H., Botti, S. & Marques, M. A. L. A non-orthogonal representation of the chemical space https://arxiv.org/abs/2406.19761 (2025).
Rappé, A. K., Casewit, C. J., Colwell, K., Goddard III, W. A. & Skiff, W. M. Uff, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 114, 10024–10035 (1992).
Chen, C. & Ong, S. P. A universal graph deep learning interatomic potential for the periodic table. Nat. Comput. Sci. 2, 718–728 (2022).
Deng, B. et al. Chgnet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nat. Mach. Intell. 5, 1031–1041 (2023).
Batatia, I. et al. A foundation model for atomistic materials chemistry. J. Chem. Phys. 163, 184110 (2025).
Barroso-Luque, L. et al. Open materials 2024 (omat24) inorganic materials dataset and models. arXiv preprint arXiv:2410.12771 (2024).
Yang, H. et al. Mattersim: A deep learning atomistic model across elements, temperatures and pressures. arXiv preprint arXiv:2405.04967 (2024).
Kim, J. et al. Data-efficient multifidelity training for high-fidelity machine learning interatomic potentials. J. Am. Chem. Soc. 147, 1042–1054 (2024).
Yin, B. et al. AlphaNet: scaling up local-frame-based neural network interatomic potentials. npj Comput Mater 11, 332 (2020).
Zhang, D. et al. Graph neural network model for the era of large atomistic models. arXiv preprint arXiv:2506.01686 (2025).
Fu, X. et al. Learning smooth and expressive interatomic potentials for physical property prediction. arXiv preprint arXiv:2502.12147 (2025).
Mazitov, A. et al. PET-MAD, a universal interatomic potential for advanced materials modeling. Nat. Commun. 16, 10653 (2025).
Liang, T. et al. Nep89: Universal neuroevolution potential for inorganic and organic materials across 89 elements. arXiv preprint arXiv:2504.21286 (2025).
Gasteiger, J., Groß, J. & Günnemann, S. Directional message passing for molecular graphs 2003.03123 (2020).
Anderson, B., Hy, T.-S. & Kondor, R. Cormorant: Covariant molecular neural networks. In Wallach, H. et al. (eds.) Advances in Neural Information Processing Systems 32, Neural Information Processing Systems Conference, 9596 (Neural Information Processing Systems Foundation, Inc., 2019).
Lubbers, N., Smith, J. S. & Barros, K. Hierarchical modeling of molecular energies using a deep neural network. J. Chem. Phys. 148, 241715 (2018).
Thomas, N. et al. Tensor field networks: Rotation-and translation-equivariant neural networks for 3d point clouds. arXiv preprint arXiv:1802.08219 (2018).
Batzner, S. et al. E (3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 13, 2453 (2022).
Satorras, V. G., Hoogeboom, E. & Welling, M. E (n) equivariant graph neural networks. In International Conference on Machine Learning, 9323–9332 (PMLR, 2021).
Unke, O. T. & Meuwly, M. Physnet: a neural network for predicting energies, forces, dipole moments, and partial charges. J. Chem. Theory Comput. 15, 3678–3693 (2019).
Schütt, K. et al. Schnet: a continuous-filter convolutional neural network for modeling quantum interactions. Adv. Neural Inf. Process. Syst. 30 (2017).
Haghighatlari, M. et al. Newtonnet: a newtonian message passing network for deep learning of interatomic potentials and forces. Digit. Discov. 1, 333–343 (2022).
Schütt, K. T., Unke, O. T. & Gastegger, M. Equivariant message passing for the prediction of tensorial properties and molecular spectra 2102.03150 (PMLR, 2021)
Gasteiger, J., Becker, F. & Günnemann, S. Gemnet: universal directional graph neural networks for molecules 2106.08903 (NIPS, 2022).
Chmiela, S. et al. Machine learning of accurate energy-conserving molecular force fields. Sci. Adv. 3, e1603015 (2017).
Pozdnyakov, S. & Ceriotti, M. Smooth, exact rotational symmetrization for deep learning on point clouds. Adv. Neural Inf. Process. Syst. 36, 79469–79501 (2023).
Nigam, J., Pozdnyakov, S., Fraux, G. & Ceriotti, M. Unified theory of atom-centered representations and message-passing machine-learning schemes. J. Chem. Phys. 156, 204115 (2022).
Batatia, I. et al. The design space of e (3)-equivariant atom-centred interatomic potentials. Nat. Mach. Intell. 7, 56–67 (2025).
Bochkarev, A., Lysogorskiy, Y., Ortner, C., Csányi, G. & Drautz, R. Multilayer atomic cluster expansion for semilocal interactions. Phys. Rev. Res. 4, L042019 (2022).
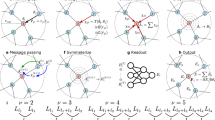
Bochkarev, A., Lysogorskiy, Y. & Drautz, R. Graph atomic cluster expansion for semilocal interactions beyond equivariant message passing. Phys. Rev. X 14, 021036 (2024).
Batatia, I., Kovacs, D. P., Simm, G., Ortner, C. & Csányi, G. Mace: higher order equivariant message passing neural networks for fast and accurate force fields. Adv. neural Inf. Process. Syst. 35, 11423–11436 (2022).
Jain, A. et al. The materials project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Schmidt, J. et al. Machine-learning-assisted determination of the global zero-temperature phase diagram of materials. Adv. Mater. 35, 2210788 (2023).
Wang, H.-C., Schmidt, J., Marques, M. A., Wirtz, L. & Romero, A. H. Symmetry-based computational search for novel binary and ternary 2D materials. 2D Mater. 10, 035007 (2023).
Barroso-Luque, L. et al. Open materials 2024 (omat24) inorganic materials dataset and models https://arxiv.org/abs/2410.12771 (2024).
Kirklin, S. et al. The open quantum materials database (OQMD): assessing the accuracy of dft formation energies. npj Comput. Mater. 1, 1–15 (2015).
Curtarolo, S. et al. Aflowlib. org: a distributed materials properties repository from high-throughput ab initio calculations. Comput. Mater. Sci. 58, 227–235 (2012).
Kaplan, A. D. et al. A foundational potential energy surface dataset for materials. arXiv preprint arXiv:2503.04070 (2025).
Kuner, M. C., Kaplan, A. D., Persson, K. A., Asta, M. & Chrzan, D. C. An r2SCAN dataset for universal machine learninginteratomic potentials. npj Comput. Mater. 11, 352 (2025).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Wang, H.-C., Botti, S. & Marques, M. A. Predicting stable crystalline compounds using chemical similarity. npj Comput. Mater. 7, 12 (2021).
Riebesell, J. et al. A framework to evaluate machine learning crystal stability predictions. Nat. Mach. Intell. 7, 836–847 (2025).
Póta, B., Ahlawat, P., Csányi, G. & Simoncelli, M. Thermal conductivity predictions with foundation atomistic models. arXiv preprint arXiv:2408.00755 (2024).
Thompson, A. P. et al. Lammps-a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Computer Phys. Commun. 271, 108171 (2022).
Togo, A., Chaput, L. & Tanaka, I. Distributions of phonon lifetimes in brillouin zones. Phys. Rev. B 91, 094306 (2015).
Togo, A., Chaput, L., Tadano, T. & Tanaka, I. Implementation strategies in phonopy and phono3py. J. Phys. Condens. Matter 35, 353001 (2023).
Kraß, H., Huang, J. & Moosavi, S. M. Mofsimbench: Evaluating universal machine learning interatomic potentials in metal-organic framework molecular modeling https://arxiv.org/abs/2507.11806 (2025).
De Jong, M. et al. Charting the complete elastic properties of inorganic crystalline compounds. Sci. data 2, 1–13 (2015).
Zheng, H. et al. Grain boundary properties of elemental metals. Acta Materialia 186, 40–49 (2020).
Tran, R. et al. Surface energies of elemental crystals. Sci. Data 3, 1–13 (2016).
Ma, P.-W. & Dudarev, S. Universality of point defect structure in body-centered cubic metals. Phys. Rev. Mater. 3, 013605 (2019).
Ma, P.-W. & Dudarev, S. Nonuniversal structure of point defects in face-centered cubic metals. Phys. Rev. Mater. 5, 013601 (2021).
Lam, S. T., Li, Q.-J., Ballinger, R., Forsberg, C. & Li, J. Modeling lif and flibe molten salts with robust neural network interatomic potential. ACS Appl. Mater. Interfaces 13, 24582–24592 (2021).
Rodriguez, A., Lam, S. & Hu, M. Thermodynamic and transport properties of lif and flibe molten salts with deep learning potentials. ACS Appl. Mater. Interfaces 13, 55367–55379 (2021).
Larsen, A. H. et al. The atomic simulation environment–a python library for working with atoms. J. Phys. Condens. Matter 29, 273002 (2017).
Menon, S. et al. From electrons to phase diagrams with machine learning potentials using pyiron based automated workflows. npj Comput. Mater. 10, 261 (2024).
Guan, X. et al. A benchmark dataset for hydrogen combustion. Sci. Data 9, 215 (2022).
Lysogorskiy, Y. et al. Performant implementation of the atomic cluster expansion. Npj Comput. Mater. 7, 97 (2021).
Bochkarev, A. et al. Efficient parametrization of the atomic cluster expansion. Phys. Rev. Mater. 6, 013804 (2022).
Peng, A. et al. LAMbench: A benchmark for large atomic models. npj Comput. Mater. 12, 62 (2026).
AI Squared. OpenLAM Benchmark Introduction. https://www.aissquare.com/openlam?tab=Benchmark&type=Introduction Accessed: 2025-07-02 (2024).
Morrow, J. D. & Deringer, V. L. Indirect learning and physically guided validation of interatomic potential models. J. Chem. Phys. 157, 104105 (2022).
Gardner, J. L. et al. Distillation of atomistic foundation models across architectures and chemical domains. arXiv preprint arXiv:2506.10956 (2025).
Mazitov, A. et al. Surface segregation in high-entropy alloys from alchemical machine learning. J. Phys. Mater. 7, 025007 (2024).
Abadi, M. et al. {TensorFlow}: a system for {Large-Scale} machine learning. In 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI 16), 265–283 (OSDI, 2016).
Kingma, D. P. & Ba, J. Adam: A method for stochastic optimization. arXiv preprint arXiv:1412.6980 (2014).
Golesorkhtabar, R., Pavone, P., Spitaler, J., Puschnig, P. & Draxl, C. Elastic: a tool for calculating second-order elastic constants from first principles. Comp. Phys. Commun. 184, 1861–1873 (2013).
Acknowledgements
The authors acknowledge funding from the Deutsche Forschungsgemeinschaft (DFG) through the CRC1394 “Structural and Chemical Atomic Complexity – From Defect Phase Diagrams to Material Properties”, project ID 409476157. The authors gratefully acknowledge the computing time made available to them on the high-performance computer Noctua2 at the NHR Center Paderborn Center for Parallel Computing (PC2). This center is jointly supported by the Federal Ministry of Research, Technology and Space and the state governments participating in the National High-Performance Computing (NHR) joint funding program (www.nhr-verein.de/en/our-partners). Calculations (or parts of them) for this publication were performed on the HPC cluster Elysium of the Ruhr University Bochum, subsidised by the DFG (INST 213/1055-1).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization and Project Administration: All authors. Y.L. and A.B. developed the software and parameterized the models. Writing - original draft: Y.L. Writing-review and editing: All authors. Resources and funding acquisition: Y.L. and R.D.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lysogorskiy, Y., Bochkarev, A. & Drautz, R. Graph atomic cluster expansion for foundational machine learning interatomic potentials. npj Comput Mater (2026). https://doi.org/10.1038/s41524-026-01979-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-026-01979-1
