
Abstract
Inherited retinal degenerations are blinding genetic disorders characterized by high genetic and phenotypic heterogeneity. In this retrospective study, we describe sixteen families with early-onset non-syndromic retinal degenerations in which affected probands carried rare bi-allelic variants in CFAP410, a ciliary gene previously associated with recessive Jeune syndrome. We detected twelve variants, eight of which were novel, including c.373+91A>G, which led to aberrant splicing. To our knowledge this is the first likely pathogenic deep-intronic variant identified in this gene. Analysis of all reported and novel CFAP410 variants revealed no clear correlation between the severity of the CFAP410-associated phenotypes and the identified causal variants. This is supported by the fact that the frequently encountered missense variant p.(Arg73Pro), often found in syndromic cases, was also associated with non-syndromic retinal degeneration. This study expands the current knowledge of CFAP410-associated ciliopathy by enriching its mutational landscape and supports its association with non-syndromic retinal degeneration.
Similar content being viewed by others

Introduction
Inherited retinal degenerations (IRDs) are a group of genetically and clinically heterogeneous disorders characterized by progressive loss of cone and rod photoreceptors. IRDs can manifest as an isolated phenotype, where only retina is affected (i.e., non-syndromic IRDs) or as a syndromic disease, where retinal degeneration is one of many signs of a multiorgan clinical manifestation. IRDs can be classified based on their onset (early vs late) and/or photoreceptor degeneration patterns (cone dystrophy (CD), cone-rod dystrophy (CRD), and rod-cone dystrophy (RCD)1.
The Cilia and Flagella Associated Protein 410 (CFAP410) gene (OMIM 603191), formerly known as C21orf2, is a ciliary gene of unclear specific function. Given its mapping position on chromosome 21, CFAP410 was initially thought to play a role in the pathogenesis of several genetic diseases including Trisomy 21 (Down syndrome), but none of these associations have been confirmed2,3,4.
Functional genomic screens for ciliary gene identification5,6 combined with mutational screening in unsolved ciliopathy patients confirmed the essential role of the CFAP410 protein in ciliogenesis. Individuals with bi-allelic pathogenic variants in this gene were reported to have Jeune syndrome (JS)6, a recessive skeletal ciliopathy (OMIM# 611263)7,8 also known as asphyxiating thoracic dystrophy and axial spondylometaphyseal dysplasia (SMDAX)9. Affected individuals usually present with shortened ribs and a narrowed chest accompanied by other skeletal abnormalities, but retinal degeneration and other non-skeletal features can be also present8.
Many ciliopathy cases harboring pathogenic CFAP410 variants have been described to date6,9,10,11,12,13,14,15,16,17. However, in 2015, Khan and colleagues described a specific phenotype of early-onset retinal dystrophy with macular staphyloma but without high myopia in three Saudi families with a history of consanguinity and carrying homozygous variants in CFAP41018. Since then, a few other non-syndromic CFAP410 cases have been reported as a consequence of mutational screens in large IRD cohorts10,11,12,13,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35. However, a conclusive association of CFAP410 mutations with non-syndromic IRD has never been reached due to the small number of non-syndromic cases. In this study, we describe fourteen new families with early-onset non-syndromic retinal degeneration and two additional cases with a milder form of JS that confirm the phenotype expansion for bi-allelic variants in CFAP410. We also report eight novel variants in this gene, six of which are pathogenic or likely pathogenic.
Results
Clinical phenotypes
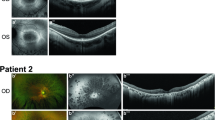
Sixteen probands (seven females and nine males) with CFAP410-associated disease had clinical phenotypes falling into four diagnostic categories: early-onset retinal dystrophy (eoRD; n = 1), cone dystrophy (CD; n = 1), cone-rod dystrophy (CRD; n = 6), and rod-cone dystrophy (RCD; n = 8) (see Fig. 1, Table 1, and Supplementary Table 1 for detailed clinical data). In most cases, the symptom onset occurred in childhood, prior to the age of 10, and at the initial clinical evaluation, the individuals were 9–71 years of age. The presenting symptom typically corresponded to the clinical diagnosis (for example, nyctalopia in RCD).

Images show fundus photos for a representative subset of individuals. Fundus autofluorescence and/or OCT imaging were available for five individuals (5, 8, 13, 14, and 16) and showed features consistent with the fundus findings and clinical diagnosis. The specific IRD phenotype of each patient is given in brackets (CD cone dystrophy, CRD cone-rod dystrophy, RCD rod-cone dystrophy, eoRD early-onset retinal dystrophy). Note the tapetal-like sheen in fundus images in proband 5 with CRD, and the morning glory disc in the left eye of proband 16 with eoRD.
Visual acuity was significantly reduced at young ages regardless of clinical diagnosis. The youngest proband with CRD (proband 5) had visual acuity of 20/100 and 20/125 when evaluated at age 9, and the youngest proband with RCD (proband 9) had visual acuity of 20/100 in each eye at age 12. Except probands of families 10 and 11, no individual in the cohort had visual acuity better than 20/80 (see Supplementary Table 1), and fourteen eyes of eight individuals had visual acuity at or beyond the threshold of legal blindness at the initial evaluation.
When available, visual field data from Goldmann kinetic perimetry showed better overall preservation of visual fields in patients with clinical diagnoses of CD/CRD whereas most with RCD had constriction sparing only the central visual fields. Full-field ERGs were available for all patients. Individuals with clinical diagnoses of CD and CRD showed varying degrees of scotopic compromise with more severe photopic dysfunction; the scotopic responses for proband 2 did show deterioration over two studies spanning 12 years. Individuals with RCD had severe generalized dysfunction of scotopic and photopic responses.
Fundus evaluation showed features that were typical for the retinal diagnosis (Fig. 1). Staphylomas were noted in two individuals (probands 4, 8). Digital OCT images were available for eight individuals and showed significant attenuation and absence of photoreceptor bands, particularly in the peripheral macula with relatively better preservation of foveal lamination. Visual acuity was lower than might be expected from the remaining structure with the structure vs. function dissociations in probands 5, 13 (OS), and 16 particularly illustrative of this observation. OCT suggested posterior staphyloma in one individual for whom it was not noted on clinical exam (proband 13).
Additional ophthalmic diagnoses included amblyopia (proband 13), bilateral pseudophakia (probands 11 and 12), history of strabismus surgery (proband 14), and nystagmus (probands 2, 16).
Skeletal abnormalities were present in two individuals: proband 14 had thoracic skeletal abnormalities requiring surgical intervention, and proband 16 had bilateral hip dysplasia corrected with hip replacement. No other individuals had skeletal abnormalities present on imaging (proband 3) or self-report. Proband 16 also had premature ovarian failure at age 30 as well as bilateral sensorineural hearing loss beginning in her 40 s, but no other systemic diagnoses of note were present in the cohort.
Rare CFAP410 variants associated with non-syndromic early-onset IRD
By analyzing data from either targeted next generation sequencing, exome sequencing (ES), or genome sequencing (GS) of a cohort of 7000 IRD cases, we identified 12 rare CFAP410 variants (V1-12, MAF < 0.0006) in 16 probands and their family members (see Fig. 2 and Table 2). No additional disease-causing variants were present in any of the currently known IRD genes36 that were able to explain the clinical phenotype.

For each family (1–16), the specific IRD phenotype diagnosed is mentioned above each pedigree (CD cone dystrophy, CRD cone-rod dystrophy, RCD rod-cone dystrophy, eoRD early-onset retinal dystrophy). Mildly syndromic families 14 and 16 are indicated with a hashtag (#). Affected male and female subjects are represented with black squares or circles, respectively. Probands are indicated by a black arrow. Novel variants are indicated in bold. First cousin marriage is indicated by a double-line. All presented variants refer to the CFAP410 transcript NM_004928.3. Bi-allelic inheritance was confirmed by familial segregation analysis (families 5 and 10), by ruling out deletion events in CFAP410 bioinformatically (families 1, 2, 6, 9, 11, 12, 13, 14, 15, 16), by analysis of NGS pair-end reads (family 8), and by cloning and by using the gnomAD v2 Variant Co-Occurrence tool (families 4 and 7). In family 3 we could not confirm bi-allelic inheritance, thus variants are indicated as [V(;)V].
The coding variants detected were truncating (p.Gln119* and p.Glu148Alafs*13), missense (p.Cys25Arg, p.Arg73Pro, p.Glu96Lys, p.Asn97Lys, p.Pro116Leu), or leading to single amino acid deletions (p.Met7del and p.Glu130del), while the non-coding variants c.96+1G>A, c.143+3A>T and c.373+91A>G were located in CFAP410 intron 2, 3 and 4, respectively.
Most of the detected variants were novel, except for c.96+1G>A20,30, p.Arg73Pro6,9,14,20,22,24,25,26,27,29,31,32,33,37, p.Glu96Lys11,24, and p.Pro116Leu9,28 which were reported in patients with syndromic and non-syndromic IRD (See Supplementary Tables 2 and 3). The p.Arg73Pro was the most commonly reported variant and also the most common in our cohort: present homozygously in eight probands and heterozygously in two (families 4 and 8, see Fig. 2). However, this variant remains extremely rare in the general population, given the allele frequency in Genome Aggregation Database (gnomAD) v4 of 0.000502338. Consanguinity was reported only in families 2 (c.218G>C, p.Arg73Pro) and 15 (c.143+3A>T), in which the parents were third and first cousins, respectively. An additional proband 16 was homozygous for the c.355C>T, p.(Gln119*) variant, though no consanguinity was noted.
Bi-allelic inheritance in the homozygous cases was confirmed by familial segregation analysis (family 10) or by ruling out deletion events in CFAP410 bioinformatically. Compound heterozygosity was confirmed by familial segregation analysis (family 5); analysis of NGS pair-end reads (family 8), by cloning and by using the gnomAD v2 Variant Co-Occurrence tool (https://gnomad.broadinstitute.org/variant-cooccurrence) (families 4 and 7) (see Supplementary Figs. 1 and 2). Unfortunately, we could not use these methods to validate the phase of the variants identified in family 3, the c.73T>C; p.(Cys25Arg) and the c.373+91A>G. Both alleles were absent from gnomAD v2 and they were too far apart (~6 kb) to be cloned in one single fragment, given the limited quality of the historical DNA samples available. Only variant c.73T>C; p.(Cys25Arg) was present in one individual in the recently released version of GnomAD v4, while variant c.373+91A>G was absent (see Table 2). However this data is too scarce to conclude definitively if these two variants are likely in cis or trans.
Novel non-coding CFAP410 variants lead to splicing defects
To investigate the effect of c.143+3A>T and c.373+91A>G on pre-mRNA splicing we generated wild-type and variant mini-gene splicing constructs, which were transfected into HEK293T cells. The effect on splicing was investigated by RT-PCR. Both variants were predicted to affect normal splicing according to multiple in silico tools, such as SpliceSiteFinder-like39, MaxEntScan40, NNSPLICE41, GeneSplicer42, Human Splicing Finder43, and SpliceAI44. Variant c.143+3A>T was predicted to disrupt the splice donor site of CFAP410 exon 3, while c.373+91A>G was predicted to generate a strong splice acceptor site in intron 4 (see Supplementary Figs. 3 and 4).
The splicing assay confirmed the presence of aberrant splicing phenotypes for both variants (see Fig. 3 and Supplementary Fig. 5). Indeed, exon 3 was skipped in the construct carrying the c.143+3A>T, while the splice acceptor created by c.373+91A>G resulted in the inclusion of a 200- base pair pseudoexon, previously predicted by SpliceAI (see Supplementary Fig. 4), in at least half of the transcripts according to our splicing assay (see Fig. 3A). Both splicing defects were classified as severe and fully penetrant, as they caused frameshift and premature stop codon in all generated transcripts (see Fig. 3).

A RT-PCR showing the formation of a pseudoexon in intron 4 (pe4) in the construct containing the CFAP410 c.373+91A>G variant compared to the wild-type (WT) band generated by the reference construct. RT-PCR reaction was performed using as input either retrotrascribed (RT+) or not retrotrascribed (RT-) RNA samples. NC, negative control. B Sanger sequencing of the splice boundaries between exon 4 and 5, confirming the breakpoint of the pseudoexon. C RT-PCR showing the skipping of exon 3 (Δ3) in the construct containing the c.143+3A>T compared to the wild-type (WT) construct, which generates both a full (e3) and truncated (e3*) version of exon 3, according to the splicing prediction D.
Protein modeling and genotype-phenotype correlation analysis
Variants in CFAP410 have been associated with both syndromic (i.e., skeletal ciliopathies) and non-syndromic forms of retinal degeneration. To investigate whether this phenotypic variability was the consequence of a specific variant localization, we plotted the known 42 CFAP410 variants reported in literature and the eight novel variants detected in our probands onto the secondary structure of the human CFAP410, a 256 amino acid protein (UniProtKB ID: O43822) (see Fig. 4 and Supplementary Table 3)6,9,10,11,12,13,14,15,16,18,19,24,25,29,30,31,32,34,45. Half of the 50 analyzed CFAP410 variants were missense, while the other half were either truncating or non-coding variants. Most of the variants were located in the N-terminal half of the protein, up to the amino acid residue 130, containing three lucine-rich repeat domains (LRR) and a leucine-rich repeat C-terminal domain (LRRCT) (see Fig. 4). The mutation tolerance at CFAP410 protein residues was analyzed using MetaDome (https://stuart.radboudumc.nl/metadome/)46, while the impact of specific missense variants on CFAP410 structure and function was predicted by tools like SIFT47, PolyPhen248, CADD Phred49 REVEL50, and EVE51. Our analyses did not reveal variants in specific regions of the protein that would explain the observed phenotypic difference between syndromic and non-syndromic cases (see Fig. 4). Nine of the CFAP410 variants detected in syndromic cases were also found in non-syndromic cases, while six were exclusive (see Fig. 4). These were: (1) p.Leu161Serfs*9, detected in one family with severe skeletal abnormalities consistent with JS6; (2) c.643-23A>T, detected homozygously in five pedigrees with JS12, SMDAX9, or other forms of skeletal dysplasia15; (3) p.Gln119* found in the mildly syndromic proband of family 16; (4) c.77+1G>C found in a severe syndromic IRD case (LOVD data and personal communication); (5) p.Thr114_Arg117dup, detected homozygously in a milder17 syndromic IRD patient; and (6) p.His211Glnfs*98 found in one case with SMDAX and CRD11 (see Fig. 4 and Supplementary Table 2).

CFAP410 secondary structure and distribution of known and novel disease variants found in affected individuals. Prediction model of the mutation tolerance landscape of the CFAP410 protein was retrieved from MetaDome webpage. Protein motifs and catalytic domain are highlighted using different colors, while variants were divided in two groups, depending whether they were found in syndromic or non-syndromic IRD patients. Known variants were retrieved from the Leiden Open Variation Database (LOVD). Variants reported in this study are in bold and novel variants are further highlighted in red. Variants p.S183* and p.A181Qfs*6, in square brackets, are part of the same complex allele as they result from the same nucleotide variant. # variants were found in mild syndromic cases. LRR, leucine-rich repeat; LRRCT, LRR C-terminal domain.
After plotting genotypes of 95 cases carrying bi-allelic CFAP410 variants, including all reported and our probands (see Fig. 5), we did not observe a clear correlation between the severity of the phyenotype (syndromic vs non-syndromic) and the variants. Most of the cases in both cohorts were either bi-allelic for the loss of function variants or were homozygous for the p.Arg73Pro variant. Other missense variants, mostly affecting the LRR and LRRCT domains were also present in syndromic and non-syndromic IRD cases (see Fig. 5). There was also no apparent correlation between a specific retinal phenotype and CFAP410 variants (see Supplementary Table 2).

For each patient, variants on alleles 1 and 2, represented as protein changes, were plotted on x and y axes. Predicted loss-of-function variants were represented as null (zero). Two larger clusters were found for cases homozygous for null alleles and for the p.Arg73Pro change, while a minor cluster included variants located in the LLRCT domain, in particular p.Tyr107His and p.Thr114_Arg117dup. Syndromic phenotype was presented in red and non-syndromic phenotype was presented in gray.
Adopting the American College of Medical Genetics (ACMG) guidelines52, ten of the identified CFAP410 variants were classified as pathogenic/likely pathogenic while p.Met7del and p.Asn97Lys were classified as variants of uncertain significance (VUSs) (Table 2).
Discussion
In this retrospective study, we describe sixteen probands with retinal degeneration associated with rare bi-allelic variants in CFAP410, a gene initially associated with recessive skeletal ciliopathies like JS and SMDAX. Fourteen probands in our cohort did not have any syndromic features, and two individuals were recognized to have systemic findings related to CFAP410 variants, noted only after genetic testing was performed. Other bi-alleleic CFAP410 cases were described in the literature with non-syndromic or mildly syndromic IRDs10,11,12,13,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35. Our study thus further supports the association of variants in CFAP410 with non-syndromic IRDs first described by Khan and colleagues18 and considerably increases the number of non-syndromic cases.
This report also expands upon prior reports of CFAP410-associated retinopathy, as cases presented here exhibited a spectrum of clinical diagnoses with CRD and RCD equally represented. Both patient-reported symptoms and assessments of retinal function segregated into these different diagnostic categories and supported the differing ways in which CFAP410 dysfunction can manifest. A notable feature, regardless of clinical diagnosis, was the early disease onset, with symptoms beginning prior to the age of 10 years in those for whom a specific age could be recalled. Two-thirds of the 36 patients described by Shinbashi et al. had symptom onset before age 1853. An additional aspect emphasized by the present cohort is the severity of central vision loss independent of clinical diagnosis: except probands of families 10 and 11, no other individuals in our cohort, including three between the ages of 9 and 13, had visual acuity better than 20/80 at the time of evaluation in our clinics. Indeed, the nystagmus observed in two patients, one with eoRD and one with CRD, is consistent with the early presence of central visual impairment. In the eight probands for whom spectral domain OCTs could be digitally reviewed, the degree of visual impairment was noted to be disproportionate to the degree of structural disruption. That is, although foveal structure was not normal in any of these patients, better visual acuity might have been anticipated. Posterior staphylomas disproportionate to the degree of myopia were present in three individuals, as previously reported by Khan and others18.
We identified eight novel variants, six of which are pathogenic/likely pathogenic, including the non-coding variant c.373+91A>G, which causes splicing defect and premature transcript truncation. Despite the spectrum of clinical variation, no genotype-phenotype correlations could be identified with regard to retinal phenotype.
The most recurrent variant in our cohort was the previously reported p.Arg73Pro change, found in eight homozygotes and two heterozygotes across clinical diagnoses. This variant is by far the most frequent change detected in CFAP410 patients (see Supplementary Table 3) and it is the only described pathogenic variant localizing in the third leucine-reach repeat domain, although very recently a similar missense variant located closeby but classified as VUS (p.Arg70Gln) has been detected homozygously in one CRD case26. Its total allele frequency is 0.0005023 in GnomAD v4, largely enriched in non-Finnish Europeans. The common origin for our cases carrying the p.Arg73Pro variant were Brittany and the British isles, particularly Ireland, suggesting a possible founder allele. The p.Arg73Pro variant is associated with a broad phenotypic spectrum (see Fig. 1)6,9,14,20,22,24,25,26,27,29,32,33,37. The proband from family 14, who was homozygous for the p.Arg73Pro variant, had thoracic skeletal abnormalities for which two surgeries were required. Homozygosity for the p.Arg73Pro variant has also been reported previously in JS, SMDAX, and other syndromic IRD cases6,9,14. However, six additional probands in our cohort, homozygous for the p.Arg73Pro variant, lacked extraocular features.
Proband from family 16, homozygous for the p.Gln119* change, suffered from bilateral hip dysplasia, asymmetric bilateral hearing loss, and early ovarian failure. The p.Gln119* change introduces a stop codon in exon 4, of the 7 exon CFAP410 gene, which most likely leads to nonsense-mediated decay (NMD)54,55,56,57,58 of the whole transcript and thus is considered a null allele. Since proband from family 16 does not have any functional CFAP410 protein, we consider her overall phenotype to be relatively mild compared to JS cases7,8. The other truncating variant detected in this study, p.Glu148Alafs*13, is located in exon 5 and is also thought to lead to transcript degradation through NMD and thus a null allele. This variant appeared in trans with the p.Arg73Pro change in the non-syndromic proband of family 4. Such genotypes were also reported in the past to lead to more severe phenotypes6,9,12,14,15.
The two non-coding variants validated in our study, c.143+3A>T and c.373+91A>G, showed a full and partial splicing defect on a mini-gene splicing assay, respectively. Both cases presented with a non-syndromic retinal degeneration (see Fig. 1 and Supplementary Fig. 6). It is important to mention that under the experimental settings of our splicing assay, namely testing the effect of a variant in a limited genomic context, the strength of the observed splicing effect is approximate and we cannot rule out that the c.143+3A>T might have a less severe molecular effect when tested in a larger genomic context.
A review of 95 previously published and our bi-allelic CFAP410 cases did not reveal a clear genotype-phenotype corrlelation and even suggested that a non-syndromic retinal degeneration is likely more common than the syndromic IRD/skeletal dysplasia in patients affected by variants in this gene. Since the actual function of CFAP410 protein remains unknown, it is still unclear what are the molecular mechanisms able to explain the phenotypic heterogeneity observed in patients carrying mutations in this gene. It has been hypothesized that this variability might be the consequence of the functional interaction of CFAP410 with two other proteins NEK1 and SPATA7, as they form a protein complex localized to photoreceptor ciliary structures in multiple species including humans6,13,18. Therefore, it seems likely that this protein complex might have different targets, some of which tissue-specific, eventually resulting in different clinical signs6,9,59. We hypothesize that other proteins may be able to partially substitute for the CFAP410 protein function, which can be facilitated by modifying variants in genes encoding these proteins and thus influencing disease severity and progression60. Such variants have been described in other ciliopathy cohorts, for example, the AHI1 variant p.(Arg830Trp), which increases seven-fold the relative risk of retinal degeneration within a nephronophthisis cohort61. Similarly, resequencing of TTC21B gene in a large group of clinically diverse ciliopathies showed that variants in this gene account as severity modifiers in ~5% of ciliopathy patients62. Collaborative resequecning of all of the published and unpublished cases may reveal such genetic modifiers of the severity of CFAP410-associated disease in the future.
In conclusion, our data validate the phenotypic expansion caused by pathogenic variants in CFAP410 and expand the mutation landscape of this gene by providing novel coding and non-coding variants in this ciliopathy gene.
Methods
Ethics statement
The study was approved by the institutional review board of all participating institutions (Committees of Protection of Persons Ile de France V for families 6, 10, 11, and 12, and Partners HealthCare System for all remaining families) and adhered to the Declaration of Helsinki. Informed consent was obtained from all individuals on whom genetic testing and further molecular evaluations were performed.
Clinical evaluation
Sixteen probands with autosomal recessive retinal degeneration were enrolled in this study. Twelve of them were ascertained from Massachusetts Eye and Ear, and other four from the National Reference Centre of Rare Diseases at Quinze-Vingts National Hospital.
Clinical evaluation was performed by experienced ophthalmologists according to previously published protocols and included functional and structural assessments63,64,65,66.
Genetic analysis
All probands analyzed in this study, except the ones of families 6, 10, 11, 12, and 16, are part of a historical cohort that underwent clinical evaluation in the Inherited Retinal Disorder Service (at MEE; Boston, MA) in the 1990s and early 2000s. Blood samples were obtained from probands, and when possible, their parents. DNA was isolated from peripheral blood lymphocytes by standard procedures. Probands of four families (5, 9, 13, 15) were sequenced using the Genetic Eye Disease (GEDi) panel, described previously67,68. The GEDi version used in this study (v6) targeted exons of 327 known and candidate IRD genes (see Supplementary Table 4)69. The NGS data from the GEDi panel was analyzed using Genome Analysis Toolkit (GATK) version 370 and annotated using the Variant Effect Predictor tool71 with additional annotations taken from the gnomAD38, the Genomic Evolutionary Rate Profiling (GERP)72, SIFT47, PolyPhen248, CADD Phred49 and retinal expression73. To detect possible copy number variations, gCNV software was used as before74. Relatedness of the families sequenced with GEDi panel was excluded using Peddy75. Exome sequencing (ES) for six probands was performed at the Center for Mendelian Genomics at the Broad Institute of the Massachusetts Institute of Technology and Harvard using methodology described previously68. WES data were aligned to hg38, and variants were called using the GATK HaplotypeCaller package version 3.5 (https://software.broadinstitute.org/gatk/). Data were displayed and analyzed with an online tool (https://seqr.broadinstitute.org)76. Genome sequencing for proband of family 3 was performed at the genomics core of the Ocular Genomics Institute. One microgram of genomic DNA purified from whole blood was fragmented to 350 bp using Covaris LE220-plus focused-ultrasonicator, followed by library preparation with KAPA HyperPrep PCRfree Kit (Roche Sequencing Solutions). Libraries were multiplexed by adding 10 bp indexes during adapter ligation (IDT for Illumina—TruSeq DNA UD Indexes v2). Library quality was assessed by fluorometric and fragmentation analysis prior to sequencing. Paired-end 150 cycle sequencing for a minimum of 30x depth of coverage was performed on a NovaSeq 6000 (Illumina).
Probands from families 6, 10, 11, and 12 had been screened applying a customized NGS panel as reported before77 updated regularly to include newly IRD-associated genes, while NGS-based testing was performed by commercial diagnostic laboratories for the proband in Family 16.
Variant validation and phasing
All presented variants refer to the CFAP410 transcript NM_004928.3. Variant segregation was performed by Sanger sequencing (see Supplementary Table 5) or analysis of NGS reads. Although the variants detected in probands of families 4 and 7 were considered in trans according to the gnomAD browser Variant Co-Occurrence tool (https://gnomad.broadinstitute.org/variant-cooccurrence), they were further phased by cloning and Sanger sequencing. Briefly, genomic DNA from the proband was amplified using Phusion (New England Biolabs) and primers spanning the region containing all variants. The amplified fragment was then cloned into the pCR2.1 plasmid, TA cloning kit (Invitrogen) and Sanger sequenced. Sanger sequencing was performed on ABI 3730xl (Applied Biosystems) using BigDye Terminator v3.1 kits (Life Technologies). Sequence analysis was done using SeqManPro (Lasergene 11, DNAStar Madison, WI, USA), in which variants were considered to be in trans when they were not present on the same clone.
Protein modeling, prediction of missense variants, and variant classification
The mutation tolerance at CFAP410 protein residues was analyzed using MetaDome (https://stuart.radboudumc.nl/metadome/)46, while the impact of specific missense variants on CFAP410 structure and function, was predicted by using five prediction algorithms: SIFT47, PolyPhen248, CADD Phred49 REVEL50, and EVE51. Variants were finally classified according to the (ACMG) guidelines52.
Data availability
Variants are available through dbGAP (phs001272.v1.p1 and phs002459.v1.p1) and ClinVar (accession numbers SCV004232444-SCV004232454).
Code availability
Software used in this study: Alamut Visual Plus, version 1.7.1; Biorender; GraphPad Prism, version 10; SeqManPro (Lasergene v11); Seqr (https://seqr.broadinstitute.org/).
References
Georgiou, M. et al. Phenotyping and genotyping inherited retinal diseases: molecular genetics, clinical and imaging features, and therapeutics of macular dystrophies, cone and cone-rod dystrophies, rod-cone dystrophies, Leber congenital amaurosis, and cone dysfunction syndromes. Prog. Retin. Eye Res. 100, 101244 (2024).
Shim, K. S. et al. Reduction of chromatin assembly factor 1 p60 and C21orf2 protein, encoded on chromosome 21, in Down syndrome brain. J. Neural. Transm. Suppl. 67, 117–128 (2003).
Scott, H. S. et al. Characterization of a novel gene, C21orf2, on human chromosome 21q22.3 and its exclusion as the APECED gene by mutation analysis. Genomics 47, 64–70 (1998).
Cheon, M. S. et al. Protein levels of genes encoded on chromosome 21 in fetal Down syndrome brain: challenging the gene dosage effect hypothesis (Part III). Amino Acids 24, 127–134 (2003).
Lai, C. K. et al. Functional characterization of putative cilia genes by high-content analysis. Mol. Biol. Cell 22, 1104–1119 (2011).
Wheway, G. et al. An siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes. Nat. Cell Biol. 17, 1074–1087 (2015).
Beraud, C., Carron, R. & Jeune, M. Asphyxiating thoracic dystrophy with familial characteristics. Arch. Fr. Pediatr. 12, 886–891 (1955).
Huber, C. & Cormier-Daire, V. Ciliary disorder of the skeleton. Am. J. Med. Genet. Part C. Semin. Med. Genet. 160 C, 165–174 (2012).
Wang, Z. et al. Axial spondylometaphyseal dysplasia is caused by C21orf2 mutations. PLoS ONE 11, 1–16 (2016).
Abu-Safieh, L. et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 23, 236–247 (2013).
De Castro-Miró, M. et al. Novel candidate genes and a wide spectrum of structural and point mutations responsible for inherited retinal dystrophies revealed by exome sequencing. PLoS ONE 11, 1–19 (2016).
Patel, N. et al. Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genet. Med. 18, 554–562 (2016).
Suga, A. et al. Identification of novel mutations in the LRR-Cap domain of C21orf2 in Japanese patients with retinitis pigmentosa and cone–rod dystrophy. Investig. Ophthalmol. Vis. Sci. 57, 4255–4263 (2016).
McInerney-Leo, A. M. et al. Homozygous variant in C21orf2 in a case of Jeune syndrome with severe thoracic involvement: extending the phenotypic spectrum. Am. J. Med. Genet. Part A 173, 1698–1704 (2017).
Maddirevula, S. et al. Expanding the phenome and variome of skeletal dysplasia. Genet. Med. 20, 1609–1616 (2018).
Kurashige, T. et al. Retinitis pigmentosa prior to familial ALS caused by a homozygous cilia and flagella-associated protein 410 mutation. J. Neurol. Neurosurg. Psychiatry 91, 220–222 (2020).
Chiu, N. et al. A homozygous in-frame duplication within the LRRCT consensus sequence of CFAP410 causes cone-rod dystrophy, macular staphyloma and short stature. Ophthalmic Genet 43, 378–384 (2022).
Khan, A. O., Eisenberger, T., Nagel-Wolfrum, K., Wolfrum, U. & Bolz, H. J. C21orf2 is mutated in recessive early-onset retinal dystrophy with macular staphyloma and encodes a protein that localises to the photoreceptor primary cilium. Br. J. Ophthalmol. 99, 1725–1731 (2015).
Jauregui, R. et al. Disease asymmetry and hyperautofluorescent ring shape in retinitis pigmentosa patients. Sci. Rep. 10, 3364 (2020).
Rodríguez-Muñoz, A. et al. Expanding the clinical and molecular heterogeneity of nonsyndromic inherited retinal dystrophies. J. Mol. Diagn. 22, 532–543 (2020).
Sharon, D. et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat. 41, 140–149 (2020).
Holtan, J. P., Selmer, K. K., Heimdal, K. R. & Bragadóttir, R. Inherited retinal disease in Norway—a characterization of current clinical and genetic knowledge. Acta Ophthalmol. 98, 286–295 (2020).
Liu, X., Tao, T., Zhao, L., Li, G. & Yang, L. Molecular diagnosis based on comprehensive genetic testing in 800 Chinese families with non-syndromic inherited retinal dystrophies. Clin. Exp. Ophthalmol. 49, 46–59 (2021).
Weisschuh, N. et al. Genetic architecture of inherited retinal degeneration in Germany: a large cohort study from a single diagnostic center over a 9-year period. Hum. Mutat. 41, 1514–1527 (2020).
Fadaie, Z. et al. Whole genome sequencing and in vitro splice assays reveal genetic causes for inherited retinal diseases. npj Genom. Med 6, 97 (2021).
Hitti-Malin, R. J. et al. Towards uncovering the role of incomplete penetrance in maculopathies through sequencing of 105 disease-associated genes. Biomolecules 14, 367 (2024).
Weisschuh, N. et al. Diagnostic genome sequencing improves diagnostic yield: a prospective single-centre study in 1000 patients with inherited eye diseases. J. Med. Genet. 61, 186–195 (2024).
Villafuerte-de la Cruz, R. A. et al. Spectrum of variants associated with inherited retinal dystrophies in Northeast Mexico. BMC Ophthalmol. 24, 1–14 (2024).
Tracewska, A. M. et al. Non-syndromic inherited retinal diseases in Poland: genes, mutations, and phenotypes. Mol. Vis. 27, 457 (2021).
Huang, L. et al. Molecular genetics of cone-rod dystrophy in Chinese patients: new data from 61 probands and mutation overview of 163 probands. Exp. Eye Res. 149, 93–99 (2016).
Zhang, Q. et al. Next-generation sequencing-based molecular diagnosis of 35 Hispanic retinitis pigmentosa probands. Sci. Rep. 6, 1–8 (2016a).
Carss, K. J. et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 100, 75–90 (2017).
Lionel, A. C. et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet. Med. 20, 435–443 (2018).
Birtel, J. et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 8, 4824 (2018).
Wang, L. et al. Application of whole exome and targeted panel sequencing in the clinical molecular diagnosis of 319 Chinese families with inherited retinal dystrophy and comparison study. Genes 9, 1–11 (2018).
Daiger S. P., Sullivan L. S., Bowne S. J., R. B. RetNet. Retinal information network. https://sph.uth.edu/retnet/ (1996).
Turro, E. et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature 583, 96–102 (2020).
Genome Aggregation Database (GnomAD). https://gnomad.broadinstitute.org.
Shapiro, M. B. & Senapathy, P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 15, 7155–7174 (1987).
Yeo, G. & Burge, C. B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394 (2004).
Reese, M. G., Eeckman, F. H., Kulp, D. & Haussler, D. Improved splice site detection in Genie. J. Comput Biol. 4, 311–323 (1997).
Pertea, M., Lin, X. & Salzberg, S. L. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res. 29, 1185–1190 (2001).
Desmet, F. O. et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67 (2009).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535–548. e24 (2019).
Gustafson, K. et al. Whole genome sequencing revealed mutations in two independent genes as the underlying cause of retinal degeneration in an ashkenazi jewish pedigree. Genes 8, 210 (2017).
Wiel, L. et al. MetaDome: Pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum. Mutat. 40, 1030–1038 (2019).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 76, 7.20.1–7.20.41 (2013).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 47, D886–D894 (2019).
Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885 (2016).
Frazer, J. et al. Disease variant prediction with deep generative models of evolutionary data. Nature 599, 91–95 (2021).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Shinbashi, M., Jewell, A., Randolph, J. & Couser, N. C21orf2 variants causing inherited retinal disease: a review of what we know and a report of two new suspected cases. Clin. Case Rep. 11, 1–7 (2023).
Losson, R. & Lacroute, F. Interference of nonsense mutations with eukaryotic messenger RNA stability. Proc. Natl. Acad. Sci. USA 76, 5134–5137 (1979).
Maquat, L. E., Kinniburgh, A. J., Rachmilewitz, E. A. & Ross, J. Unstable beta-globin mRNA in mRNA-deficient beta o thalassemia. Cell 27, 543–553 (1981).
Peltz, S. W., Brown, A. H. & Jacobson, A. mRNA destabilization triggered by premature translational termination depends on at least three cis-acting sequence elements and one trans-acting factor. Genes Dev. 7, 1737–1754 (1993).
Maquat, L. E. Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 5, 89–99 (2004).
Amrani, N., Sachs, M. S. & Jacobson, A. Early nonsense: mRNA decay solves a translational problem. Nat. Rev. Mol. Cell Biol. 7, 415–425 (2006).
Fang, X. et al. The NEK1 interactor, C21ORF2, is required for efficient DNA damage repair. Acta Biochim. Biophys. Sin. 47, 834–841 (2015).
Kousi, M. et al. Genetic modifiers and oligogenic inheritance. Cold Spring Harb. Perspect. Med. 5, 1–22 (2015).
Louie, C. M. et al. AHI1 is required for photoreceptor outer segment development and is a modifier for retinal degeneration in nephronophthisis. Nat. Genet. 42, 175–180 (2010).
Davis, E. E. et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat. Genet. 43, 189–196 (2011).
Scott, H. A. et al. Expanding the phenotypic spectrum in RDH12-associated retinal disease. Cold Spring Harb. Mol. Case Stud. 6, 1–14 (2020).
Moye, A. R. et al. Mutations in ARL2BP, a protein required for ciliary microtubule structure, cause syndromic male infertility in humans and mice. PLoS Genet. 15, 1–28 (2019).
Haer-Wigman, L. et al. Non-syndromic retinitis pigmentosa due to mutations in the mucopolysaccharidosis type IIIC gene, heparan-alpha-glucosaminide N-acetyltransferase (HGSNAT). Hum. Mol. Genet. 24, 3742–3751 (2015).
Men, C. J. et al. The importance of genetic testing as demonstrated by two cases of CACNA1F-associated retinal generation misdiagnosed as LCA. Mol. Vis. 23, 695–706 (2017).
Consugar, M. B. et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet. Med. 17, 253–261 (2015).
Zampaglione, E. et al. The importance of automation in genetic diagnosis: lessons from analyzing an inherited retinal degeneration cohort with the Mendelian Analysis Toolkit (MATK). Genet. Med. 24, 332–343 (2022).
Retinal Information Network (RetNet). https://sph.uth.edu/retnet/home.htm.
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010).
McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17, 1–14 (2016).
Davydov, E. V. et al. Identifying a high fraction of the human genome to be under selective constraint using GERP. PLoS Comput. Biol. 6, 1–13 (2010).
Farkas, M. H. et al. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 Mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genom. 14, 1–14 (2013).
Zampaglione, E. et al. Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations. Genet. Med. 22, 1079–1087 (2020).
Pedersen, B. S. & Quinlan, A. R. Who’s who? Detecting and resolving sample anomalies in human DNA sequencing studies with peddy. Am. J. Hum. Genet. 100, 406–413 (2017).
Pais, L. S. et al. seqr: a web-based analysis and collaboration tool for rare disease genomics. Hum. Mutat. 43, 698–707 (2022).
Audo, I. et al. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J. Rare Dis. 7, 1–17 (2012).
Acknowledgements
This work was supported by grants from the National Eye Institute [R01EY012910 (E.A.P.), R01EY026904 (K.M.B./E.A.P.) and P30EY014104 (MEEI core support)], the Foundation Fighting Blindness (EGI-GE-1218-0753-UCSD, K.M.B.), and the Curing Kids Foundation (K.M.B.). Exome sequencing and analysis were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1HG008900 and in part by National Human Genome Research Institute grant R01 HG009141. The authors would like to thank the patients and their family members for their participation in this study and the Ocular Genomics Institute Genomics Core members for their experimental assistance. The authors would like to thank the Exome Aggregation Consortium, the Genome Aggregation Database (gnomAD) and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about and http://gnomad.broadinstitute.org/about. We thank the clinical laboratory geneticist Dr. Haer-Wigman, PhD and Dr. Pfundt, PhD from the Department of Human Genetics, Radboud University Medical Center, Nijmegen, The Netherlands, for their help with the clinical interpretation of some CFAP410 variants. DNA samples from the French centers originate from NeuroSensCol DNA bank, part of the BioCollections network for research in neuroscience (PI: JA Sahel, coPI I Audo, partner with Centre Hospitalier National d’Ophtalmologie des Quinze-Vingts, INSERM and CNRS). Funding from this group came from LABEX LIFESENSES (ANR-10-LABX-65) supported by French state funds managed by the Agence Nationale de la Recherche within the Investissements d’Avenir program (ANR-11-IDEX-0004-0) and IHU FOReSIGHT (ANR-18-IAHU-0001), and from the National Reference Centers for Rare Diseases Program.
Author information
Authors and Affiliations
Contributions
R.S. performed most of the experiments, data analysis, and led the manuscript writing. P.G., C.P., E.M.P., S.M., X.Z, J.L.D., J.A.S., E.A.P., I.A., R.M.H, analyzed clinical data and reviewed the manuscript. S.D.T., E.O.H., J.H., J.N., C.C., A.A., performed part of the genetic analysis. K.M.B. C.Z. and I.A. guided the experimental design, aided in variant analysis, and contributed to writing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sangermano, R., Gupta, P., Price, C. et al. Coding and non-coding variants in the ciliopathy gene CFAP410 cause early-onset non-syndromic retinal degeneration. npj Genom. Med. 9, 58 (2024). https://doi.org/10.1038/s41525-024-00439-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41525-024-00439-3
