
Abstract
Oro-Facial-Digital Syndrome (OFDS) and Joubert syndrome are ciliary disorders. Fifteen individuals of consanguineous Bedouin kindred presented with global developmental delay with no speech, and a clear OFDS phenotype, combined with Joubert syndrome, with MRI showing hypoplastic corpus callosum and molar tooth sign. Renal and liver function tests and ultrasound were unremarkable. Within a 0.5 Mb disease-associated locus (LOD score 6.2), whole genome sequencing identified a single variant: CEP83 NG_051825.2:g.5774dupG, (NM_016122.3):c.-278dupG. Patient fibroblasts showed aberrantly long cilia, and alternative splicing of CEP83 5’UTR, skipping most of exon 1 of the canonical transcript, and frameshift, abrogating CEP83 mRNA and protein expression. CEP83 acts in primary cilium assembly. CEP83 biallelic missense or in-frame deletions, with presumed residual function, were previously associated with early-onset nephronophthisis culminating in end-stage renal disease. We now demonstrate that a biallelic complete loss-of-function CEP83 variant culminates in elongated primary cilia, causing OFDS with Joubert-like features without evident renal involvement.
Similar content being viewed by others


Introduction
Cilia are microtubule-based sensory organelles crucial in signal transduction during development and in maintaining cellular homeostasis1. Primary cilia are pivotal in embryonic patterning and organogenesis2 acting as signaling hubs and sensors integrating extracellular cues, and are thus central to cellular communication and function3. Disruption in their structure or activity, often due to genetic variants, results in ciliary dysfunction. This can lead to many clinical manifestations, as evidenced in the diverse presentations of Joubert syndrome, Oro-Facial-Digital Syndrome (OFDS), and nephronophthisis, each distinguished by its phenotypic and genotypic peculiarities : over 40 genes are known to be associated with primary ciliary dysfunction, mostly through autosomal recessive inheritance patterns4. The specific genes implicated in the various subtypes of Joubert syndrome, OFDS, and nephronophthisis, contribute to the distinct phenotypic features observed in patients5. These genes encode proteins that act in the formation, maintenance, and function of primary cilia, and their variants cause abnormal ciliary structure and function, with widespread developmental consequences6.
Previous studies have shown that few rare cases of early-onset nephronophthisis with variable intellectual disability are associated with biallelic missense variants in CEP83, encoding a component of the distal appendage (DAP) involved in primary cilium assembly7. Functional studies have shown altered DAP composition and ciliary defects in affected individuals’ fibroblasts and tubular renal cells7. We now show that a non-coding null-variant in CEP83 causes a different ciliopathy: Joubert syndrome with OFDS, yet no evident renal involvement. We further demonstrate through molecular and microscopy studies, that the 5’UTR donor-gain variant causes alternative splicing of the 5’UTR, skipping most of exon 1 of the canonical transcript, resulting in a frameshift and abrogating CEP83 expression - both mRNA and protein, culminating in abnormally long cilia.
Results
Clinical studies
Fifteen individuals of a single inbred large Israeli Arab Bedouin kindred presented with a consistent OFDS phenotype with neurological sequelae. All were born at term (normal birth weights) following uneventful pregnancies. Five of the affected individuals were available for deep phenotyping: all had global developmental delay with practically no speech, as well as delayed motor development, achieving walking at 5–10 years of age. All had craniofacial features characterized by low-set/large posterior-rotated ears, hypertelorism, tented eyebrows with synophris, flat nasal bridge, median cleft or pseudo-cleft tented upper lip, high-arched cleft palate (sub-membranous cleft in some), hypodontia/enamel dysplasia, and tethered lobed tongue with multiple tongue cysts/nodules/hamartomas (Fig. 1). Digit malformations consisted of post-axial polydactyly, brachydactyly, and broad/duplicated angulated first toes (Fig. 1C,G,H,K,L). Micro-penis was evident in affected males. Head circumference was within normal range. Brain MRIs consistently showed hypoplastic corpus callosum and a molar tooth sign (Fig. 2). Nystagmus and strabismus were evident in one of the five well-phenotyped individuals. Hepatic function tests done at ages 1–4 years were normal. Notably, renal function tests and ultrasound were unremarkable with no signs of polyuria, polydipsia or iron-resistant anemia. As renal involvement was assessed at an early age in most, and only few were available for ultrasound, late-onset nephronophthisis cannot be ruled out. Detailed phenotypes of affected individuals are given in Table 1 and Table 2.

Clinical characteristics of individual V-15: A, B Front and side views showing craniofacial features (hypertelorism, low set ears posteriorly rotated, low anterior hairline, pseudocleft of the upper lip). C Left foot showing duplicated hallux. D Oro-dental features showing hamartoma of the tongue and hypodontia. Clinical characteristics of individual V-3: E, H Front and side views showing craniofacial features (hypertelorism, low set posteriorly-rotated ears, low anterior hairline, pseudo-cleft of the upper lip); G, H X-ray of right foot showing duplicated hallux & angulated first toe of the right foot. Clinical characteristics of individual V-8: I, J Front and side views showing craniofacial features (hypertelorism, low set ears posteriorly rotated, low anterior hairline, pseudocleft of the upper lip). K X-ray showing post-axial polydactyly. L Post-axial polydactyly, right hand.

A–D MRI images of individual V-15. E–H MRI images of individual V-3. I–L MRI images of individual V-8. All demonstrated molar tooth sign, ventriculomegaly, cleft of the soft palate, and intracerebral cyst. The MRI findings in each image are indicated by arrows.
Genetic studies
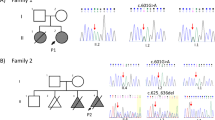
Karyotype (done for individuals F1-V-3, F2-II-1, Fig. 3A) and chromosomal microarray analyses (done for individuals F1-V-3, V-8, V-11, V-15, and F2-II-1, Fig. 3A) did not detect any abnormalities (data not shown). Genome-wide homozygosity mapping, examining SNP data from all available members of families 1 and 2, revealed a single unique segment of homozygosity exclusively shared among all affected individuals. This ~500 Kbp chromosome 12 locus (between SNPs rs2361355 and rs892494 positioned respectively at 94236253 and 94723104 per GRCh38/hg38 assembly) segregated within the kindred as expected for autosomal recessive heredity. Maximal multipoint LOD score was 6.28 (Fig. 3B, C). Whole genome sequencing analysis of individuals V-8 and V-11, done using both our in-house VARista software8 and the commercial Franklin software, identified only a single rare variant (Fig. 3D) within the narrow-shared locus: CEP83 NG_051825.2:g.5774dupG, (NM_016122.3):c.-278dupG (chr12:94459679, A > AC). This variant in the 5’-UTR of CEP83, a known ciliopathy-related gene, was not found in the gnomAD, TOPMed Bravo, ExAC and 1000 Genomes databases, and was predicted by splice AI9 to cause alternative splicing due to donor-gain of a splice site in the first exon of multiple transcripts of the gene. The CEP83 variant and its appropriate segregation within all available family members were verified by Sanger sequencing; genotypes of tested individuals are given in supplementary Table 1.

A Pedigrees of studied consanguineous Bedouin kindreds. Affected individuals are marked in black. Subjects available for this study are marked with asterisks. B LOD score analysis plot of chromosome 12 using SUPERLINK-Online. C Homozygosity Mapper plot showing single homozygosity locus shared by affected individuals, marked in red (black arrow). D Chromatograms obtained by Sanger sequencing of unaffected wildtype, unaffected carrier and affected subjects demonstrating the CEP83:c.-278dupG variant (black rectangle).
Exon splicing studies
Splicing experiments focusing on the canonical transcript of CEP83 (NM_016122.3) were conducted. Primary fibroblasts, derived from skin punch biopsies of both subjects and controls, were cultured to examine a predicted novel donor-gain site of exon 1: cDNA of fibroblasts from affected individuals and controls underwent PCR with primers designed to amplify sequences surrounding the predicted novel donor site and its anticipated affected sequence in exon 1 (Fig. 4A). The PCR amplicons verified the predicted alternative splicing in the mutant cells (Fig. 4B). Subsequently, each PCR product was isolated from the gel and underwent Sanger sequencing, confirming the predicted effect on the mutant mRNA: skipping of 124 bp in exon 1, attributed to alternative splicing induced by the gain of a new donor site (Fig. 4C).

A Schematic presentation of the effect of the CEP83 donor-gain variant on Pre-mRNA splicing; the variant marked by black arrow. B Agarose gel electrophoresis (2% agarose) of PCR-amplified products from cDNA derived from fibroblasts of affected homozygous (HOM) individuals and wildtype (WT) controls, demonstrating full transcript 259 (wild-type)/260 (mutant) bp amplicon in contrast with 136 bp mutant amplicon due to alternative splicing of exon 1. C Sanger sequencing of PCR amplicons from cDNA of fibroblasts of affected individuals demonstrate both full and alternative splicing transcripts. D Illustration of primer design to RT-qPCR and relative quantification of CEP83 expression RT-qPCR in patient fibroblasts compared to the wildtype controls; 2-tailed Mann-Whitney test (*p < 0.05, **p < 0.005, and ***p < 0.0005, by Mann-Whitney test). E Western blot analysis of CEP83 protein in patient fibroblasts compared to wildtype controls. CEP83 protein levels given relative to Actin levels. This figure was created in BioRender. Jean, M. (2025) https://BioRender.com/8r0ggrv.
CEP83 mRNA and protein expression studies
To assess the impact of the CEP83 variant on nonsense-mediated mRNA decay (NMD), RT-qPCR and western blot analyses were performed on primary fibroblasts from both mutant and control subjects. The RT-qPCR was designed to compare the overall CEP83 transcript levels comparing patients and controls by focusing on the junction between exons 3 and 4 (Fig. 4D). Markedly lower levels of CEP83 mRNA were seen in the mutant fibroblasts compared to the wild-type (WT) counterparts (Fig. 4D). In line with those findings, western blot analysis demonstrated a similar reduction in CEP83 protein expression in mutant fibroblasts relative to WT controls (Fig. 4E). Thus, the 5’ UTR CEP83 variant reduces CEP83 expression at both the mRNA and protein levels.
Cilia microscopy studies
To test the possible effects of the reduced CEP83 expression on cilia structure, we used confocal microscopy: fixed primary fibroblasts were stained with γ-Tubulin to mark the centrosome and with acetylated α-Tubulin to highlight primary cilia. Additionally, DAPI was employed to identify DNA. Mutant fibroblasts of affected individuals exhibited longer cilia compared to their wild-type (WT) counterparts (Fig. 5): Prism software delineated a significant alteration in cilia length, with the distribution analysis showing longer cilia in mutant samples.

A Confocal imaging of wildtype (WT) and homozygous mutant primary fibroblasts showing staining for acetylated α-Tubulin (green), γ-Tubulin centrosome marker (red) and DAPI (blue); scale bar 10 μm; zoom-in of white rectangle below (scale bar 5μm). B Primary cilia length measurements in patient fibroblast (n = 2) compared to wildtype controls (n = 2); sample size WT column n = 50, Mutant column n = 58. Measurements were done in two separate independent experiments. Two tailed t-test (****p-value < 0.0001) C Cilia length distribution analysis categorized in groups of short (less than 3 μm) in blue, medium (3–4 μm) in light blue, long (4–6 μm) in orange, and extra-long (more than 6 μm) in pink.
Discussion
Ciliopathies, including OFDS, Nephronophthisis, Bardet Biedl syndrome (BBS), Meckel-Gruber syndrome, and Joubert syndrome, encompass a heterogeneous group of monogenic disorders caused by structural or functional disruption of cilia or abnormal cilia biogenesis10 Unraveling the underlying variants and molecular pathways of these diseases is crucial for delineating their complex etiology as well as normal cilia structure and function11,12.
OFDS presents a diverse array of genetically-inherited disorders characterized by developmental anomalies of the face (hypertelorism, low-set ears), the oral cavity (lingual hamartoma, abnormal frenula, lobulated tongue), and digits (brachydactyly and polydactyly). With at least 19 subtypes, OFDS showcases a mosaic of genetic and phenotypic heterogeneity, reflecting a broader spectrum of ciliopathies - disorders arising from dysfunctional cilia13. Joubert syndrome is another ciliopathy closely related to OFDS, characterized by the distinctive ‘molar tooth sign’ visible on brain MRI, representing a complex brainstem malformation14. This syndrome shares overlapping phenotypic features with OFDS, notably the abnormalities in the development of facial structures and digits, underscoring the interconnected nature of these disorders and the importance of primary cilia in their pathogenesis15. Nephronophthisis is a hereditary kidney disorder associated with defective ciliary function. This ciliopathy is the most common genetic cause of end-stage renal disease (ESRD) in children and young adults. The disease typically manifests as chronic tubulointerstitial nephritis, leading to symptoms such as polyuria, polydipsia, secondary enuresis, and ultimately progressing to renal failure. Genetically, nephronophthisis is highly heterogeneous, with variants in over 20 genes (NPHP1-NPHP20) implicated in disease causation. All NPHP genes encode proteins localized to the primary cilium or its basal body, taking part in various aspects of ciliary structure and/or function, including signaling and maintenance. Given its link to ciliary function, nephronophthisis shares molecular and clinical features with other ciliopathies, including Joubert syndrome and Meckel-Gruber syndrome12. The inheritance pattern of most ciliopathies is typically autosomal recessive, though there are rare cases with autosomal dominant transmission11.
Centrosomal Protein 83 (CEP83) is a centriolar protein crucial for the formation of primary cilia, particularly impacting the structure and function of distal appendage proteins (DAPs), complex structures composed of proteins anchoring the primary cilia to the cell membrane - vital for ciliogenesis and cellular processes16. CEP83 is the most upstream protein in the DAP assembly pathway: CEP89, SCLT1, CEP164, LRRC45, and FBF1, downstream of CEP83, are all required for ciliary assembly17. Disruption in the DAP complex leads to compromised ciliary assembly, and variants in genes encoding DAP proteins manifest in multi-organ phenotypes, including nephronophthisis and OFDS. CEP83 dysfunction is related to nephronophthisis and abnormal cortical development, influencing neuron differentiation and kidney lineage, highlighting its significance in fundamental biological processes and developmental disorders7,18,19.
Previous studies have shown that biallelic variants in CEP83 are associated with early-onset nephronophthisis. Severe tubulointerstitial lesions (microcystic tubular dilatations/atrophic tubules and interstitial fibrosis) were present in all previously reported individuals with biallelic CEP83 variants, reaching ESRD at age 1–4 years. The nephronophthisis was isolated in some of the cases; however, in approximately 50% of cases, there were associated neurological alterations, including speech delay, intellectual disability, and/or hydrocephalus. Few cases presented with ophthalmologic defects (strabismus/retinal degeneration) or with liver alterations, namely hepatic cytolysis and cholestasis, or severe portal septal fibrosis with mild thickening of arterial walls and an increase in the number of biliary canaliculi on liver biopsy. All these cases were associated with biallelic missense or in-frame deletions in CEP83, with presumed residual activity, Functional studies showed that CEP83 mutations disrupt DAP composition and cause ciliary defects in fibroblasts and renal cells. Notably, a single previously reported case carrying a homozygous frameshift variant leading to a truncated protein (p.Glu669Aspfs∗14) exhibited a more severe phenotype of multiple organ involvement, with ESRD at 1 year of age, hydrocephalus, intellectual disability, and craniofacial features7.
The ciliopathy phenotype we describe in our patients is well in line with our demonstration of aberrantly long cilia in patients’ fibroblasts, similar to the aberrant cilia length we and others have shown in other ciliopathies20. Notably, the consistent disease phenotype shared by the 15 affected individuals we describe differs significantly from that previously delineated for CEP83 variants. All our patients presented with oral, facial, and digital phenotypic elements of OFDS, as well as severe neurological elements and a molar tooth sign on MRI - a hallmark of Joubert syndrome. Thus, the disease we delineate is very different from that shown with previously reported CEP83 variants: the classical molar tooth sign found in all our patients has not been previously reported in CEP83 variants; moreover, while ESRD has been reported by the age of 4 years in all previous cases, all our 15 patients (some of whom are already well into their twenties) lack clinical evidence of nephronophthisis, not to speak of ESRD. However, nephronophthisis may initially present with non-specific ultrasonographic findings before progressing to more definitive structural abnormalities, Thus, as renal evaluation in most was partial and done at an early age, late-onset nephronophthsis cannot be ruled out. Further follow-up renal studies (bi-annual renal function tests and ultrasound with possible biopsies in cases of abnormal findings) of all patients into later ages is warranted. Moreover, given the role of primary cilia in hepatic and ocular development and function, as well as previous reports of strabismus and retinal degeneration in patients with CEP83 variants, thorough follow-up studies of hepatic function tests and ophthalmological evaluation for all 15 affected individuals are warranted. With a LOD score of 6.2 and a single variant within the small 0.5 Mb disease-associated locus, there is no doubt that the variant we demonstrate in the cilia-associated CEP83 gene, affecting ciliary length, is the disease-causing variant. How, then, can one explain the fact that our 15 cases have a disease phenotype significantly different than that with the previously reported CEP83 variants? One might suggest that the different phenotypes are due to the extent of loss-of-function secondary to the CEP83 variants: while previously-described variants are suggested to cause a partial loss-of-function, we demonstrate in patients’ fibroblasts that the variant we describe culminates in NMD and complete loss of expression of both CEP83 mRMA and protein. However, at least one previously reported patient7 had a homozygous truncation variant that is expected to cause full loss of function, yet had no reported molar tooth sign, and did have ESRD by one year of age. We demonstrate in our patients’ fibroblasts that the CEP83 variant causes skipping of 124 bp in exon 1, attributed to alternative splicing induced by the gain of a new donor site. However, it is plausible that in other, embryonic, tissues, the 5’-UTR variant has other effects on different cell types, such as cells in the developing embryonic brain and kidneys. The 5′ untranslated region contains many elements such as upstream ORFs (uORFs), microRNA binding sites, internal ribosome entry sites, and structural components involved in the regulation of mRNA stability, pre-mRNA splicing, and translation initiation21. Thus, dysregulation of cis-regulatory elements or secondary structures within the 5′UTR may cause a change in gene expression. For instance, it is plausible that the 5’-UTR variant we describe does affect the developing brain but not the kidneys, hence the molar tooth sign on one hand, yet the lack of a discernable renal disease. In fact, 5’-UTR variants have been specifically shown to affect translation and protein production in the causation of neurodevelopmental disorders22. Altogether, we expand our understanding of CEP83-related ciliopathies, demonstrating that a CEP83 loss-of-function variant can culminate in aberrantly elongated cilia, causing a unique syndrome of OFDS with elements of Joubert syndrome, with no clear evidence of the previously described nephronophthisis.
Methods
Clinical assessment
The study was approved by the Soroka Medical Center institutional review board (approval No. 5071 G) and written informed consent was obtained from all participating family members or their legal guardians. Consent to participate and consent to publish was obtained from all relevant participating family members or their legal guardians. The authors have obtained written consent to publish the details and images. We have complied with all relevant ethical regulations including the Declaration of Helsinki. All participating family members underwent genetic evaluation and phenotypic assessment by senior pediatricians, neurologists, and clinical geneticists.
Genomic DNA extraction
Genomic DNA was extracted using the E.Z.N.A SQ blood DNA Extraction Kit (Omega) from whole blood samples of the affected individuals and unaffected parents and siblings (Fig. 3A).
Linkage analysis
Genome-wide linkage analysis to all available DNA samples from family 1 (Fig. 3A) was performed using Illumina OmniExpress Beadchip with more than 750 K SNP loci per sample (Illumina, SanDiego, CA, USA). Homozygosity mapping analysis was done using Homozygosity-Mapper (http://www.homozygositymapper.org/)23. Fine mapping and haplotype reconstruction were performed for all available DNA samples using polymorphic markers as previously described24. All physical positions mentioned are according to the GRCh38/hg38 genome assembly. Multipoint LOD score at the shared locus was calculated using SUPERLINK ONLINE SNP 1.1 (http://cbl-hap.cs.technion.ac.il/superlink-snp/)25.
Whole-genome sequencing (WGS) analysis
Whole-genome sequencing was performed for subjects V-8 and V-11 (Fig. 3A) by Macrogen® using Agilent SureSelect V7-postcapture library construction enrichment kit and Illumina NovaSeq6000 system with 150 bp paired-end reads. Raw data reads were aligned to GRCh38 reference genome using BWA-MEM26. Variant calling was done using GATK. Data were analyzed using the online interpretation tool Franklin by genoox® (franklin.genoox.com) and our in-house variant analysis tool, VARista, https://VARista.link8. We excluded variants observed with an allele frequency of ≥1% in the Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org/), Exome Aggregation Consortium (ExAC, https://exac.broadinstitute.org/) or the international genome sample resource (IGSR, https://www.internationalgenome.org/), as well as variants that appeared in a homozygous state in our in-house next generation sequencing database of over 400 ethnically -matched controls.
Segregation analysis
Segregation analysis of the CEP83:c.-278dupG A > AC variant within the studied kindreds was done through Sanger sequencing, using the following primers: Forward 5’-ACCTCTCCCGGGATACTTCAC-3’; Reverse 5’ -TACATACCAGACGCACGCAC-3’ (253 bp amplicon; annealing temperature 60 °C; extension time 16 s).
Cell culture
A 4-mm human skin punch biopsy was dissected and prepared for the derivation of fibroblast culture, following routine methods, and primary fibroblasts were grown in cell culture in standard media as previously described27.
Reverse transcription and real-time quantitative PCR
Total RNA was extracted from cultured fibroblasts using GENzol™ Tri RNA Pure Kit (Geneaid Biotech Ltd.). Single-stranded cDNA libraries were prepared using UltraScript™ cDNA Synthesis Kit (PCR Biosystems Ltd.). Primers were used for CEP83 exon 2-3 amplification (259/260 bp amplicon, Fig. 4B): Forward 5’ - GTTCGGCCCTCTTCCTCTCG-3’; Reverse 5’ - GCCAAATTATACCTTCCTCTAAAGTAG-3’. Quantitative Real-time PCR (qRT-PCR) was done using a Hy-SYBR Power mix (hylabs). CEP83 transcript levels were normalized to Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) housekeeping gene. Primers were used for CEP83 exon 3–4 amplification (112 bp amplicon, Fig. 4D): Forward 5’ -GACAGGTTCTCAGTCGGAGTTCC-3’; Reverse 5’ -ATTCTGCAACCTTGTGTGTTCAGC-3’. Primers used for GAPDH amplification (87 bp amplicon): Forward 5’-TCGACAGTCCCGCATC-3’; Reverse 5’ -CCGTTGACTCCGACCTTC-3’. The annealing temperature was 62 °C, the extension time was set for 20 s, and the PCR reaction repeats were 40 cycles.
Statistical analyses were done using GraphPad Prism 8 (GraphPad Software). An unpaired 2-tailed Mann-Whitney test was used to compare experimental groups. P values of less than 0.05 were considered statistically significant. All data are presented as the mean ± SD.
Western blot
Protein lysates were extracted from fibroblasts of homozygote patients (V11, V15) and ethnicity-matched healthy controls using radioimmunoprecipitation assay (RIPA) buffer [50 mM tris-HCl (pH 8), 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM dithiothreitol (DTT), and 1:100 protease inhibitor mixture (Sigma-Aldrich)]. After extraction, total protein sample concentration was measured using Pierce™ BCA Protein Assay Kit (#23225, Thermo Scientific™). Each lane was loaded with the same amount of total protein on SDS-PAGE (8% acrylamide). Blocking was done with phosphate-buffered saline with 0.1% Tween 20 (PBST) mixed with 10% skim milk for 1 hr. After blocking, membranes were incubated overnight at 4 °C with primary antibodies anti-CEP83 (1:1000, HPA038161, Sigma-Aldrich) and Anti-β-Actin (1:2000, A5441, Sigma-Aldrich) and washed three times with PBST for 5 min; finally, membranes were incubated with secondary antibody Peroxidase AffiniPure Goat Anti-Mouse IgG (H + L) (1:10000, 115-035-062, Jackson ImmunoResearch) for 1 hr at room temperature and washed with PBST (once for 15 min and twice for 5 min). Blots were developed using WesternBright Quantum HRP substrate chemiluminescence reagent (K-12045-D20, Advansta). Relative amounts of protein were normalized to β-Actin with ImageJ software.
Immunofluorescence
Fibroblasts were seeded on coverslips in ibidi µ-Slide 8 Well (Cat.No:80826) and maintained 48 h, in low serum media (0.5% FBS) to induce ciliation. Cells were subsequently fixed on ice with 4% PFA/PBS for 5 min. An additional step of a 5 min incubation in chilled methanol (−20°C) was conducted for γ-tubulin detection. Cells were permeabilized in 0.1% Triton X-100/PBS for 15 min and maintained 1 h in blocking solution (4% goat serum/PBS/0.05% Tween 20). Slides were kept overnight at 4°C with primary antibodies diluted in blocking buffer. The next day, cells were washed with PBS/0.05% Tween 20, and fluorescence-conjugated secondary antibodies (1:500; ThermoFisher [A32787, A32814]) were added for 1 h at room temperature. Primary antibodies: acetylated-α tubulin (1:200; Abcam [EPR16772]), γ-tubulin (1:2,000; Abcam [GTU-88]). Nuclei were stained with DAPI (1:5000; BioLegend [422801]). Slides were used for image acquisition in a Zeiss LSM880 Airyscan microscope.
Image analysis and statistics
Images were analyzed in QuPath ver. 0.4.4 software. Cilia were annotated manually using a line tool and their length was measured. Ciliated and non-ciliated cells were classified manually and labeled by a built-in multipoint tool. The resulting data were exported as a CSV table and statistics were calculated using Graph Pad prism version 8 software. The statistical difference in cilia length comparing control and mutant cells was calculated using t-test. The normal distribution of the data was checked by the Shapiro-Wilks test.
Data availability
The data cannot be shared openly because of privacy considerations but are available from the corresponding author upon request.
References
Fliegauf, M., Benzing, T. & Omran, H. When cilia go bad: Cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 8, 880–893 (2007).
Gerdes, J. M., Davis, E. E. & Katsanis, N. The vertebrate primary cilium in development, homeostasis, and disease. Cell 137, 32–45 (2009).
Singla, V. & Reiter, J. F. The primary cilium as the cell’s antenna: Signaling at a sensory organelle. Science (1979) 313, 629–633 (2006).
Louie, C. M. & Gleeson, J. G. Genetic basis of Joubert syndrome and related disorders of cerebellar development. Hum. Mol. Genet 14, 235–242 (2005).
Mitchison, H. M. & Valente, E. M. Motile and non-motile cilia in human pathology: from function to phenotypes. J. Pathol. 241, 294–309 (2017).
Badano, J. L., Mitsuma, N., Beales, P. L. & Katsanis, N. The ciliopathies: An emerging class of human genetic disorders. Annu Rev. Genomics Hum. Genet 7, 125–148 (2006).
Failler, M. et al. Mutations of CEP83 cause infantile nephronophthisis and intellectual disability. Am. J. Hum. Genet 94, 905–914 (2014).
Hadar, N. et al. VARista: a free web platform for streamlined whole-genome variant analysis across T2T, hg38, and hg19. Hum. Genet. https://doi.org/10.1007/s00439-024-02671-4 (2024).
Jaganathan, K. et al. Predicting splicing from primary sequence with deep learning. Cell 176, 535–548.e24 (2019).
Tobin, J. L. & Beales, P. L. The nonmotile ciliopathies. Genet. Med. 11, 386–402 (2009).
Hilgendorf, K. I., Myers, B. R. & Reiter, J. F. Emerging mechanistic understanding of cilia function in cellular signalling. Nat. Rev. Mol. Cell Biol. https://doi.org/10.1038/s41580-023-00698-5 (2024).
Srivastava, S., Molinari, E., Raman, S. & Sayer, J. A. Many genes-one disease? Genetics of nephronophthisis (NPHP) and NPHP-associated disorders. Front Pediatr. 5, 1–15 (2018).
Valente, E. M., Rosti, R. O., Gibbs, E. & Gleeson, J. G. Primary cilia in neurodevelopmental disorders. Nat. Rev. Neurol. 10, 27–36 (2014).
Maria, B. L. et al. Molar tooth sign in Joubert syndrome: Clinical, radiologic, and pathologic significance. J. Child Neurol. 14, 368–376 (1999).
Brancati, F., Dallapiccola, B. & Valente, E. M. Joubert Syndrome and related disorders. Orphanet J. Rare Dis. 5, 1–10 (2010).
Yang, T. T. et al. Super-resolution architecture of mammalian centriole distal appendages reveals distinct blade and matrix functional components. Nat. Commun. 9, 1–11 (2018).
Mansour, F., Boivin, F. J., Shaheed, I. B., Schueler, M. & Schmidt-ott, K. M. The role of centrosome distal appendage proteins (DAPs) in nephronophthisis and ciliogenesis. Int. J. Mol. Sci. 22,12253 (2021).
Mansour, F. et al. The centrosomal protein 83 (CEP83) regulates human pluripotent stem cell differentiation toward the kidney lineage. Elife 11, 1–28 (2022).
Shao, W. et al. Centrosome anchoring regulates progenitor properties and cortical formation. Nature 580, 106–112 (2020).
Wormser, O. et al. SCAPER localizes to primary cilia and its mutation affects cilia length, causing Bardet-Biedl syndrome. Eur. J. Hum. Genet. 27, 928–940 (2019).
Ryczek, N., Łyś, A. & Makałowska, I. The functional meaning of 5′UTR in protein-coding genes. Int. J. Mol. Sci. 24, 2976 (2023).
Plassmeyer, S. P. et al. A massively parallel screen of 5′UTR mutations identifies variants impacting translation and protein production in neurodevelopmental disorder genes. medRxiv (2023).
Seelow, D., Schuelke, M., Hildebrandt, F. & Nürnberg, P. HomozygosityMapper - An interactive approach to homozygosity mapping. Nucleic Acids Res. 37, 593–599 (2009).
Safran, A. et al. Hyperinsulinism/hyperammonemia syndrome caused by biallelic SLC25A36 mutation. https://doi.org/10.1002/jimd.12594 (2023).
Silberstein, M. et al. Online system for faster multipoint linkage analysis via parallel execution on thousands of personal computers. Am. J. Hum. Genet. 78, 922–935 (2006).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Vangipuram, M., Ting, D., Kim, S., Diaz, R. & Schüle, B. Skin punch biopsy explant culture for derivation of primary human fibroblasts. J. Vis. Exp. 9–11. https://doi.org/10.3791/3779 (2013).
Acknowledgements
We thank the patients and their families for their efforts and collaboration in the study. Figure 3 was created in BioRender. Jean, M. (2025) https://BioRender.com/8r0ggrv. The study was funded in part by the Israel Science Foundation (grant no. 2463/23; OSB); The Morris Kahn Family Foundation (OSB); The Israel Ministry of Science, Technology and Space, through the National Knowledge Center for Rare/Orphan Diseases, Ben-Gurion University of the Negev, Beer-Sheva, Israel (OSB). The Kadar-Sheba prize (OSB).
Author information
Authors and Affiliations
Contributions
Conceptualization: M.M.J., H.F., A.Y., O.S.B. Investigation: M.M.J., A.Y., T.E.B., V.D., A.M., M.E.S., A.A.T., N.A., T.P., A.S., O.F., N.H., D.L., I.S., K.E.A., H.F., O.S.B. Funding acquisition: O.S.B. Supervision: O.S.B. Writing the manuscript: M.M.J., O.S.B., with comments and consent of all authors. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jean, M.M., Yunis, A., Elbaz-Biton, T. et al. A ciliopathy combining Joubert syndrome and Oro-Facial-Digital syndrome caused by bi-allelic 5’-UTR loss-of-function CEP83 variant. npj Genom. Med. 10, 66 (2025). https://doi.org/10.1038/s41525-025-00514-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41525-025-00514-3
