
Abstract
A serious consequence of marine biofouling on metallic structures is the insidious localized corrosion at the attachment sites of fouling organisms, such as barnacles. Albeit known, this phenomenon is poorly understood and currently mitigated using cost- and labor-intensive methods. In this work, we study the contribution to biofouling corrosion by a protein contained in the adhesive cement that barnacles secrete to attach to immersed substrates. We synthesize a specific cement protein of 20 kDa (CP20) from the barnacle Megabalanus rosa and study its corrosion behavior independently of the animal. Our results show that CP20 accelerates the corrosion rate of a marine-grade, mild steel from 0.7 to 1.6 mm year−1. Through chemical analysis of the corrosion products, protein adsorption studies on the metal surface, and cyclic voltammetry, we elucidate an intricate corrosion mechanism that relies on the strong adhesive properties of CP20 and its electrochemically active disulfide groups. Our results have far-reaching implications on the prediction and mitigation of biocorrosion in marine applications. Moreover, the protein-induced corrosion mechanism unveiled in our study may be extended to other scenarios to understand the degradation of metal alloys used in food storage and biomedical implants.
Similar content being viewed by others

Introduction
Virtually any structure that is submerged in seawater suffers from the colonization of microorganisms and macroorganisms—a phenomenon that is referred to as biofouling1,2,3. Besides increasing hydrodynamic drag of ship hulls—which results in higher fuel consumption and CO2 emission4,5—a deleterious consequence of biofouling on metallic structures is the accelerated corrosion of materials at sites of fouling attachment6,7,8,9,10,11. Masked by the organisms, this localized corrosion is difficult to detect and quantify. Thus, it may cause unexpected failure of subsea structures with serious environmental consequences12,13. Inadequate understanding of the corrosion rates and mechanisms associated with biofouling forces the marine industry to rely on cost- and labor-intensive mitigation measures, such as frequent scraping of metallic structures or regular assessment of protective coatings.
Some of the most common and resilient macrofouling organisms are barnacles14. Barnacles are capable of permanently attaching to immersed solid infrastructures by means of cement proteins that firmly fix the calcite base of the animal’s plate to foreign substrates15. Previous investigations of this type of biofouling have reported contrasting corrosion mechanisms. Some have indicated that barnacle cement proteins indirectly affect metal dissolution by limiting seawater flow underneath the animal, thus providing a suitable environment for crevice corrosion7,16. Others have postulated the possibility of a more active role of cement proteins, albeit no electrochemical mechanisms could be identified17.
Here, we directly investigate the corrosion activity of CP20—a cement protein with a molecular weight of 20 kDa from the species Megabalanus rosa (M. rosa)—which has been suggested to play a key role in promoting adhesion at the cement/substrate interface18,19. We select CP20 amongst all cement proteins in M. rosa owing to its high content of the amino acid cysteine (17 mol.%)20, which is a good candidate to promote redox activity in saline conditions. We find that recombinantly produced CP20 accelerates the corrosion rate of AH36 alloy—a mild steel commonly used in marine applications—up to 1.6 mm year−1, with visible pitting corrosion appearing just a few minutes after immersion of metal coupons in the protein-rich solution. To study the mechanisms by which the protein adsorbs to and enhances corrosion rates of the metal alloy, we compare CP20’s behavior to that of two other model corrosive proteins—bovine serum albumin (BSA) and lysozyme (LYZ). Our experiments demonstrate that the high protein-induced corrosion (PIC) rate of CP20 stems from the combination of its strong adhesive properties and its propensity to oxidize iron (Fe), which results from the reduction of intramolecular disulfide bonds contained within CP20. Our findings shed light on the underlying mechanisms of PIC and stimulate innovative strategies for the prevention and prediction of biofouling corrosion. Because many other proteins contain similar redox-active moieties to those that govern CP20’s corrosion behavior, we believe that our findings may be extended to other applications—such as food storage and biomedical implants.
Results
Time-resolved corrosion study
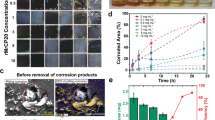
To quantify PIC rate on mild steel, we immerse AH36 steel coupons in a 4.6 µM of folded recombinant CP20 in tris-buffered saline (TBS) and image the sample surface at different points in time. We select AH36 steel because it is ubiquitous in shipbuilding and offshore structures due to its high strength and low cost21,22, which makes it a relevant case study material for a wide range of marine applications. We decide to use a protein concentration of 4.6 µM in all our experiments (unless otherwise stated) based on the concentration study presented in the Supplementary Information (Supplementary Fig. 1). Noteworthily, this concentration may be representative of that secreted naturally by barnacles23. Figure 1a shows the time series of the immersion test as seen through the optical microscope. The dark contrast that is visible on the sample surface corresponds to corrosion products. Our time-resolved corrosion study shows that corrosion products are clearly visible by the naked eye after only 30 min of immersion, and that they grow and coalesce with time. However, we confirm the formation of corrosion pits as early as 8 min after coupon immersion by means of scanning electron microscopy (SEM) (Fig. 1b).

a Optical micrographs showing the initiation and growth of corrosion pitting sites at immersion time t = 0, 0.5, 1.5, 3, and 4 h; b Secondary electron micrograph of a pit initiating as early as 8 min; c Secondary electron micrographs of corrosion sites before (left) and after (right) removal of corrosion products, respectively; d XPS spectra from the corrosion tubercle and uncorroded surface, displaying the nitrogen (N1s) peak at the corrosion tubercle associated with the protein; and e time-resolved corrosion rate of mild steel by EIS. Error bars represent the standard deviation of triplicate measurements.
To investigate the extent of PIC damage underneath the corrosion products, we clean the surface of the tested samples in a bath of dilute hydrochloric acid (HCl) and directly image the affected sites. Figure 1c shows one such site after 4 h immersion. We observe a stark resemblance in geometry between the corrosion products and the underlying pits. We compare the area fraction corresponding to the corrosion products to that showing pitting damage and find a very good match (Supplementary Information, Supplementary Fig. 1e inset). Accordingly, we use the corrosion product area fraction as a measure of pitting area fraction.
To confirm that this accelerated corrosion is indeed caused by CP20, we compare the elemental composition at corrosion sites with that of other non-corroded regions of the sample by means of X-ray photoelectron spectroscopy (XPS). When we scan the corroded areas (Fig. 1d), we record a substantially stronger nitrogen peak (N1s, at 399.7 eV) with 3% standard deviation, which confirms the accumulation of proteins at these sites24,25. Indeed, AH36 steel does not contain nitrogen (Supplementary Information, Supplementary Table 1).
We repeat the time-resolved corrosion study on a different batch of samples to perform electrochemical impedance spectroscopy (EIS) and quantify corrosion damage as a function of immersion time. The corrosion rates from the measurements are shown in Fig. 1e. The trend shows an increased extent of corrosion versus immersion time that is in agreement with the qualitative observations presented in Fig. 1a.
Comparison between CP20 and other corrosive proteins
To establish the aggressiveness of CP20, we compare the mass loss in our metal samples when immersed in a bath of CP20, BSA, and LYZ. We choose BSA and LYZ because these proteins are well known to induce corrosion, albeit in different applications and environments26,27,28,29. Moreover, unlike CP20, neither BSA nor LYZ has been described as an adhesive protein in the literature. Thus, the comparison between these proteins provides an opportunity to investigate the relationships between protein adhesive properties and corrosion behavior.
To quantify mass loss upon immersion in different solutions, we measure the Fe mass contained in corrosion products that adhere to the sample surface, as well as that dissolved in solution using inductively coupled plasma mass spectrometry (ICP-MS; see “Methods” section). The results, shown in Fig. 2a, indicate that CP20 accelerates Fe removal by ~50% compared to the other proteins for the same immersion time and solution concentration. Surprisingly, the difference in corrosion rate between BSA, LYZ, and TBS buffer (which is protein free) is statistically insignificant. Visual inspection of steel coupons after 4 h immersion confirms that CP20 yields the most severe corrosion amongst the different solutions tested (Fig. 2a). By contrast, minimal corrosion pitting is detected on samples immersed in BSA and LYZ. We also confirm these trends by EIS and linear polarization resistance (LPR) measurements, both of which are presented in Supplementary Information (Supplementary Fig. 2c).

a Iron release from AH36 steel in various solutions after 4 h; and b QCM adsorption–desorption profiles of BSA, LYZ, reduced CP20, and pristine CP20 solutions on Fe. Error bars represent the standard deviation of triplicate measurements.
We assess the adsorption properties of the three proteins using a quartz crystal microbalance (QCM) to understand if PIC is correlated to the protein’s adhesive properties. This experiment (detailed in “Methods” section) involves an adsorption and a desorption phase. During the adsorption phase, we plot the adsorbed mass on the QCM while flowing the protein-rich solution. In the desorption phase, we flow protein-free buffer to remove weakly bonded proteins from the QCM surface. We calculate the mass adhering to the QCM via the Sauerbrey relation30, and plot the adsorption and desorption profiles of CP20, BSA, and LYZ in Fig. 2b. We commence flowing the protein-rich solution after 10 min from starting the measurement to ensure a stable baseline. While all proteins adsorb to the QCM (as evidenced by the rapid increase of measured adsorbed mass), CP20 shows the highest adsorptivity. Moreover, CP20 shows no significant desorption, which starts at the inflection points in the graph in Fig. 2b. We estimate that protein desorption reaches a steady state after 25 mins, when the curves in Fig. 2b plateau. This steady-state value indicates the quantity of proteins that is irreversibly attached to the metal surface of the QCM.
PIC mechanism associated to CP20
The results shown in Fig. 2 (and Supplementary Fig. 1) are proof of the aggressiveness of CP20, and suggest an interplay between adhesion and corrosion in PIC. To investigate this relationship, we compare the electrochemical activity of all three proteins via cyclic voltammetry. The results, plotted in Fig. 3a, show that all proteins exhibit an oxidation peak at ~1.2 V (corresponding to the oxidation of the gold working electrode) and the corresponding reduction peak at ~0.4 V. Importantly, only CP20 shows an additional oxidative peak at ~1.4 V. To elucidate the origin of this additional oxidative peak, we compare peak currents at ~1.2 V and ~1.4 V under variable scan rates (50, 100, 150, and 200 mVs−1). The results are shown in Fig. 3b as a Randles-Sevcik plot. While peak current at ~1.2 V increases linearly with scan rate (indicating the diffusion-controlled oxidation of the gold electrode)31,32, the peak current at ~1.4 V disappears at higher scan rates. This difference suggests that the additional peak at ~1.4 V is related to an electron transfer process occurring via surface-adsorbed species32,33. Based on these measurements and the comparison of cyclic voltammetry curves from different proteins34, we attribute this peak to the oxidation of the disulfide bonds in the CP20 structure.

a Cyclic voltammograms of various protein solutions with magnified image in the inset (the curve from CP20 is shifted by +10 µA for better visualization of the peak at 1.4 V); b Randles-Sevcik plot of peaks at 1.2 and 1.4 V; c square-wave voltammograms of pristine and reduced CP20; and d charge transfer resistance (blue dots) and corrosion rates (gray bars) of AH36 mild steel after 4 h exposure to buffer, CP20, TCEP, and reduced CP20 in TBS. Error bars represent the standard deviation of triplicate measurements.
This distinctive feature in the electrochemical signature of CP20 hints at disulfide bonds in the amino acid cysteine to be the direct cause of the accelerated corrosion rate promoted by this protein. To corroborate this hypothesis, we reassess the corrosion rate of our steel samples in the presence of CP20 after reducing all disulfide bonds into free thiols using a solution containing tris(2-carboxyethyl)phosphine (TCEP). Figure 3c shows how the square-wave voltammogram of CP20 changes before and after reduction. The complete absence of the peak at ~1.4 V after exposing CP20 to TCEP confirms the cleavage of disulfide bonds. We then use a solution of CP20 (4.6 µM) and TCEP (in excess) for a 4 h immersion test on AH36 steel sample and compare the resulting corrosion rate to that measured using a bath of buffer, CP20 alone, and pure TCEP solution (used as a control experiment). We compute the corrosion rates from charge transfer resistance values, Rct, obtained from EIS measurements via the relationship detailed in “Methods” section. The results are plotted in Fig. 3d. CP20 yields a corrosion rate of 1.6 mm year−1, which is significantly higher than that recorded for the buffer solution (0.7 mm year−1). We compute the probability of finding such a large difference across three immersion tests performed to yield a p-value of 0.003, indicating that this result is statistically significant. By contrast, the corrosion rate drops to ~0.2 mm year−1 when using reduced CP20. Noteworthily, the addition of TCEP into the buffer solution does not seem to alter the corrosion rate of the steel (~0.7 mm year−1).
The foregoing results confirm that disulfide groups in CP20 are directly responsible for the accelerated corrosion rate associated with CP20. To investigate the relationships between the corrosive and adhesive properties of CP20, we measure the adsorption and desorption of reduced CP20 using QCM. Interestingly, we find no appreciable difference in the adsorption behavior of the reduced and pristine protein (see Fig. 2b). This result suggests that, besides the fraction of free thiols, adsorption is strongly linked to the high surface charge of CP20.
Discussion
PIC has been a long-standing problem in applications, such as biomedical implants35,36,37 and food processing38,39. In the context of biofouling, however, the role of PIC on metal degradation is still under debate7,10. One of the main reasons is that previous investigations could not decouple the effects of proteins from those of the animal (e.g., barnacles) on the corrosion process16,17 (see Fig. 4a).

a Cross section illustration of an adult barnacle, M. rosa, attached to a metal substrate; b Structure of CP20, redrawn from ref. 43; c CP20 diffuses proximally to the metal surface (on the left) and adsorbs strongly on the mild steel via its thiol functional groups (colored in red) and/or the positively charged patches on its surface (colored in blue). Redox-active disulfide bonds (colored in green) are also present in the vicinity of the adsorption site; and d CP20 drives metal–protein complexation, as shown in UV absorption studies, via the favorable redox reaction involving the reduction of disulfide bonds and oxidation of Fe, resulting in PIC. The autocatalytic nature of pitting accelerates the pitting corrosion at this site, accumulating corrosion products in the form of ferric or ferrous hydroxides. Inset: amino acid sequence for structure in b, with connected cysteine residues (C, in green) representing disulfide bonds.
Our study unequivocally indicates that metal degradation can be directly induced by cement proteins even in the absence of the animal. The time-resolved study presented in Fig. 1 is a simple yet effective method to isolate the contribution of CP20 to the corrosion of mild steel. The results show that PIC is localized to where proteins adhere to the metal surface (Fig. 1b, c), and that corrosion occurs very rapidly after immersion of the metal sample in the protein-rich solution. It is known that the time required for proteins to adhere to metal surfaces is on the order of 10 min28. While our measurements on CP20 confirm these adsorption kinetics (see Fig. 2b), we find evidence that corrosion starts earlier (within 8 min, as shown in Fig. 1a). With time, the extent of corrosion damage increases significantly as more and more corrosion sites appear. However, after 30 min we do not detect the initiation of new corrosion sites. Rather, the ones previously formed grow in size and eventually coalesce, suggesting the establishment of a steady state. This trend is confirmed by the Nyquist plots in Supplementary Fig. 2a, whose capacitive loop increases from t = 0.5–2.5 h and then plateaus at t > 2.5 h. Given that there is an excess of proteins in solution, the lack of new corrosion sites formation cannot be attributed to protein depletion. Instead, we believe that the autocatalytic nature of pitting cathodically protects uncorroded regions of the metal surface40 as the size of the pits keeps increasing.
Not only does CP20 promote rapid, localized corrosion of mild steel; it also yields a significantly higher corrosion rate compared to other known corrosive proteins. The results plotted in Fig. 2a indicate a ~50% higher dissolution of Fe when the sample is immersed in a CP20 solution bath compared to solutions containing BSA and LYZ. Incidentally, BSA and LYZ do not alter the corrosion susceptibility of AH36 steel and produce a total mass loss similar to that of steel immersed in a buffer solution (Fig. 2a). This result is surprising if compared to other works focused on PIC of steel28,41. However, we note that most of these studies used high-alloy steels, which form a protective passive layer on the surface. Indeed, an earlier study by Hedberg et al. reported a lack of corrosion enhancement in pure Fe when exposed to BSA solutions27.
The higher corrosion rate induced by CP20 stems from an intricate corrosion mechanism, which relies on the interplay between its strong adhesive properties and the electrochemically active functional groups contained in the protein. Among all tested proteins, CP20 has the highest adsorption and the lowest desorption (Fig. 2b). Its maximum adsorbed mass as measured by QCM is 11 and 17 times higher than that of LYZ and BSA, respectively. Protein desorption, which is defined as \(D = \% \frac{{{\rm Maximum}\,{\rm mass}} - {{\rm Final}\,{\rm mass}}}{{\rm Maximum}\,{\rm mass}}\), measures the irreversibility of the adsorption process. In other words, D provides a means to quantify the likelihood of an adsorbed protein to desorb. CP20 shows the lowest desorption (8%) compared to BSA (26%) and LYZ (89%). These results demonstrate the high adhesive power of CP20, which had only been postulated previously20.
Several studies have suggested a connection between PIC and protein adhesive strength28,42. However, this connection has not been proven conclusively. Our comparison between pristine and reduced CP20 shows that, although protein adhesion is required to trigger PIC (which is in agreement with previous literature6,7,8,9,10), it is not the cause of it. Indeed, the value of D that we measure after reduction of CP20 is 9%, just 1% higher than that of pristine CP20 (Fig. 2b). Yet, the corrosion rate of steel decreases from 1.6 to 0.2 mm year−1 after the protein is reduced. These findings suggest that adhesion and corrosion are governed by two distinct mechanisms.
In a recent study43, we unveiled the tertiary structure of CP20 via multidimensional NMR measurements and showed that 12 out of the 32 cysteine molecules contribute to stabilizing the protein’s structure by forming disulfide bonds (see Fig. 4b). Conversely, 20 cysteine residues remain as free thiols. Besides these free thiols, we also found positively charged pockets, which cover the protein surface and are thus in direct contact with the substrate to which it adsorbs. It is noteworthy that these charges remain even upon protein reduction44. Thus, we believe that both free thiols and positively charged pockets45 are directly responsible for the low D associated with both pristine and reduced CP20 on the negatively charged metal substrate, as shown in Fig. 4c.
The corrosive behavior of CP20 comes instead from the cleavage of the electrochemically active disulfide groups, which drives the oxidation of Fe at the metal surface, as shown in Fig. 4d. The standard electrode potential for the half-reaction of Fe reduction is −0.44 V, while that associated with the reduction of disulfide bonds is −0.23 V (refs. 46,47). Accordingly, disulfide bonds will spontaneously oxidize the metallic Fe found in their proximity into Fe2+. Such reaction can easily occur through the porous native oxide layer on mild steel, which offers little protection against electrolyte seepage48,49,50. These ions can then react with oxygen under neutral or alkaline conditions to form ferric and ferrous hydroxides11 and metal–protein complex in the form of corrosion products35. We confirm the formation of this complex by means of UV spectroscopy (see Supplementary Information, Supplementary Fig. 3). Consequently, these corrosion products lower Fe2+ concentration in solution, shift the dynamic equilibrium of the Fe oxidation reaction toward the Fe2+ species, and increase the corrosion rate of the metal substrate. We believe that the above sequence of events promotes the initiation of corrosion pits on the metal substrate, which cascades due to the autocatalytic nature of pitting and leads to the accumulation of corrosion products on the surface.
When combining CP20 with TCEP, we cleave all disulfide bonds in the protein. As a result, reduced CP20 lacks the capacity of oxidizing Fe and thus yields a corrosion rate (~0.2 mm yr−1) that is even lower than that recorded for the buffer solution (~0.6 mm year−1). This result confirms that the corrosion behavior of CP20 stems from the protein’s redox-active cysteine groups. Moreover, the surprising low corrosion rate of reduced CP20 suggests that cleaving disulfide bonds may even inhibit the corrosion of mild steel in seawater. We attribute this phenomenon to the retained adhesive power of reduced CP20, which may cover the metal surface45,51 and act as a protective film.
Cysteines are known to exhibit redox activity in other biological adhesive proteins, such as those contained in mussel adhesive complex52. In such systems, however, disulfide bond formation serves as the oxidation-resistant group. By contrast, our work indicates that cysteine-rich proteins can exhibit strong corrosion inducing activities—a mechanism that could also be relevant for extracellular proteins of biofilms secreted by bacteria in the context of microbially induced corrosion53,54.
Our study provides a new perspective on the prevention and mitigation of biocorrosion in marine applications, which currently rely on cost- and labor-intensive procedures, such as frequent scraping of metallic structures or regular assessment of protective coatings. For instance, the accelerated corrosion rates we measure may be used to quantify and predict the onset of PIC during the early stages of biofouling of metal structures used in marine applications. Moreover, our results suggest that cysteine-rich proteins in the reduced state (i.e., containing only free thiols) may be used as protective thin films against biocorrosion. Finally, the electrochemical corrosion mechanism unveiled here may be relevant to biomedical implants, since cysteine-rich proteins stabilized by disulfide bonds are present in plasma and could exhibit similar electrochemical activity toward metallic surfaces41.
Methods
Sample preparation
We acquire slabs of AH36 steel from JFE steel corporation through Sembcorp Marine, Singapore. The chemical composition of the AH36 steel is provided in Supplementary Table 1. We machine 10 × 10 × 2 mm coupons from the slabs using wire electrical discharge machining. We embed the coupons in high-edge retention, two-part, cold mount epoxy (Uni lab & Co. Gmbh) so that only the top 10 × 10 mm surface is exposed. We mechanically grind these samples using a sequence of grit paper and then polish them to a mirror-like finish, using a colloidal silica suspension (Ø = 0.04 µm) to mitigate any effects of surface roughness on protein adsorption55,56. We perform microstructural analysis of the steel samples by means of X-ray diffraction (XRD) and electron backscatter diffraction (EBSD) using a JOEL JSM7600F scanning electron microscope. These measurements show that the samples consist of ferrite, exhibit a weak crystallographic texture, and average grain size of 10 µm (see Supplementary Information, Supplementary Fig. 4).
Preparation of protein (CP20)-rich solution
A low-copy kanamycin-resistant pJ431 vector with cloned cDNA of CP20 (Uniprot Q9GRC4) and C terminal his-tag was acquired from DNA 2.0 (Newark, California) and subsequently transformed into BL21 (DE3) competent Escherichia coli cells. A preculture was prepared by inoculating a single colony of transformants in Luria–Bertani (LB) medium with 50 μg mL−1 kanamycin at 37 °C overnight. Recombinant expression of CP20 was initiated by adding appropriate volume of preculture to 1 L LB (kanamycin) medium at 37 °C and shaken for 3–4 h to reach mid-log phase. Afterward, isopropyl thio-β-D-galactoside (1 mM) was added for induction and the cells were cultivated at 20 °C for 16 h. The cells were then pelleted at 8 K r.p.m. for 15 min and broken down by sonication in binding buffer (50 mM Tris, 300 mM NaCl, 5 mM 2-mercaptoethanol, 10 mM imidazole at pH 8.3) on ice. The crude lysate was then centrifuged at 18 K r.p.m. for 60 min at 4 °C. CP20 was then purified from supernatant using affinity chromatography (Ni-NTA affinity column) using binding buffer and elution buffer (500 mM imidazole in 50 mM Tris, 500 mM NaCl, 5 mM 2-mercaptoethanol pH 8.3). The eluted CP20 was further purified to homogeneity using size-exclusion chromatography (Superdex 75, 16/600), using isocratic elution in 20 mM Tris, 150 mM NaCl, at pH 8.3 and concentrated using Vivaspin column (M.W. cutoff 5 kDa).
The protein-rich solution is then diluted to 4.6 µM for PIC tests, in a TBS solution adjusted to 7.6 pH. The TBS solution contains 20 mM of Tris(hydroxymethyl) aminomethane (Thermo Fisher Scientific, Invitrogen) and 150 mM of NaCl (Sigma-Aldrich) in ultrapure water (18.2 MΩ cm, MilliporeTM). We determine protein concentration using a NanoDropTM 2000c Spectrophotometer (Thermo Fisher Scientific). We confirm Nanodrop measurements using the Bradford method57. We acquire BSA and LYZ from Sigma-Aldrich and USB Corporation, respectively.
Corrosion test and damage quantification
We study the corrosion behavior of AH36 steel in protein solutions using classical immersion test. We place the samples in glass beakers, add 3 mL of protein solution, and place them on a circulating orbital shaker. We then collect the protein solution for analysis at different intervals in time and wash the samples with DI water. After drying the samples, we dissolve the corrosion product on the sample surface in 1% HCl solution under ultrasonication. We add hexamethylenetetramine as a corrosion inhibitor during the dissolution of corrosion products to curb additional Fe dissolution, which could otherwise yield overestimates in the measurements of Fe mass loss58. All corrosion tests were repeated at least three times. We quantify corrosion damage in four different ways: (1) by measuring Fe mass loss via ICP-MS (7700 series, Agilent Technologies); (2) by measuring the pitting area fraction over the sample surface; and (3) by computing the charge transfer resistance (Rct) of material via EIS; and (4) by measuring the corrosion current (icorr) via LPR.
We prepare the samples for mass spectroscopy analysis by dissolving Fe precipitates in 2 mL of nitric acid (37 wt.%), filtered through a 0.2 µm membrane. This filtrate is then diluted to 100 mL. To determine the percentage of surface area pitted, we first remove the corrosion products and binarize photographs of the sample surface using ImageJ (1.48 v). The image processing method we employ is similar to those previously presented in the literature59,60,61.
The electrochemical experiments are carried out using an in-house setup (refer to Supplementary Information, Supplementary Fig. 5). The setup consists of connecting the back of the epoxy-embedded sample to an electrode via an insulated electrical wire. Subsequently, a hollow glass cylinder is affixed on a plastic plate with epoxy adhesive. A hole then is made on the plastic plate to thread the wire through. The setup is placed on an orbital shaker. For each experiment, 3 mL of solution is introduced in the glass and left standing for 4 h. The tests are performed using a three-electrode cell equipped with an Ag/AgCl reference electrode. We use a twisted platinum-coated titanium wire as a counter electrode and the metal sample as the working electrode.
For the EIS measurements, we set the amplitude of the AC signal perturbation to 10 mV within the frequency range of 100 kHz–30 mHz. The LPR measurements are conducted within ±20 mV of open-circuit potential at a scan rate of 167 mV s−1. All measurements are repeated three times at 296 K in air. Based on the features observed in the Bode plots (not shown here) and the single loop (i.e., single time constant) seen in Nyquist curves, we select the modified Randles-CPE equivalent circuit (EC)29,62 as a fitting model for all measurements. EIS and LPR measurements are carried out using a Biologic potentiostat interfaced with analysis software (EC-Lab® v.11.20). By combining Eq. (1) (ref. 63) and Eq. (2) (ref. 64), we arrive at the relationship that corrosion rate is related to charge transfer resistance, as expressed in Eq. (3).
Here, Rct is the charge transfer resistance, CR is the corrosion rate in mm year−1, R is the gas constant, T is temperature, K is a constant (3272)65, Ew is the equivalent weight of steel, F is the Faraday’s constant, and ρ is the density of Fe.
Quartz crystal microbalance (QCM)
We employ 5-MHz piezoelectric quartz crystals sensors (Q-Sense) coated with Fe (QSX 319 sensors). Prior to the measurement, we prepare the sensor surface by immersion in a 1% HellmanexTM III solution at room temperature for 3 h, followed by rinsing with ultrapure DI water, drying with nitrogen gas, sonicating in 99% ethanol for 10 min, rinsing with ultrapure water, and drying with nitrogen gas (adapted from Q-sense protocol C-II). We mount the sensors into the Q-Sense E4 system and keep them at 296 K. All measurements are run in duplicates. After measuring the fundamental frequency, TBS is pumped into the flow cell. After attaining a stable resonant frequency, we inoculate the proteins (250 µL, 4.6 µM) into the flow cell at a constant flow rate of 100 μL min−1 using a peristaltic pump (Ismatec). Subsequently, protein desorption is studied under buffer flow. This step is continued until stable frequencies are attained. We clean the setup after each measurement to avoid cross-contamination by pumping through 2% Hellmanex III solution (20 mL) followed by ultrapure water (100 mL).
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Natalio, F. et al. Vanadium pentoxide nanoparticles mimic vanadium haloperoxidases and thwart biofilm formation. Nat. Nanotechnol. 7, 530 (2012).
Natarajan, K. A. In Biotechnology of Metals (ed. K. A. Natarajan) 355–393 (Elsevier, 2018).
Tesler, A. B. et al. Extremely durable biofouling-resistant metallic surfaces based on electrodeposited nanoporous tungstite films on steel. Nat. Commun. 6, 8649 (2015).
Schultz, M. P., Bendick, J. A., Holm, E. R. & Hertel, W. M. Economic impact of biofouling on a naval surface ship. Biofouling 27, 87–98 (2011).
Munk, T., Kane, D. & Yebra, D. M. In Advances in Marine Antifouling Coatings and Technologies (eds Claire Hellio & Diego Yebra) 148–176 (Woodhead Publishing, 2009).
LaQue, F. L. & Clapp, W. F. Relationships between corrosion and fouling of copper-nickel alloys in sea water. Trans. Electrochem. Soc. 87, 103–125 (1945).
Eashwar, M., Subramaniam, G., Chandrasekaran, P. & Balakrishnan, K. Mechanism for barnacle-induced crevice corrosion in stainless steel. Corrosion 48, 608–612 (1992).
Palanichamy, S., Subramanian, G. & Eashwar, M. Corrosion behaviour and biofouling characteristics of structural steel in the coastal waters of the Gulf of Mannar (Bay of Bengal), India. Biofouling 28, 441–451 (2012).
Blackwood, D. J., Lim, C. S., Teo, S. L. M., Hu, X. & Pang, J. Macrofouling induced localized corrosion of stainless steel in Singapore seawater. Corros. Sci. 129, 152–160 (2017).
Shide, M. The corrosive effect of barnacles on low alloy steels. Chin. J. Oceanol. Limn. 7, 271–273 (1989).
Coetser, S. E. & Cloete, T. E. Biofouling and biocorrosion in industrial water systems. Crit. Rev. Microbiol. 31, 213–232 (2005).
Abraham, G. J., Kain, V. & Dey, G. K. In Materials Performance Vol. 48, 64–69 (NACE, 2009).
Srivastava, S. K. & Katarki, M. V. In Materials Performance Vol. 47, 52–56 (NACE International, Houston, 2008).
Terlizzi, A. & Faimali, M. In Biofouling (eds Simone, D. & Jeremy, C. T.) 170–184 (Blackwell Publishing Ltd, 2009).
Kamino, K. Mini-review: barnacle adhesives and adhesion. Biofouling 29, 735–749 (2013).
Luciana, V. Rd. M., Leila, Y. R., Lucio, S. & Ricardo, C. Evaluation of biocorrosion on stainless steels using laboratory-reared barnacle Amphibalanus amphitrite. Anti-Corros. Methods Mater. 61, 402–408 (2014).
Sangeetha, R., Kumar, R., Doble, M. & Venkatesan, R. Barnacle cement: an etchant for stainless steel 316L? Colloids Surf. B 79, 524–530 (2010).
So, C. R. et al. Self-assembly of protein nanofibrils orchestrates calcite step movement through selective nonchiral interactions. ACS Nano 9, 5782–5791 (2015).
So, C. R. et al. Molecular recognition of structures is key in the polymerization of patterned barnacle adhesive sequences. ACS Nano 13, 5172–5183 (2019).
Kamino, K. Novel barnacle underwater adhesive protein is a charged amino acid-rich protein constituted by a Cys-rich repetitive sequence. Biochem. J. 356, 503–507 (2001).
Qin, Q., Tian, M., Pu, J. & Huang, J. Constitutive investigation based on the time-hardening model for AH36 material in continuous casting process. Adv. Mech. Eng. 10 (2018).
ASTM Standard A131/A131M. Standard Specification for Structural Steel for Ships, pp. 1–7 (ASTM International, 2014).
Dickinson, G. H. et al. Barnacle cement: a polymerization model based on evolutionary concepts. J. Exp. Biol. 212, 3499–3510 (2009).
Jansen, R. J. J. & van Bekkum, H. XPS of nitrogen-containing functional groups on activated carbon. Carbon 33, 1021–1027 (1995).
Gromiha, M. M. In Protein Bioinformatics (ed M. Michael Gromiha) 1–27 (Academic, 2010).
Wagener, V., Faltz, A.-S., Killian, M. S., Schmuki, P. & Virtanen, S. Protein interactions with corroding metal surfaces: comparison of Mg and Fe. Faraday Discuss. 180, 347–360 (2015).
Hedberg, Y. et al. Surface-protein interactions on different stainless steel grades: effects of protein adsorption, surface changes and metal release. J. Mater. Sci. Mater. Med 24, 1015–1033 (2013).
Lundin, M. et al. Adsorption and protein-induced metal release from chromium metal and stainless steel. J. Colloid Interface Sci. 366, 155–164 (2012).
Karimi, S., Nickchi, T. & Alfantazi, A. Effects of bovine serum albumin on the corrosion behaviour of AISI 316L, Co–28Cr–6Mo, and Ti–6Al–4V alloys in phosphate buffered saline solutions. Corros. Sci. 53, 3262–3272 (2011).
Scholz, F., Bard, A. J. & Inzelt, G. Electrochemical Dictionary 559 (Springer, Berlin, Heidelberg, 2008).
Jia, H. et al. Electrospun kraft lignin/cellulose acetate-derived nanocarbon network as an anode for high-performance sodium-ion batteries. ACS Appl. Mater. Interfaces 10, 44368–44375 (2018).
Elgrishi, N. et al. A practical beginner’s guide to cyclic voltammetry. J. Chem. Educ. 95, 197–206 (2018).
Zong, W. et al. Cyclic voltammetry: a new strategy for the evaluation of oxidative damage to bovine insulin. Protein Sci. 19, 263–268 (2010).
He, L.-S., Zhang, G. & Qian, P.-Y. Characterization of two 20kDa-cement protein (cp20k) homologues in Amphibalanus amphitrite. PLOS ONE 8, e64130 (2013).
Hedberg, Y. S. Role of proteins in the degradation of relatively inert alloys in the human body. npj Mater. Degrad. 2, 26 (2018).
Talha, M., Ma, Y., Kumar, P., Lin, Y. & Singh, A. Role of protein adsorption in the bio corrosion of metallic implants – a review. Colloids Surf. B 176, 494–506 (2019).
Rozali, A. A., Masdek, N. R. N., Murad, M. C., Salleh, Z. & Koay, M. H. The effect of protein concentration on corrosion of nitinol alloy. Materialwiss. Werkstofftech. 49, 489–493 (2018).
Atapour, M. et al. Metal release from stainless steel 316L in whey protein - and simulated milk solutions under static and stirring conditions. Food Control 101, 163–172 (2019).
Jellesen, M. S., Rasmussen, A. A. & Hilbert, L. R. A review of metal release in the food industry. Mater. Corros. 57, 387–393 (2006).
Fontana, M. G. Corrosion engineering (McGraw-Hill, 1986).
Hedberg, Y. et al. Interaction of albumin and fibrinogen with stainless steel: influence of sequential exposure and protein aggregation on metal release and corrosion resistance. Corrosion 73, 1423–1436 (2017).
Hedberg, Y., Hedberg, J., Liu, Y. & Wallinder, I. Complexation- and ligand-induced metal release from 316L particles: importance of particle size and crystallographic structure. Biometals 24, 1099–1114 (2011).
Mohanram, H., Kumar, A., Verma, C. S., Pervushin, K. & Miserez, A. Three-dimensional structure of Megabalanus rosa Cement Protein 20 revealed by multi-dimensional NMR and molecular dynamics simulations. Philos. T. R. Soc. B 374, 20190198 (2019).
Stenesh, J. In Biochemistry (ed J. Stenesh) 27–45 (Springer US, 1998).
Raman, S. et al. Adhesive barnacle peptides exhibit a steric-driven design rule to enhance adhesion between asymmetric surfaces. Colloids Surf. B 152, 42–48 (2017).
Bard, A. J., Parsons, R., Jordan, J., International Union of, P. & Applied, C. Standard Potentials in Aqueous Solution (M. Dekker, 1985).
Yi, M. C. & Khosla, C. Thiol-disulfide exchange reactions in the mammalian extracellular environment. Annu Rev. Chem. Biomol. Eng. 7, 197–222 (2016).
Zhang, Z. et al. Inhibition of carbon steel corrosion in phase-change-materials solution by methionine and proline. Corros. Sci. 111, 675–689 (2016).
El Mendili, Y., Abdelouas, A. & Bardeau, J. F. Insight into the mechanism of carbon steel corrosion under aerobic and anaerobic conditions. Phys. Chem. Chem. Phys. 15, 9197–9204 (2013).
AlAbbas, F. M. et al. Influence of sulfate reducing bacterial biofilm on corrosion behavior of low-alloy, high-strength steel (API-5L X80). Int. Biodeterior. Biodegrad. 78, 34–42 (2013).
Zhang, F. & Pan, J. Recent development of corrosion protection strategy based on mussel adhesive protein. Front. Mater. 6, 1–7 (2019).
Yu, J. et al. Mussel protein adhesion depends on interprotein thiol-mediated redox modulation. Nat. Chem. Biol. 7, 588 (2011).
Kip, N. & van Veen, J. A. The dual role of microbes in corrosion. ISME J. 9, 542–551 (2015).
Dominique, T. & Wolfgang, S. In Corrosion Mechanisms in Theory and Practice, Third Edition Corrosion Technology 737–776 (CRC, 2011).
Rechendorff, K., Hovgaard, M. B., Foss, M., Zhdanov, V. P. & Besenbacher, F. Enhancement of protein adsorption induced by surface roughness. Langmuir 22, 10885–10888 (2006).
Tugba, A., Cansu, C., Ahmet, S. & Seniha, G. F. Which is more effective for protein adsorption: surface roughness, surface wettability or swelling? Case study of polyurethane films prepared from castor oil and poly(ethylene glycol). Polym. Int. 62, 1202–1209 (2013).
Bradford, M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 (1976).
ASTM Standard G1. Standard Practice for Preparing, Cleaning, and Evaluating Corrosion Test Specimens (ASTM International,1999).
Codaro, E. N. et al. An image processing method for morphology characterization and pitting corrosion evaluation. Mater. Sci. Eng. A 334, 298–306 (2002).
Ebrahimi, N., Noël, J. J., Rodríguez, M. A. & Shoesmith, D. W. The self-sustaining propagation of crevice corrosion on the hybrid BC1 Ni–Cr–Mo alloy in hot saline solutions. Corros. Sci. 105, 58–67 (2016).
Paolinelli, L. D., Brown, B., Simison, S. N. & Nesic, S. Inhibition of CO2 corrosion of carbon steel with 1% Cr. Mater. Chem. Phys. 136, 1092–1102 (2012).
Hassani, S. et al. Improving the corrosion and tribocorrosion resistance of Ni–Co nanocrystalline coatings in NaOH solution. Corros. Sci. 51, 2371–2379 (2009).
Denuault, G., Sosna, M. & Williams, K.-J. In Handbook of Electrochemistry (ed. Cynthia, G. Z.) Ch. 11, 431–469 (Elsevier, 2007).
Brossia, S. In Handbook of Environmental Degradation of Materials 3rd edn, (ed, Myer, K.) 27–50 (William Andrew Publishing, 2018).
El-Sayed Shehata, O. Effect of acetamide derivative and its Mn-complex as corrosion inhibitor for mild steel in sulphuric acid. Egypt. J. Chem. 60, 243–259 (2017).
Acknowledgements
This work was supported by the COE RIE 2020 Seed Funding from Nanyang Technological University, by the US Office of Naval Research—Global (ONR-G), grant no. N62909-17-1-2045, and by the Singapore National Research Foundation under the Marine Science Research and Development Program (MSRDP-P29). We give credit to Liu Weiling from the Facility of analysis characterization testing and simulation (FACTS) at NTU for XPS analysis. We thank Talgat Sailov and Enrico Marsili from Singapore Centre for Environmental Life Sciences Engineering at NTU for fluorescence confocal microscopy and discussions on cyclic voltammetric techniques, respectively.
Author information
Authors and Affiliations
Contributions
M.S. and A.M. planned and supervised the project. M.S., A.M, and V.K.M. conceived the experimental design. H.M. expressed and purified CP20 for all experiments. M.B, V.K.M., and R.D.W. performed the mass spectrometry analysis. V.K.M. and A.L. performed and analyzed the EIS measurements. V.K.M. performed and analyzed the corrosion tests, QCM, CV, UV–visible, XRD, and EBSD measurements. V.K.M, H.M., M.S., and A.M contributed to the manuscript writing. All authors contributed to the discussion and manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Murugan, V.K., Mohanram, H., Budanovic, M. et al. Accelerated corrosion of marine-grade steel by a redox-active, cysteine-rich barnacle cement protein. npj Mater Degrad 4, 20 (2020). https://doi.org/10.1038/s41529-020-0124-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-020-0124-z
This article is cited by
-
Barnacle cement protein as an efficient bioinspired corrosion inhibitor
Communications Materials (2024)
-
Microstructure analysis on complex surfaces enables digital quality control of metal parts
npj Computational Materials (2024)
