
Abstract
Previous studies have established that rare biallelic SYNJ1 mutations cause autosomal recessive parkinsonism and Parkinson’s disease (PD). We analyzed 8165 PD cases, 818 early-onset-PD (EOPD, < 50 years) and 70,363 controls. Burden meta-analysis revealed an association between rare nonsynonymous variants and variants with high Combined Annotation-Dependent Depletion score (> 20) in the Sac1 SYNJ1 domain and PD (Pfdr = 0.040). A meta-analysis of EOPD patients demonstrated an association between all rare heterozygous SYNJ1 variants and PD (Pfdr = 0.029).
Similar content being viewed by others


Parkinson’s disease (PD) is a complex condition influenced by multiple genes and environmental factors, with specific single-gene mutations accounting only for 1–2% of cases, excluding GBA11. Early onset of PD (EOPD; < 50 years) represents about 3–14% of all PD cases depending on population2, and has been linked to mutations in several genes (i.e. PRKN, PINK1, DJ-1, LRRK2, SNCA, GBA1)2. Considering variants in these genes only, the frequency of genetically associated PD in early-onset cases reaches up to 20%3,4.
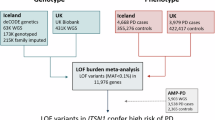
Synaptojanin 1, encoded by SYNJ1, is a lipid phosphatase abundantly expressed in brain tissues5 which plays an important role in synaptic trafficking and in autophagic clearance6,7,8. The protein consists of three functionally distinct domains: Sac1, 5-phosphatase, and proline rich domains (PRD)9. Previous studies suggested that biallelic SYNJ1 mutations cause autosomal recessive, early-onset parkinsonism and EOPD10,11,12,13. In the current study, we sought to examine the role of rare SYNJ1 variants in six non-familial cohorts with a total of 8,165 PD cases (including 818 EOPD) and 70,363 controls (detailed in Supplementary Table 1).
The average coverage of the SYNJ1 gene in the four cohorts sequenced at McGill was > 4000X, with 100% of nucleotides covered at >30X (Supplementary Table 2). From these cohorts, 23–75 rare variants were included in the analysis, depending on the cohort (detailed in Supplementary Table 3). In the AMP-PD cohort, 523 rare variants were included in the analysis and 440 rare variants were included in the UKBB cohort (coding and functional variants detailed in Supplementary Table 4).
Burden analysis with SKAT-O suggested a possible association between all rare variants and PD in the AMP-PD cohort (P = 1.42E-05; Pfdr = 2.98E-04), with nominal associations that did not survive false discovery rate (fdr) correction in the Columbia cohort (P = 0.030; Pfdr = 0.126) and the McGill cohort (P = 0.009; Pfdr = 0.095). No association was found in the meta-analysis of all six cohorts after false discovery rate correction (P = 0.025; Pfdr = 0.131). We also found a nominal association between non-synonymous variants and PD in the Columbia cohort (P = 0.012; Pfdr = 0.084; Table 1).
We then analyzed only EOPD (age at onset < 50 years) and found an association between all rare variants and PD in the AMP-PD cohort (P = 3.48E-05; Pfdr = 7.31E-04) and nominal association that did not survive FDR correction in the McGill cohort (P = 0.027; Pfdr = 0.189). In the meta-analysis, which included only EOPD patients and controls, all rare variants in SYNJ1 were associated with PD (P = 2.80E-03; Pfdr = 0.029). The association between non-synonymous variants and PD was nominally significant in the Columbia cohort (P = 0.044; Pfdr = 0.231) but not in the meta-analysis (Table 1).
To analyze variants within specific functional domains of SYNJ1 (Sac1, 5-phosphatase, PRD), we divided the gene regions into these domains and then repeated SKAT-O analysis. We found an association between the Sac1 domain of SYNJ1 and PD for non-synonymous variants with high Combined Annotation Dependent Depletion (CADD) scores in the UKBB cohort (P = 0.006; Pfdr = 0.082), which did not survive fdr correction. However, this association became significant in the meta-analysis (P = 0.002; Pfdr = 0.04; Supplementary Table 5). The association with the Sac1 domain of SYNJ1 was nominally significant in the EOPD group (P = 0.02; Pfdr=0.37) and remained significant in the group with PD onset after 50 years (P = 0.001; Pfdr = 0.04). We observed that this association was primary driven by the p.A195T SYNJ1 variant in the UKBB cohort (Odds ratio (OR) = 4.87; 95% confidence interval (CI) 1.76–13.46, P = 0.002, with a minor allele frequency (MAF) in cases of 0.001 and 0.0002 in both UKBB controls and gnomAD population of European ancestry). This variant is classified as a Variant of Uncertain Significance (VUS) in ClinVar and is likely benign according to the ACMG classification but is noted for its high CADD score. This variant was also detected in one patient from the Pavlov and Human Brain cohort and was not reported in any other studied cohorts.
We did not find any homozygous or compound heterozygous carriers of pathogenic variants (previously reported in association with PD11, defined as pathogenic or likely pathogenic by ClinVar or variants with CADD score > 20) and deletions in none of the studied cohorts.
In the current study, we found an association between all rare heterozygous variants in SYNJ1 and heterozygous variants with high CADD score in the Sac1 domain of SYNJ1, and the risk of PD in some of the analyzed cohorts. These findings suggest that SYNJ1 could be associated with sporadic PD. Additional studies are required to determine whether this potential association holds in other cohorts. The association we found of rare heterozygous SYNJ1 variants with EOPD was more convincing, yet here too, additional studies are needed. We did not identify biallelic carriers in our analysis, although private bi-allelic SYNJ1 variants were previously associated with EOPD and atypical parkinsonism (detailed in ref. 11),
Multiple genes that are involved in the autophagy-lysosomal pathway are also associated with PD14. From the biological point of view, SYNJ1 has a role in two pathways relevant to PD: synaptic trafficking and autophagic clearance7,8. Recent functional studies demonstrated that mutations in SYNJ1 destabilize dopaminergic neurons potentially due to defective clathrin uncoating, disrupting lipid metabolism and synaptic function15,16. However, SYNJ1 overexpression can counteract these effects, highlighting its potential therapeutic potential in PD15,16.
Our study has several limitations. In some of our cohorts, we had significant differences in sex and age between PD patients and controls. We adjusted for the effects of these covariates in our analysis to address this limitation. Additionally, in our study we predominantly included participants with European ancestry. Furthermore, we used different types of sequencing data and quality control procedures were performed independently for each of the cohorts. Thus, it could potentially create differences in variant enrichment between cohorts. To partially overcome this limitation, we analyzed each cohort separately and conducted meta-analyses of the different cohorts, rather than joint analysis of all cohorts. Finally, due to the limitations of the SKAT-O meta-analysis method, we were unable to assess heterogeneity across cohorts, which may influence the interpretation of our meta-analysis results.
To conclude, we found that rare heterozygous SYNJ1 variants were potentially associated with EOPD and variants in the Sac1 domain are associated with sporadic PD. Larger studies in cohorts of different ethnic backgrounds are needed to replicate our results.
Methods
Population
The study population comprised 8165 PD patients and 70,363 controls from six cohorts, including 818 EOPD (all demographic characteristics detailed in Supplementary Table 1). Four cohorts were collected at McGill university: (1) a cohort of French/French-Canadian from Quebec, Canada and Montpellier, France, (2) a cohort from Columbia University (New York, NY), (3) a cohort from Sheba Medical Center (Israel) and (4) a cohort from Pavlov and Human Brain institutes (Russia)17. The Columbia cohort comprises patients and controls of varied racial and ethnic origin (European, Ashkenazi [AJ] descent and a minority of Hispanics and blacks). Patients and controls in the Sheba cohort, which was recruited in Israel, are of full AJ ancestry (all four grandparents are AJ). The Pavlov and Human Brain institute cohort was collected from the North-Western region of Russia and mainly with East-European ancestry. Additionally, we performed the analysis in the Accelerating Medicines Partnership – Parkinson Disease (AMP-PD) initiative cohorts (https://amp-pd.org/; detailed in the Acknowledgment) and the UK biobank (UKBB) cohort. We only included participants of European ancestry from both cohorts, and we excluded any first and second-degree relatives from the analysis. All PD patients were diagnosed by movement disorder specialists according to the UK brain bank criteria18 or the MDS clinical diagnostic criteria19. The ethics committee of McGill University gave ethical approval for this work.
Standard protocol approvals, registrations, and patient consents
All local IRBs approved the protocols and informed consent was obtained from all individual participants before entering the study.
Targeted next-generation sequencing by molecular inversion probes
The entire coding sequence of the SYNJ1 gene, including exon-intron boundaries (±50 bps) and the 5′ and 3′ untranslated regions (UTRs), was targeted using molecular inversion probes (MIPs) as described earlier20. The full protocol is available at https://github.com/gan-orlab/MIP_protocol. The Genome Quebec Innovation Centre’s Illumina NovaSeq 6000 SP PE100 platform was used to sequence the library. Alignment was carried out to hg19 reference genome21 with coordinates for SYNJ1 chr21:34,001,069-34,100,351. Genome Analysis Toolkit (GATK, v3.8) was used for post-alignment quality checking and variant calling22. We applied standard quality control procedures as described before23. In brief, using the PLINK program version 1.9 and GATK, v3.8, we carried out quality control by eliminating variants and samples with poor quality. SNPs and samples with genotyping rate lower than 90% were excluded. The analyses only included variants with 30x minimal depths of coverage, having a MAF less than 1% and a minimum quality score (GQ) of 30.
Detection of copy-number variations (CNVs)
To detect copy-number variations (CNVs) from MIPs, we utilized the ExomeDepth R package24, which has been validated for CNV detection in MIPs25. Briefly, ExomeDepth selects reference samples that show high correlation with the test samples and employs a hidden Markov model to call CNVs. We applied quality control measures to our MIPs library, excluding probes with less than 100x coverage and genes where over 10% of probes had insufficient coverage. Additionally, we removed low-quality samples with less than 50x coverage. CNVs in SYNJ1 were called following parameters from previous MIPs analyses25, including GC correction and a transition probability of 1e-06. The reference set consisted solely of control samples, and test samples were excluded when calling CNVs in the control group.
Whole-exome and whole-genome sequencing data
The genetic data in AMP-PD and UKBB were aligned to the human reference genome hg38 and we used the appropriate coordinates to extract the SYNJ1 data (chr21:32,628,759-32,728,039). Quality control procedures were performed as previously described in detail for AMP-PD26 and UKBB27 cohorts.
Statistical analysis
To analyze rare variants (MAF < 0.01), we applied the optimized sequence Kernel association test (SKAT-O, R package)28 in each cohort separately, followed by meta-analysis using the metaSKAT package29. We performed separate analyses for all rare variants, non-synonymous variants, and variants with high pathogenicity scores of ≥ 20 (CADD v1.6)30. For domain-based analysis, domains boundaries were decided by the widest intervals of each domain based on a combination of estimates from publicly available domain annotation resources. These resources are SuperFamily, Pfam, Smart, Gene3D, PANTHER, Conserved Domains Database, and PROSITE31,32,33,34. We adjusted for sex and age in all analyses. For the Colombia cohort specifically, we also adjusted for ethnicity, as this cohort includes individuals of European, AJ, Hispanic, and Black descent. FDR correction was applied to all p-values.
Data availability
Data used in the preparation of this article were obtained from the AMP PD Knowledge Platform (https://www.amp-pd.org) and UKBB. Access to these datasets is available for eligible researchers upon request. The code utilized in this study is accessible at https://github.com/gan-orlab/SYNJ1. We provided the variants used for burden analyses in the supplementary tables.
References
Blauwendraat, C., Nalls, M. A. & Singleton, A. B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178 (2020).
Riboldi, G. M., Frattini, E., Monfrini, E., Frucht, S. J. & Di Fonzo, A. A Practical Approach to Early-Onset Parkinsonism. J. Parkinson’s Dis. 12, 1–26 (2022).
Alcalay, R. N. et al. Frequency of Known Mutations in Early-Onset Parkinson Disease: Implication for Genetic Counseling: The Consortium on Risk for Early Onset Parkinson Disease Study. Arch. Neurol. 67, 1116–1122 (2010).
Westenberger, A. et al. Relevance of genetic testing in the gene-targeted trial era: the Rostock Parkinson’s disease study. Brain 147, 2652–2667 (2024).
Miranda, A. M. et al. Excess synaptojanin 1 contributes to place cell dysfunction and memory deficits in the aging hippocampus in three types of Alzheimer’s disease. Cell Rep. 23, 2967–2975 (2018).
Fasano, D. et al. Alteration of endosomal trafficking is associated with early-onset parkinsonism caused by SYNJ1 mutations. Cell Death Dis. 9, 385 (2018).
Choudhry, H., Aggarwal, M. & Pan, P.-Y. Mini-review: Synaptojanin 1 and its implications in membrane trafficking. Neurosci. Lett. 765, 136288 (2021).
George, A. A. et al. Synaptojanin 1 is required for endolysosomal trafficking of synaptic proteins in cone photoreceptor inner segments. PloS one 9, e84394 (2014).
Chang-Ileto, B. & Di Paolo, G. In Encyclopedia of Neuroscience (ed L. R. Squire) 809-814 (Academic Press, 2009).
Olgiati, S. et al. PARK20 caused by SYNJ1 homozygous Arg258Gln mutation in a new Italian family. neurogenetics 15, 183–188 (2014).
Lesage, S. et al. Clinical Variability of SYNJ1-Associated Early-Onset Parkinsonism. Front. Neurol. 12, 648457 (2021).
Xie, F. et al. A novel homozygous SYNJ1 mutation in two siblings with typical Parkinson’s disease. Parkinsonism Relat. Disord. 69, 134–137 (2019).
Kumar, S. et al. Novel and reported variants in Parkinson’s disease genes confer high disease burden among Indians. Parkinsonism Relat. Disord. 78, 46–52 (2020).
Senkevich, K. & Gan-Or, Z. Autophagy lysosomal pathway dysfunction in Parkinson’s disease; evidence from human genetics. Parkinsonism Relat. Disord. 73, 60–71 (2020).
Jacquemyn, J. et al. Parkinsonism mutations in DNAJC6 cause lipid defects and neurodegeneration that are rescued by Synj1. npj Parkinson’s Dis. 9, 19 (2023).
Ng, X. Y. et al. Mutations in Parkinsonism-linked endocytic proteins synaptojanin1 and auxilin have synergistic effects on dopaminergic axonal pathology. npj Parkinson’s Dis. 9, 26 (2023).
Gan-Or, Z. et al. The Quebec Parkinson Network: A Researcher-Patient Matching Platform and Multimodal Biorepository. J. Parkinsons Dis. 10, 301–313 (2020).
Hughes, A. J., Ben-Shlomo, Y., Daniel, S. E. & Lees, A. J. What features improve the accuracy of clinical diagnosis in Parkinson’s disease. A clinicopathologic study 42, 1142–1142 (1992).
Postuma, R. B. et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 30, 1591–1601 (2015).
Rudakou, U. et al. Targeted sequencing of Parkinson’s disease loci genes highlights SYT11, FGF20 and other associations. Brain 144, 462–472 (2021).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Bencheikh, B. O. A. et al. Variants in the Niemann-Pick type C gene NPC1 are not associated with Parkinson’s disease. Neurobiol. Aging 93, 143.e1–143.e4 (2020).
Plagnol, V. et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 28, 2747–2754 (2012).
Yu, E. et al. Analysis of Heterozygous PRKN Variants and Copy-Number Variations in Parkinson’s Disease. Mov. Disord. 36, 178–187 (2021).
Iwaki, H. et al. Accelerating medicines partnership: Parkinson’s disease. genetic resource. Mov. Disord. 36, 1795–1804 (2021).
Carson, A. R. et al. Effective filtering strategies to improve data quality from population-based whole exome sequencing studies. BMC Bioinforma. 15, 125 (2014).
Lee, S. et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet 91, 224–237 (2012).
Lee, S., Teslovich, T. M., Boehnke, M. & Lin, X. General framework for meta-analysis of rare variants in sequencing association studies. Am. J. Hum. Genet 93, 42–53 (2013).
Rentzsch, P., Schubach, M., Shendure, J. & Kircher, M. CADD-Splice-improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med 13, 31 (2021).
Pandurangan, A. P., Stahlhacke, J., Oates, M. E., Smithers, B. & Gough, J. The SUPERFAMILY 2.0 database: a significant proteome update and a new webserver. Nucleic acids Res. 47, D490–D494 (2019).
Mi, H. et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 45, D183–D189 (2017).
Marchler-Bauer, A. et al. CDD: a Conserved Domain Database for protein classification. Nucleic Acids Res. 33, D192–D196 (2005).
Sigrist, C. J. et al. New and continuing developments at PROSITE. Nucleic Acids Res 41, D344–D347 (2013).
Acknowledgements
We would like to thank the participants in the different cohorts for contributing to this study. This research used the NeuroHub infrastructure and was undertaken thanks in part to funding from the Canada First Research Excellence Fund, awarded through the Healthy Brains, Healthy Lives initiative at McGill University, Calcul Québec and Compute Canada. This research has been conducted using the UK Biobank Resource under Application Number 45551. Data used in the preparation of this article were obtained from the Accelerating Medicine Partnership® (AMP®) Parkinson’s Disease (AMP PD) Knowledge Platform. For up-to-date information on the study, visit https://www.amp-pd.org. The AMP® PD program is a public-private partnership managed by the Foundation for the National Institutes of Health and funded by the National Institute of Neurological Disorders and Stroke (NINDS) in partnership with the Aligning Science Across Parkinson’s (ASAP) initiative; Celgene Corporation, a subsidiary of Bristol-Myers Squibb Company; GlaxoSmithKline plc (GSK); The Michael J. Fox Foundation for Parkinson’s Research; Pfizer Inc.; Sanofi US Services Inc.; Verily Life Sciences and AbbVie. ACCELERATING MEDICINES PARTNERSHIP and AMP are registered service marks of the U.S. Department of Health and Human Services. Genetic data used in preparation of this article were obtained from the Fox Investigation for New Discovery of Biomarkers (BioFIND), the Harvard Biomarker Study (HBS), the Parkinson’s Progression Markers Initiative (PPMI), the Parkinson’s Disease Biomarkers Program (PDBP), the International LBD Genomics Consortium (iLBDGC), and the STEADY-PD III Investigators. BioFIND is sponsored by The Michael J. Fox Foundation for Parkinson’s Research (MJFF) with support from the National Institute for Neurological Disorders and Stroke (NINDS). The BioFIND Investigators have not participated in reviewing the data analysis or content of the manuscript. For up-to-date information on the study, visit michaeljfox.org/news/biofind. The Harvard Biomarker Study (HBS) is a collaboration of HBS investigators [full list of HBS investigators found at https://www.bwhparkinsoncenter.org/biobank/ and funded through philanthropy and NIH and Non-NIH funding sources. The HBS Investigators have not participated in reviewing the data analysis or content of the manuscript. PPMI is sponsored by The Michael J. Fox Foundation for Parkinson’s Research and supported by a consortium of scientific partners: [list the full names of all of the PPMI funding partners found at https://www.ppmi-info.org/about-ppmi/who-we-are/study-sponsors]. The PPMI investigators have not participated in reviewing the data analysis or content of the manuscript. For up-to-date information on the study, visit www.ppmi-info.org. The Parkinson’s Disease Biomarker Program (PDBP) consortium is supported by the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health. A full list of PDBP investigators can be found at https://pdbp.ninds.nih.gov/policy. The PDBP investigators have not participated in reviewing the data analysis or content of the manuscript. The Study of Isradipine as a Disease-modifying Agent in Subjects With Early Parkinson Disease, Phase 3 (STEADY-PD3) is funded by the National Institute of Neurological Disorders and Stroke (NINDS) at the National Institutes of Health with support from The Michael J. Fox Foundation and the Parkinson Study Group. For additional study information, visit https://clinicaltrials.gov/ct2/show/study/NCT02168842. The STEADY-PD3 investigators have not participated in reviewing the data analysis or content of the manuscript. Genome sequence data for the Lewy body dementia case-control cohort were generated at the Intramural Research Program of the U.S. National Institutes of Health. The study was supported in part by the National Institute on Aging (program #: 1ZIAAG000935) and the National Institute of Neurological Disorders and Stroke (program #: 1ZIANS003154). ZGO is supported by the Fonds de recherche du Québec - Santé (FRQS) Chercheurs-boursiers award, and is a William Dawson Scholar. The access to part of the participants for this research has been made possible thanks to the Quebec Parkinson’s Network (http://rpq-qpn.ca/en/). K.S. is supported by Parkinson Canada Movement Disorders clinical fellowship. This work was financially supported by grants from the Michael J. Fox Foundation, the Canadian Consortium on Neurodegeneration in Aging (CCNA), the Canada First Research Excellence Fund (CFREF), awarded to McGill University for the Healthy Brains for Healthy Lives initiative (HBHL). The Columbia University cohort is supported by the Parkinson’s Foundation, the National Institutes of Health (K02NS080915, and UL1 TR000040) and the Brookdale Foundation.
Author information
Authors and Affiliations
Contributions
K.S. was responsible for the design and conceptualization of the study, acquisition, and analysis of data, and drafting or revising the manuscript for intellectual content. Z.G.O. was responsible for the design and conceptualization of the study, acquisition, and analysis of data, and drafting or revising the manuscript for intellectual content. S.C.P., C.C., E.Y., J.A., J.A.R., F.A., D.S., C.W., O.M., Y.D., N.D., I.M., A.T., A.E., S.P., L.G., S.H.B., and R.N.A. were involved in the acquisition and analysis of data and in drafting or revising the manuscript for intellectual content. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
Z.G.O. received consultancy fees from Lysosomal Therapeutics Inc. (LTI), Idorsia, Prevail Therapeutics, Ono Therapeutics, Denali, Handl Therapeutics, Neuron23, Bial Biotech, Bial, UCB, Capsida, Vanqua bio, Congruence Therapeutics, Takeda, Jazz Pharmaceuticals, Guidepoint, Lighthouse and Deerfield.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Senkevich, K., Parlar, S.C., Chantereault, C. et al. Are rare heterozygous SYNJ1 variants associated with Parkinson’s disease?. npj Parkinsons Dis. 10, 201 (2024). https://doi.org/10.1038/s41531-024-00809-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41531-024-00809-9
