
Abstract
Treatment with L-3,4-dihydroxyphenylalanine (L-Dopa) compensates for decreased striatal dopamine (DA) levels and reduces Parkinson’s disease (PD) symptoms. However, during disease progression, L-Dopa-induced dyskinesia (LID) develops virtually in all PD patients, making the control of PD symptoms difficult. Thus, understanding the mechanisms underlying LID and the control of these motor abnormalities is a major issue in the care of PD patients. From experimental and clinical studies, a complex cascade of molecular and cellular events emerges, but the primary determinants of LID are still unclear. Here, with a translational approach, including four animal models and a wide cohort of PD patients, we show that striatal DA denervation is the major causal factor for the emergence of LID, while α-synuclein aggregates do not seem to play a significant role. Our data also support the concept that maladaptive basal ganglia plasticity is the main pathophysiological mechanism underlying LID.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, affecting about 3% of the population over 65 years of age1. The typical hallmarks of this disease are degeneration of dopaminergic neurons in the substantia nigra, causing striatal dopamine (DA) deficiency, and intracellular inclusions containing aggregates of α-synuclein (α-syn)2,3.
The most effective treatment for PD is L-3,4-dihydroxyphenylalanine (L-Dopa), a precursor of DA, which restores the DA levels in the brain. L-Dopa is very efficient during the first few years of administration4. However, as PD progresses, involuntary movements, named L-Dopa-induced dyskinesia (LID), appear. Most PD patients develop LID during the disease, some just after a few years of L-Dopa treatment5,6.
Pathogenetic mechanisms of LID include phasic stimulation of DA receptors, non-physiological L-Dopa-to-DA conversion in serotoninergic neurons, hyperactivity of corticostriatal glutamatergic transmission, and abnormal activities of multiple receptors on DA-releasing axons7,8.
In recent years, several progresses in the knowledge of LID mechanisms led to the identification of many molecular and electrophysiological features. A complex cascade of intracellular events underlies the pathophysiology of LID; among these, aberrant plasticity in the cortico-basal ganglia system plays a key role9. LID is characterized by the loss of the ability to reverse synaptic plasticity and an associated increase in excitatory neuronal inputs to a range of cortical and subcortical brain areas10. This pathological condition has been observed in rodents and non-human primates as well as in dyskinetic PD patients11,12,13,14,15. In particular, control and non-dyskinetic rats show synaptic depotentiation, measured as a reversal of long-term potentiation (LTP) recorded from striatal spiny projection neurons (SPNs) in response to subsequent low-frequency stimulation (LFS). Conversely, dyskinetic rats do not show this form of homeostatic plasticity11. In PD patients with LID, few neurophysiological studies using transcranial magnetic stimulation (TMS) documented alterations in the processes of synaptic plasticity at the level of the primary motor cortex (M1)16,17,18. Interestingly, through a theta burst stimulation (TBS) paradigm, a loss of the ability to display depotentiation as a response to a protocol that reverses the LTP-like plasticity in M1 has been observed in dyskinetic PD patients12. Although the lack of bidirectional control of synaptic plasticity seems to be an interesting pathophysiological factor for developing LID, a direct correlation between impairment of depotentiation in M1 and dyskinesia severity has not been described in humans.
Independent risk factors for developing LID have been analyzed in a large cohort of de-novo PD patients enrolled in the Parkinson’s Progression Markers Initiative (PPMI)19. The following risk factors were identified: cumulative L-Dopa exposure, female gender, the severity of motor and functional impairment, non-tremor dominant clinical phenotype, genetic risk score, anxiety, and marked caudate asymmetric pattern at 123I-FP-CIT (trade name DaTSCAN®) SPECT at baseline19. However, the mechanistic link among these risk factors has not been established. Moreover, in this cohort, total cerebral spinal fluid (CSF) α-syn emerged as a potential interesting factor for LID, suggesting that further investigations are warranted for exploring the role of α-syn in the appearance of LID19.
This complex scenario, emerging from experimental and clinical studies, raises crucial questions. First, among various risk factors, is striatal DA denervation the major determinant of LID or do L-Dopa treatment duration and dosage play a significant role? Second, might α-syn exert a critical role in the emergence of LID? Third, is the lack of depotentiation a key pathophysiological mechanism of LID?
To provide answers to these crucial questions, we designed an experimental approach including four rodent models, two of which were α-syn-based, and a cross-sectional study analyzing clinical, biochemical, and neurophysiological features in a cohort of more than one hundred PD patients.
Results
Different degree of denervation in 6-OHDA-full and -partial models
Significant insight into disease mechanisms in PD has been obtained using an experimental model of complete striatal DA depletion by inducing a full lesion of the medial forebrain bundle (MFB) through a unilateral intracerebral infusion of 6-hydroxydopamine (6-OHDA). To determine the level of dopaminergic neuronal loss in the substantia nigra pars compacta (SNpc) of 6-OHDA models, we performed an unbiased stereology analysis of the Tyrosine Hydroxylase positive (TH+) neurons. In 6-OHDA fully-lesioned animals, in the SNpc of the lesioned side (Lesion), we found an almost complete TH+ neuronal loss (~99%), compared to the unlesioned side (Control) (Fig. 1B). The reduced number of dopaminergic neurons in SNpc was associated with nearly complete degeneration of dopaminergic fibers in the dorsolateral striatum (Fig. 1C). TH densitometric quantification revealed that the full lesion resulted in a massive reduction (~97%) of TH+ fibers compared to the intact side (Fig. 1C).

A Scheme representing the timeline of the experimental procedures. Rats were unilaterally injected in the left hemisphere with 6-OHDA in the MFB. After 2 weeks from the injection, rats were subjected to an apomorphine test (APO-Test). Fully-lesioned (Full) animals were treated for 3 weeks with L-Dopa. Twenty min after the last L-Dopa treatment, rats were sacrificed for electrophysiological and immunohistochemical analysis. B Quantification of the number of TH+ cells in the SNpc after 3 weeks of L-Dopa treatment in the contralateral (Control) and ipsilateral (Lesion) hemispheres to the lesion (n = 4 rats; unpaired t-test Lesion vs. Control; t = 8.029, df = 6, ***P < 0.001). C Optical density (O.D.) of TH+ fibers in the contralateral and ipsilateral striatum after L-Dopa treatment (n = 5 rats; unpaired t-test Lesion vs. Control; t = 24.95, df = 8, ***P < 0.001). D Experimental plan of the procedures. Rats were unilaterally injected in the left hemisphere with 6-OHDA in the MFB. After 2 weeks from the injection, rats were subjected to an apomorphine test. Partially-lesioned (Partial) animals were divided into two groups: the first group was subjected to L-Dopa for 3 weeks, while the second group was treated for 6 weeks. Twenty min after the last L-Dopa administration, animals were sacrificed for electrophysiological and immunohistochemical analysis. E Quantification of the number of TH+ cells in the SNpc after L-Dopa treatment in the contralateral (Control) and ipsilateral (Lesion) hemispheres to the lesion (n = 5 rats; unpaired t-test Lesion vs. Control; t = 9.596, df = 8, ***P < 0.001). F Optical density of TH+ fibers in the contralateral and ipsilateral striatum of the lesion after L-Dopa treatment (n = 4 rats; unpaired t-test Lesion vs. Control; t = 13.93, df = 6, ***P < 0.0001).
Since early clinical symptoms of PD are only detected when nearly 60% of dopaminergic neurons are lost, causing a marked decrease in the striatal DA levels20, we used an experimental paradigm of partial DA depletion obtained after a 6-OHDA injection in MFB lesion21. In these animals, the stereological analysis demonstrated that in the SNpc of the lesioned side (Lesion), the percentage of dopaminergic cell loss was lower than in fully-lesioned animals (Fig. 1E). Compared to the unlesioned side (Control), in the SNpc of the lesioned side we found that >60% of TH+ neurons were lost (64%; Fig. 1E). Similarly, in the striatum of the lesioned side, TH densitometric quantification revealed that the partial lesion resulted in a 72% reduction of TH+ fibers compared to the intact side (Fig. 1F).
A partial dopamine denervation is not associated with an overt dyskinetic phenotype
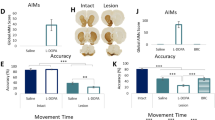
Similarly to PD patients, DA replacement with chronic L-Dopa treatment in the 6-OHDA-Full model leads to the onset of motor side effects and the appearance of a dyskinetic phenotype11,22. As previously reported, abnormal involuntary movements (AIMs) were scored as an index of dyskinetic phenotype starting 20 min after L-Dopa injection (10 mg/kg i.p. twice a day), every 20 min for 180 min on alternate days for the entire treatment schedule (3 weeks)11,23. As already demonstrated, our results indicate that 3 week-treatment with L-Dopa induced a severe dyskinetic behavior in fully-denervated animals (Fig. 2A), and this was evident in all three behavioral components analyzed (Fig. 2A). To investigate the effects of chronic treatment with L-Dopa in animals with a partial DA denervation (6-OHDA-Partial model), 1 month after the apomorphine test, partially-lesioned animals were divided into two groups, and subjected to a different treatment timing: 3 and 6 weeks of L-Dopa (Fig. 1D). AIMs score shows that these two groups of animals develop irrelevant or absent dyskinetic motor effects after both L-Dopa treatments, as shown by the Session time and behavioral components graphs (Fig. 2B and Suppl. Fig. 2A, B). This data reveals that the lack of dyskinetic phenotype in the partially-lesioned group could be attributable to the low degree of denervation.

A Left: AIMs scores analysis of all session times. Right: evaluation of AIMs distinct components, limb, axial and orolingual. B AIMs scores analysis of Partial rats 3 W (dark blue circle) and Partial rats 6 W (light blue circle) treated with L-Dopa (Two-way ANOVA Partial rats 3 W vs. Partial rats 6 W, Interaction: F(8,1197) = 1.170, P > 0.05 Bonferroni’s post-hoc test). Right: evaluation of AIMs distinct components in both experimental groups (unpaired t-test for limb, axial and orolingual of Partial rats 3 W vs. Partial rats 6 W, P > 0.05). C Effect of L-Dopa treatment on corticostriatal LTP and synaptic depotentiation of SPNs. Left: Time course of EPSPs amplitude in response to HFS (n = 6, paired t-test pre- vs. post‐HFS, t = 8.671, df = 9, ***P < 0.001) and LFS (n = 6, paired t-test post‐HFS vs. post‐LFS, t = 1.013, df = 6, P > 0.05) protocols in Full +3W L-Dopa (green circle). Green solid line shows EPSPs amplitude in response to HFS in SPNs of Full model without treatment (C) Right: Time course of EPSPs amplitude in response to HFS (n = 5, paired t-test pre- vs. post‐HFS, t = 12.190, df = 9, ***P < 0.0001) and LFS (n = 5, paired t-test post‐HFS vs. post‐LFS, t = 11.570, df = 7, ###P < 0.0001) protocols. The blue solid line shows EPSP amplitude in response to HFS in SPNs of the Partial model without treatment. Representative traces of EPSPs from SPNs before (baseline) and after HSF and LFS. The scale bar is 5 ms/10 mV for all traces. D Linear regression and correlation analysis between the depotentiation, expressed as % change of EPSP amplitude and AIMs score (Coefficients of correlation: r = 0.892, F (1,8) = 65.88, ***P < 0.001) in 6-OHDA- Full and -Partial Models following a chronic 3 week L-Dopa administration.
This marked dyskinetic phenotype observed in fully-denervated animals was reflected in electrophysiological analysis of corticostriatal plasticity in SPNs, showing that striatal LTP was restored by chronic L-Dopa treatment (Fig. 2C, left panel). After the induction of LTP by high-frequency stimulation (HFS), a LFS was delivered to produce a reversal of synaptic strength to pre-LTP levels. The LFS could not reestablish baseline synaptic responsiveness in dyskinetic animals (Fig. 2C, left panel). Contrary to fully-lesioned rats, exhibiting a complete loss of LTP (Fig. 2C, green solid line), SPNs from partially-lesioned animals show only a short-term potentiation21,24, as reported in Fig. 2C (blue solid line). Interestingly, electrophysiological analysis of corticostriatal plasticity reveals that daily chronic L-Dopa administration fully reverses the synaptic plasticity deficits in this experimental model (Fig. 2C, right panel). In agreement with behavioral data, SPNs showed the capacity to express depotentiation after LFS (Fig. 2C, right panel), similarly to a non-dyskinetic animal.
Accordingly, we found a significant positive correlation between the depotentiation, expressed as % change of EPSP amplitude, and the AIMs score in 6-OHDA animals, Full and Partial (r = 0.892, p = 0.0002). This result highlights that rats presenting a dyskinetic phenotype (6-OHDA-Full Model) are characterized by a neuronal synaptic impairment in the bidirectional plasticity, i.e. the loss of depotentiation, the appearance of LID (Fig. 2D).
Chronic L-Dopa in α-syn-PFF animals is not sufficient to induce dyskinetic behavior
In recent years, the α-synuclein-preformed-fibrils (α-syn-PFF) animal model has emerged as a faithful paradigm to reproduce human PD25. The striatal injection of α-syn-PFF model allows for studying the early stages of the disease with a time-dependent α-syn-PFF spread from the striatum to SNpc25.
In the α-syn-PFF model treated with L-Dopa for 3 weeks (Fig. 3A), we found a lower TH+ neuronal loss compared to fully-lesioned and partially-lesioned animals. Indeed, compared to the SNpc of the unlesioned side (Control), in the SNpc of the lesioned side (Lesion) >35% of TH+ neurons were lost (Fig. 3B). Similarly, in the striatum, TH-immunolabeling was quite diffuse and quantification revealed that in the α-syn-PFF animals, the lesioned side showed a 38% reduction of TH+ fibers compared to the intact side (Fig. 3C), confirming their substantial integrity.

A Scheme representing the experimental setting. Rats were unilaterally injected in the left hemisphere with α-syn-PFFs in the dorsal striatum (dST). After 22 weeks, animals were treated with L-Dopa for 3 weeks. Twenty min after the last L-Dopa administration, animals were sacrificed for immunohistochemical analysis. B Quantification of the number of TH+ cells in the SNpc after 3 weeks of L-Dopa treatment in the contralateral (Control) and ipsilateral (Lesion) hemispheres to the lesion (n = 8 rats; unpaired t-test Lesion vs. Control; t = 3.230, df = 14, **P < 0.01). C Optical density of TH+ fibers in the contralateral and ipsilateral striatum of the lesion after L-Dopa treatment (n = 8 rats; unpaired t-test Lesion vs. Control; t = 10.26, df = 14, ***P < 0.001). D Experimental design and timeline. Rats were injected in dST with the α-syn-PFFs or PBS and into the SNpc with AAV2/6-hα-syn or AAV2/6-synapsin-GFP. At 18- and 33 weeks post-surgery, animals were treated with L-Dopa twice daily for 5 and 3 weeks, respectively. Rats were sacrificed at 36 weeks of surgery for immunohistological analysis. E Left, representative coronal brain images of TH-immunostained SNpc of Control (PBS + AAV2/6-synapsin-GFP-injected) and Lesion (α-syn-PFF + AAV2/6-hα-syn-injected) groups. Right, the stereological cell counts plot shows a significant reduction of SNpc TH+ neurons (n = 4 rats; unpaired t-test Lesion vs. Control; t = 4.339, df = 3.26, *P < 0.05). F Left, coronal brain images of the TH-immunostained dST of Control and α-syn-PFF + AAV-hα-syn injected (Lesion) rats. Scale bar, 50 µm. Right, quantification of TH- immunoreactivity in the dST (n = 4 rats; unpaired t-test Lesion vs. Control; t = 4.793, df = 3.29, *P < 0.05). Data are presented as mean ± SEM of the optical density as a percentage of the control group.
Double lesion with nigral injection of AAV overexpressing α-syn plus striatal injection of PFFs does not favor the induction of dyskinetic behavior by L-Dopa
Preclinical models of PD, combining viral human α-synuclein (AAV-hα-syn) overexpression with the injection of α-syn-PFFs, represent a robust approach for the study of PD related h-α-syn etiology, as they recapitulate some of the pathological hallmarks observed in humans26. Here, we tested the insurgence of LID in this experimental model. To this end, Control and α-syn-PFF + AAV-hα-syn rats were treated with L-Dopa (10 mg/Kg) plus benserazide (6 mg/Kg) i.p. twice a day and AIMs rating was evaluated (Fig. 3D). No significant AIMs rating was scored after 5 weeks of L-Dopa treatment at 23 weeks post-surgery as well as after 3 weeks of L-Dopa treatment at 36 weeks post-surgery. After 36 weeks from surgery, injected rats were evaluated for SNpc dopaminergic neuronal loss by the stereological cell count of TH+ neurons. In the SNpc, the number of TH+ cells was significantly reduced in the α-syn-PFF + AAV-hα-syn rats compared to the Control, with almost 72% spared TH+ neurons (Fig. 3E). Accordingly, in the dorsolateral striatum, the TH-immunoreactivity quantification demonstrated a significant reduction of the TH+ fibers compared to Control (79%) (Fig. 3F).
Reduction in striatal dopamine active transporter (DAT) immunoreactivity is observed in dyskinetic animals
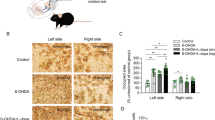
To further explore the correlation between the extent of nigrostriatal degeneration and the emergence of a dyskinetic phenotype, we investigated DAT immunostaining in the dorsolateral striatum of the animals of different PD models used in this study. The densitometric analysis showed a significant reduction of DAT+-immunolabelling in fully-lesioned rats, compared with the intact side (Fig. 4A). This data is in line with a near full TH+ cell loss in the SNpc observed in fully-lesioned rats.

Left, confocal images of coronal brain sections showing DAT-immunostaining in the dST of Full (A), Partial (B), α-syn-PFF (C) and α-syn-PFF + AAV-hα-syn (D) models. Scale bar, 50 µm. Right, quantification of DAT immunoreactivity in the dST of all experimental groups (unpaired t-test Lesion vs. Control, Full (n = 5 rats): t = 22.36, df = 8, ***P < 0.0001, Partial (n = 4 rats): t = 6.619, df = 6, ***P < 0.001, α-syn-PFF (n = 7 rats): t = 5.221, df = 13, ***P < 0.001 and α-syn-PFF + AAV-hα-syn (n = 4 rats): t = 5.80, df = 6, **P < 0.01). Data are presented as mean ± SEM of the optical density as a percentage of the Control. E Linear regression of XY pairs describes the correlation between DAT+ terminals in dST and AIMs score (Coefficients of correlation: 6-OHDA Full Model r = 0.953, F(1,3) = 61.23, **P < 0.01; 6-OHDA Partial Model r = 0.961, F(1,2) = 48.83; *P < 0.05; α-syn-PFF Model r = 0.169, F(1,5) = 1.013 and α-syn-PFF + AAV-hα-syn Model r = 0.856, F(1,2) = 11.88, both P > 0.05).
In the partially-lesioned group, DAT+ terminals were also significantly reduced (Fig. 4B), even to a lesser extent than in the fully-lesioned model.
The decrease in DAT-immunofluorescence was less severe in the PFF than in the 6-OHDA model, with a 38% reduction compared to the intact side (Fig. 4C). This is in line with the moderate dopaminergic cell degeneration induced by α-syn-PFFs injections at this time point.
Notably, striatal DAT-immunofluorescence was markedly reduced in the double α-syn-PFF + AAV-hα-syn rats (Fig. 4D) despite a modest SNpc cell loss.
Finally, we tested whether AIMs expression correlates with the degree of neurodegeneration of dopaminergic terminals. Linear regression and correlation analysis indicated that an inverse correlation exists between the intensity of AIMs and the number of spared DAT+ dopaminergic terminals (as % of Control) only in the fully-lesioned model (Fig. 4E), supporting that the degree of DA denervation is the main determinant of L-Dopa-induced AIMs.
Clinical, molecular, and neurophysiological characterization of PD patients with and without LID
To explore the potential translational implications of these data, we consecutively enrolled 174 patients and collected their clinical-demographical data. Venipuncture for serum collection was performed in 104 patients, and 54 patients underwent lumbar puncture for CSF analysis.
Clinical and demographical data are summarized in Table 1. Overall, patients had a mild parkinsonism (Movement Disorders Society-Unified Parkinson’s Disease Rating Scale, MDS-UPDRS III score of 21 ± 12) with a mean disease duration of 7 ± 5 years and L-Dopa equivalent daily dose (LEDD) intake of 617 ± 412 mg. LID was observed in 78 patients (Dysk), while 96 patients were defined as non-dyskinetic (Non-Dysk). As expected, patients with LID had a longer disease duration, higher LEDD intake, younger age and age at onset and worse MDS-UPDRS III scores.
Uptake values of DaTSCANⓇ were available for 50 patients. DaTSCAN binds with high affinity to the DAT on the presynaptic nigrostriatal terminals. It is the only agent approved by the European Medicines Agency and the Food and Drug Administration for detecting dopaminergic neuron terminal loss in the striatum in patients with parkinsonism. In addition to supporting a clinical diagnosis, semiquantitative analysis of the uptake values has been studied as a disease progression biomarker in PD. Lower uptake values seem to be associated with higher motor-related disability and non-motor symptoms such as cognitive impairment, psychosis, and depression. However, the relationship between DAT binding and PD progression remains unclear27. Interestingly, in our cohort, patients with LID showed higher putaminal dopaminergic denervation at diagnosis, measured with semiquantitative analysis of DaTSCAN uptake values (Fig. 5A, B). This association also remained significant after adjusting for age, LEDD, and disease duration (p = 0.032) in a logistic regression model. Moreover, in a multiple regression model, DaTSCAN uptake values in both putamen and caudate, along with age of onset, significantly predict the amount of LID, quantified with Unified Dyskinesia Rating Scale (UDysRS) score, while disease duration and LEDD do not seem to play a role, as shown in supplementary table (R square 0.79, p = 0.021).

A Example of 123I-FP-CIT (DaTSCANⓇ) SPECT imaging in a patient with PD who developed LID. R indicates the right side of the brain. The bar on the right side shows the color scale used for the uptake values (white 100%, on top, and black 0% on the bottom). In the six cuts, the asymmetrically reduced striatal uptake values are shown. B Differences in the putamen and caudate uptake values in dyskinetic (Dysk) and non-dyskinetic (Non-Dysk) patients (unpaired t-test Non-Dysk vs. Dysk DaTSCAN: Putamen, t = 5.863, df = 48, ***P < 0.001 and Caudate, t = 1.02, df = 48, P > 0.05). C Patients with and without LID share similar values of total-α-syn in serum (unpaired t-test Non-Dysk vs. Dysk, t = 0.271, df = 102, P > 0.05), and CSF (unpaired t-test Non-Dysk vs. Dysk, t = 0.306, df = 52, P > 0.05). D In Non-Dysk patients, cTBSc0 facilitates motor evoked potentials. cTBS150 given at 1 min after cTBSc0 reverses the potentiation following cTBSc0 by returning it to baseline level. Error bars refer to the standard error of the measurements (Two-way ANOVA of the time points after application of cTBS150: F3.93 = 24.74, P < 0.001). MEPs size was assessed after the end of TBS and every 5 min for 20 min after the end of TBS (respectively T1, T2, T3, T4 and T5). E In Dysk patients, cTBSc0 facilitates motor evoked potentials, while cTBS150 given at 1 min after cTBSc0 does not modify the facilitation produced by cTBSc0. Error bars refer to the standard error of the measurements (Two-way ANOVA of the time points after application of cTBS150: F3.93 = 0.006, P = 0.9). MEPs size was assessed after the end of TBS and every 5 min for 20 min after the end of TBS (respectively T1, T2, T3, T4 and T5). F The amount of depotentiation induced with cTBS150 given at 1 min after cTBSc0 (measured as percent change of normalized motor evoked potential -MEP- amplitudes between T1 and time points after cTBS150) inversely correlates to patients’ clinical dyskinesia scores (part III-UDysRS). Patients with more severe dyskinesia (higher scores) undergo lower depotentiation following cTBS150 than patients with less severe dyskinesia (lower scores).
Other factors considered in this study are the serum and CSF levels of α-syn. CSF t-α-syn is measurable with immunoassays with good analytical validity28. In our study, we measured both serum and CSF levels of t-α-syn to explore the possible role of this protein in LID development. However, no differences were found in t-α-syn levels in patients with and without LID in both serum and CSF (Fig. 5C).
A subgroup of 30 patients, 15 without LID and 15 with LID, also underwent neurophysiological examination with TMS. Different TMS protocols have been studied in PD, demonstrating that patients show altered cortical excitability as compared to controls, but also more specific alterations, recently reviewed29, which could be of diagnostic or prognostic utility. Here, we found a significant depotentiation in Non-Dysk patients and a not significant depotentiation in the group of Dysk patients (Fig. 5D, E). The amount of depotentiation was also different between Dysk and Non-Dysk patients when corrected with LEDD and disease duration in a logistic regression model (p = 0.031). Furthermore, we found a significant negative correlation between the amplitude of depotentiation and the score of UDysRS part III in Dysk patients (r = −0.67, p = 0.006), i.e., the less depotentiation, the more severe the LID (Fig. 5F).
Discussion
In the present study, we have taken this approach to provide answers to three key questions that, at present, represent major matters of debate in the perspective of discovering novel therapeutic strategies to alleviate LID. Firstly, we found that striatal DA denervation is a primary cellular and clinical determinant of LID, while the duration and amount of L-Dopa therapy represent secondary factors. Secondly, we observed that, although misfolded α-syn plays a fundamental role in PD pathogenesis and the progression of DA denervation, this protein does not directly influence the induction and maintenance of LID. Thirdly, our preclinical and clinical data show the lack of homeostatic physiological processes, such as synaptic depotentiation, as a key factor coupled with LID occurrence, both at striatal and cortical levels, and this synaptic abnormality correlates with the severity of LID.
Several complex molecular, biochemical, and synaptic changes have been reported in LID, such as the excessive release of DA from serotonergic terminals, supersensitivity of postsynaptic dopaminergic receptors, changes of intracellular signaling in striatal neurons, and abnormal cortico-striatal plasticity11,30. Although these studies provided relevant information concerning LID, they did not clarify, among the various mechanisms, the key cellular and clinical determinants of this phenomenon. So far, insufficient effort has been made to answer key pathophysiological questions utilizing a combined experimental approach, including multiple animal models and a large and well-selected cohort of PD patients.
In the present study, experiments in which DA denervation was achieved with the neurotoxin 6-OHDA clearly show that the appearance of LID is highly dependent on the level of nigrostriatal denervation. Compared to the unlesioned side, in the SNpc of the lesioned side, we found an almost complete dopaminergic cell loss (~99%). The reduced number of dopaminergic neurons in the SNpc was associated with almost complete degeneration of dopaminergic fibers in the dorsolateral striatum, measured with TH-immunolabelling (~97%). In line with our previous findings, these results indicate that a 3 week treatment with L-Dopa induces severe dyskinetic behavior in fully-denervated animals in all three behavioral components analyzed (limb, axial, and orolingual). In contrast with fully-denervated animals, in the partially-lesioned animals in which, however, nigral and striatal denervation was observed (respectively 64% and 72%), irrelevant or absent dyskinetic motor effects developed after an even more prolonged L-Dopa treatment (6 weeks). This evidence supports the concept that, in the development of LID, the degree of denervation is more important than the treatment duration. In addition, we observed that the link between LID and DA denervation was not only evident by using TH-immunostaining, but also by the analysis of striatal DAT+ terminals. An inverse correlation between the intensity of AIMs and the number of spared DAT+ dopaminergic terminals was found in the fully-lesioned model, but not in the other models. As previously demonstrated31, in the presence of a full DA denervation, the dosage of L-Dopa may be relevant in LID induction. Indeed, we provided experimental evidence to support a direct correlation between the daily dosage of L-Dopa (20 mg/kg/day as in the present study vs. 50 mg/kg/day) and the induction of motor complications. We found that while the low L-Dopa dose induced LID only in half of the rats, the high dose caused motor complications in all the treated animals.
Clinical studies reported a dose-dependent increase in peak-dose dyskinesia when L-Dopa is administered orally in PD patients6. Evidence suggests that LID develops with the same frequency and severity irrespective of whether L-Dopa treatment was started in the disease course32. In particular, an interesting study was performed on L-Dopa initiation in sub-Saharan African PD patients. These patients, having a longer delay in receiving L-Dopa (4.2 years) had a shorter LID-free period when compared with Italian patients (1.8 years)33. This latter finding further supports the hypothesis that the natural history of the disease, coupled with the degree of DA denervation rather than the duration of L-Dopa therapy, regulates the onset of LID. In line with these findings, also in our cohort, including more than one hundred PD patients, subjects with LID had a longer disease duration, higher LEDD intake, and worse MDS-UPDRS III scores. Interestingly, the degree of putaminal dopaminergic denervation, measured with DaTSCANⓇ SPECT at diagnosis, was associated with the development of LID, and this association was also significant when corrected with age, LEDD, and disease duration. All these observations show that disease severity and the consequent DA denervation are more important than the duration of L-Dopa treatment for developing LID in PD patients34.
Over the past 25 years, many studies have suggested a significant pathogenic role of α-syn in familial and sporadic PD forms35. PD is defined as a synucleinopathy, along with dementia with Lewy bodies and multiple system atrophy36. However, the possible role of this protein in the pathogenesis of LID has never been deeply investigated. A solid rationale to address this issue is the evidence that an alteration in the function of N-methyl-D-aspartate (NMDA) receptor subunits has been reported in LID37 and also in α-syn-related models of PD, where α-syn itself regulates NMDA-mediated synaptic currents38. The activation of the NMDA receptors, in conjunction with the pulsatile stimulation of D1 DA receptors, is instrumental for the induction and maintenance of LID39,40,41, suggesting that a functional overactivity of NMDA receptors plays a role in LID15,42,43,44. Moreover, the altered ratio of synaptic GluN2A/GluN2B-containing NMDA receptors has been observed in the striatum of L-Dopa-treated dyskinetic rats and monkeys and post-mortem tissues from dyskinetic PD patients, supporting that abnormal NMDA subunit composition might represent a synaptic trait of LID37. In a previous study, we found that also α-syn regulates NMDA-mediated synaptic currents and impairs LTP in SPNs, producing deficits in visuospatial learning. These alterations were associated with a reduced function of the GluN2A NMDA receptor subunit, indicating it as a selective target of α-syn38. These preclinical findings are in agreement with a positron emission tomography study using a marker of activated NMDA receptors, which found that dyskinetic patients had higher striatal uptake compared to patients without LID, suggesting that dyskinetic patients may have abnormal glutamatergic transmission in motor areas45.
Based on this evidence, in the present study, we have investigated whether, in two α-syn-related rodent models of PD with chronic L-Dopa treatment, the induction of LID could be different compared to models induced by the neurotoxin 6-OHDA. Interestingly, we found that chronic L-Dopa in α-syn-PFF animals is not sufficient to induce dyskinetic behavior either in rats receiving intrastriatal injection of this protein or in animals with a double lesion obtained by nigral injection of AAV overexpressing α-syn plus striatal injection of PFFs. Two reasons might account for the failure to induce LID in these models. The first and more reasonable explanation is that the degree of nigral and striatal DA denervation measured in these α-syn-related rodent models is insufficient to reach the threshold for the induction of dyskinetic behavior. This denervation is even lower than the one observed in the partially-lesioned model. A second reason might be the selective decrease of the GluN2A-mediated currents mediated by α-syn in SPNs38. The α-syn-mediated reduction of the GluN2A responses might partially counteract the increased GluN2A/GluN2B ratio, a specific molecular trait of the dyskinetic behavior37.
In addition to the preclinical findings, we investigated the possible link between α-syn and LID at the clinical level. No difference was observed in t-α-syn levels in patients with and without LID in both serum and CSF. Several reasons might account for this lack of correlation. Firstly, serum t-α-syn levels might not reflect the brain levels of this protein. Secondly, we only measured t-α-syn while it is possible that other forms of this protein, such as oligomeric and phosphorylated forms, may be altered, leading to a pathogenetic role in LID. CSF α-syn seed amplification assays have been successfully applied to detect fibrillary/oligomeric α-syn in vivo46. However, these assays are not yet quantitative and mainly provide a dichotomous outcome, making them not suitable to determine associations within a group of PD patients46. Nevertheless, our preclinical data seems to exclude this hypothesis since, in our animal models, both oligomeric and phosphorylated forms are present in brain tissues, particularly in the nigrostriatal system25,38.
Overall, our preclinical and clinical findings do not support a direct role of α-syn in the development and maintenance of LID, while the major determinant seems to be the degree of nigrostriatal DA denervation.
One of the pathophysiological features of experimental models of PD is the lack of LTP and LTD at cortico-striatal synapses as a consequence of striatal DA reduction47. This defective plasticity might account for some of the motor symptoms observed in rodent models of PD48. Recovery of synaptic plasticity by L-Dopa therapy is associated with recovery in synaptic strength at the cortico-striatal synaptic transmission and with motor improvement25. In a rat model of PD and LID, the restored LTP can undergo depotentiation only in animals without LID. The impairment of the ability of corticostriatal synapses to undergo depotentiation of LTP has been considered important in the genesis of LID11. Abnormalities of synaptic plasticity in dyskinetic animals are related to changes in the D1–PKA–DARPP-32 signaling pathway that inhibits the activity of protein phosphatase-1 (PP-1), a molecule involved in depotentiation at corticostriatal synapses11. Activation of D1 receptors causes the PKA-induced phosphorylation on Thr34 of DARPP-32. This event in turn inhibits PP-1. This view aligns with the concept that homeostatic synaptic scaling is a form of synaptic plasticity that adjusts the strength of all excitatory synapses up or down to avoid storing unessential information49. The preclinical findings of our study introduce a new concept in this scenario. We show that while chronic L-Dopa treatment causes the complete lack of depotentiation in fully-lesioned animals, in partially-lesioned rats this homeostatic mechanism is preserved, indicating that a total loss of endogenous DA is necessary to activate this complex molecular cascade.
Neuromodulation of the cortical motor areas is used to assess the plastic responsiveness of the motor network in humans17,18,50. De novo PD patients show reduced LTP-like response in the M1 after intermittent (iTBS) or continuous TBS (cTBS)16 or paired-associative stimulation (PAS)51. Patients with more advanced PD showed weak or absent LTP-like response in M1 after PAS in the OFF status, which usually improved in the ON status17. Interestingly, while the metaplastic LTP induced by cTBS plus muscle contraction can be restored by L-Dopa in all PD patients, irrespective of the presence of LID, its depotentiation can be achieved by cTBS only in non-dyskinetic patients12.
In line with these previous findings, the results of the present study show that PD patients with LID are unresponsive to the depotentiation protocol. This association was significant also when corrected with disease duration and LEDD. However, from our study, it emerges the novel observation that impairment in depotentiation correlates with dyskinesia severity, reinforcing the concept that the loss of depotentiation is the key synaptic mechanism responsible for the appearance of LID. Thus, it is likely that a gradual impairment in depotentiation progressively affects physiological motor learning and programming, causing the onset of increasing involuntary excessive movements until the development of clinically manifest dyskinesia.
Prevention and reduction of LID remain critical unmet needs in managing PD patients. In the present study, using preclinical and clinical investigations, we took an innovative and ambitious approach to provide answers to some unsolved questions in the field. Our data support the idea that nigrostriatal DA denervation remains the major determinant in the pathogenesis of LID, while duration and dosage of L-Dopa treatment represent secondary factors. Thus, although avoiding excessive dosage is mandatory in the clinical setting, L-Dopa treatment must be considered without delay when necessary to improve the quality of life of PD patients, even if a rationale use of this drug is required in the clinical setting, considering each patient’s therapeutic window.
We believe that, although α-syn does not seem to play a direct influence in the induction and maintenance of LID, the misfolding of this protein might indirectly contribute to LID development by triggering the neurodegeneration of nigral DA neurons and the consequent striatal denervation. For this reason, research on disease-modifying drugs targeting α-syn, aiming at slowing the progression of the disease, could also prevent the appearance of LID.
Finally, from our study, the concept emerges that the lack of homeostatic plasticity in both striatal and cortical regions is a constant pathophysiological feature of LID in both experimental models and in PD patients.
Methods
Animals and housing conditions
All animal procedures were performed in accordance with the Animal Care and Use Committee and the Guidelines of The European Union Council (2010/6106/UE) and approved by the Italian Ministry of Health (Prot. Number: 177/2021-PR). The animals were housed in two per cage with free access to water and food and were kept under constant temperature, humidity and a 12 h light/dark cycle.
6-OHDA nigrostriatal lesion
Adult male Wistar rats were deeply anesthetized with Zoletil (an anesthetic drug; 50/50 mg/ml tiletamine and zolazepam) and Rompun (a sedative drug; 20 mg/ml xylazine hydrochloride) and were injected into the left MFB with 4 μL of 6-OHDA (3 μg/μL, in saline containing 0.1% ascorbic acid) (coordinates were calculated in mm from the bregma and the dura surface of the rats: anteroposterior (AP) − 4.4 mm, mediolateral (ML) + 1.2 mm, and dorsoventral (DV) − 7.8 mm), according to the stereotaxic atlas of rat brain52,53. 6-OHDA solution was delivered by a microinjection pump with a rate of 0.38 μL/min, and the cannula was left in place for 5 min after the end of the injection. After surgery, rats were allowed to recover from anesthesia in a temperature-controlled chamber and then placed in individual cages.
Two weeks after the injection, rats were tested with 0.05 mg/kg subcutaneous apomorphine, and contralateral turns to the lesion were counted for 40 min. Animals that displayed apomorphine-induced rotation of over 200 turns, were chosen for the study as fully denervated (Full); rats showing from 10 – 100 turns in 40 min were considered partially denervated and put in the Partial group11. Successfully, 8 Full and 10 Partial rats were subjected to treatment, behavioral test, and immunohistochemical experiments.
Intrastriatal injection of α-syn-PFFs
A-syn-PFFs were prepared as described previously38,54. Eight male Wistar rats (2–3 months old) underwent stereotaxic surgery for intrastriatal α-syn-PFFs-injection. After anesthesia with a mix of Zoletil and Rompum, rats received a unilateral injection of α-syn-PFFs (1 mg/ml dissolved in PBS) using a 10 μl Hamilton microsyringe fitted with a 26-gauge steel cannula. The volume injected was 1 μl (coordinates from bregma: AP, +1.0 mm; ML, −3.0 mm; DV, −5.0 mm)38. After the surgical procedure, the animals were monitored until awake and then returned to their cage. Twenty-two weeks after surgery the animals were used for the experiments.
Injection of α-syn-PFFs and AAV2/6-hα-syn
Eight male Wistar rats (Charles River Laboratories) 2–3 months old were used at the beginning of the experiments. Surgical procedures were performed on deeply anesthetized animals with a mix of Zoletil and Rompum. Animals were subjected to unilateral injections (1 µL) of 30 nM α-syn-PFFs (1 mg/ml dissolved in PBS) or PBS in the striatum (stereotaxic coordinates from bregma: AP, −1.0; ML, −3.0; DV, –5.0), and, sequentially, were injected with AAV2/6-human-α-syn or AAV2/6-synapsin-GFP (control virus) (4.0 × 109 genome copies, 2 µL) in the ipsilateral SNpc (stereotaxic coordinates from bregma: AP, −5.3; ML, −2; DV, −7.2). After surgery, the animals were monitored until awake and returned to their cage. After 18 weeks from surgery, Control (PBS + AAV2/6-synapsin-GFP) and α-syn-PFF + AAV2/6-hα-syn-injected rats were used for the experiments55.
Experimental design
The 34 animals used in the study were divided into five groups. 6-OHDA-lesioned rats were divided into three groups: (1) Full plus 3 weeks of L-Dopa (Full + 3W L-Dopa, n = 8 rats); (2) Partial plus 3 weeks of L-Dopa (Partial + 3W L-Dopa, n = 5 rats) and (3) Partial plus 6 weeks of L-Dopa (Partial+ 6W L-Dopa, n = 5 rats). The fourth and fifth groups were represented by α-syn-PFFs-injected rats treated with 3 weeks of L-Dopa (α-syn-PFF, n = 8 rats) and α-syn-PFFs plus AAV2/6- hα-syn-injected rats subjected to 5 and 3 weeks of L-Dopa (α-syn-PFF + AAV- hα-syn, n = 8 rats).
Behavioral assessment
AIMs were assessed according to the previous methods11,56. Briefly, all animals treated with L-Dopa [10 mg/kg L-Dopa (Sigma-Aldrich, USA) plus 6 mg/kg benserazide (Sigma-Aldrich, USA), intraperitoneal (i.p.) twice a day; dissolved in 0.9% NaCl saline solution] were evaluated for axial, limb, and orolingual movements on alternate days (three times a week) for 3, 5 or 6 weeks, as reported in the experimental design. At an interval of 20 min, for 160 min, AIMs were rated for 60 s. A severity score from 0 to 4 was assigned to each category of AIMs, as previously described11,56. AIMs were measured on days 3, 5, 8, 10, 12, 15, 17, and 19 of treatment. At last, the total AIMs score for each session was obtained by summing the score collected for each time point.
Experimental electrophysiology
Animals were sacrificed to obtain corticostriatal slices for intracellular (thickness 280 μm) and whole-cell patch-clamp (thickness 240 μm) electrophysiological recordings. Corticostriatal slices were inserted in a recording chamber with a continuously flowing Krebs’ solution, composed in mM: 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 10 D (+)-glucose, and 25 NaHCO3. The Krebs solution is maintained at RT with constant flow, 2.5–3 ml/min, bubbled with a 95% O2–5% CO2 gas mixture. Patch pipettes had a resistance of 5–9 MΩ when filled with an internal solution containing: 125 K+-gluconate, 10 NaCl, 1 CaCl2, 2 MgCl2, 10 HEPES, 2 MgATP, and 0.3 Na3GTP, adjusted to pH 7.3 with KOH. Differently, intracellular pipettes had a resistance of 30–60 MΩ when filled with 2 M KCl. The stimulation electrode was placed within the white matter and the recording electrode was in the dorsolateral striatum. LTP was induced in an Mg2 + -free Krebs solution condition by application of a HFS protocol (3 trains of 100 Hz, 3 s of duration, and 20 s of interval). LFS protocol (10 min at 2 Hz) was applied to induce the depotentiation of LTP, as previously reported11. All the experiments were performed in the continuous presence of Picrotoxin (50 μM), to block GABAA-mediated potentials. During the experiment, variations of the access resistance >20% led to the rejection of the experiment. Both techniques were used for recording the synaptic plasticity of striatal SPNs. If the values obtained from the two electrophysiological techniques were similar, they were normalized and plotted together.
Immunostaining and confocal microscopy
Animals were deeply anesthetized by intraperitoneal (i.p.) injections of Rompun and Zoletil. For both immunohistochemical and immunofluorescence procedures, rats were perfused with saline followed by 4% paraformaldehyde in a 0.1 M pH 7.4 phosphate buffer (PB). Brains were dissected, post-fixed, and cryoprotected in 30% sucrose. Then, they were frozen with dry ice and cut into 30–40 μm sections with a Cryostat (Leica, Germany). For immunohistochemistry, primary and secondary antibody solutions, as well as ExtrAvidin solutions, were prepared in PB and 0.3% Triton X-100. Each incubation step was followed by three 5 min rinses in PB. For immunoperoxidase staining, sections were incubated for 48 h at 4 °C in PB and 0.3% Triton with a mouse anti-TH (1:1000, Merck Millipore; MAB318) and then incubated for 2 h with a biotinylated donkey anti-mouse antibody (1:200, Sigma; B-7264) at RT. Further, sections were incubated for 1 h in an ExtrAvidin solution (1:500; Sigma; E2886) at RT. As chromogen, 3,3′ diaminobenzidine 0.05% was used (SK-4105, Vector). Finally, sections were mounted on chrome-alum–coated slides, air-dried, dehydrated and coverslipped. For immunofluorescence staining, sections were incubated 48 h at 4 °C with the following primary antibodies: rat anti-DAT (1:1000; Millipore; MAB369) and mouse anti-TH (1:600; Millipore; MAB318). Following incubation, slices were washed three times for 5 min with PB before the incubation with a cocktail of secondary antibodies, including anti-rat Alexa Fluor 555 (1:200, ThermoFisher; A78945) and anti-mouse Alexa Fluor 488 (1:200, ThermoFisher; A21202). After washing, sections were mounted in gelatin-coated slides, air-dried, and coverslipped with Fluoromount (Sigma, F4680).
Densitometric analyses of fluorescence images
The TH and DAT immunofluorescence (O.D.) at the level of dorsolateral striatum was performed by densitometric analyses. Specifically, after background subtraction, TH- and DAT-associated signals were quantified by using two fields of predetermined area (180 × 180 μm2) randomly positioned for each section. For each animal, 6 regularly spaced rostro-caudally sections were considered. Ipsilateral and contralateral striatum were considered. Analysis was done by using the ImageJ software (http://rsb.info.nih.gov/ij/). The F/A ratio defines the mean fluorescence of (F) normalized to the total surface (A).
Stereological analysis of dopaminergic neurons
To assess the extent of neuronal loss in SNpc following the different damages, stereological cells count of TH+ neurons, identified by a clear nuclear profile, was performed. To better appreciate the effects of the lesions, the amount of neurons TH+ in the lesioned side was expressed as a percentage compared with the contralateral side and control animals. Stereological analysis was performed on brain sections containing the SNpc. By using the Stereo Investigator System (MicroBrightField Europe e.K., Magdeburg, Germany) a three-dimensional optical fractionator counting probe (x, y, z dimension of 50 × 50 × 10 μm, respectively) was applied to obtain unbiased estimates of total TH+ cells according to published procedures57,58. The SNpc was outlined using the 4x objective and the 100x oil immersion objective was used for marking the neuronal cells. The total number of SNpc was estimated according to the formula given below:
where SQ represents the total number of neurons counted in all optically sampled fields of the SNpc, ssf is the section sampling fraction, asf is the area sampling fraction, and tsf is the thickness sampling fraction.
For each animal, 10 sections covering the rostro-caudal extension of the SNpc were counted. Data collecting for stereology was done by an experimenter blind to the group analyzed.
Patients, sample collection and ELISA assay
We retrospectively and prospectively collected data from PD patients followed up at the Movement Disorders outpatient service, at Gemelli Hospital, Rome, Italy, between September 2021 and April 2023. Inclusion criteria were the diagnosis of a clinically defined idiopathic PD according to the Movement Disorders Society criteria59 and age between 18 and 80 years old. The study was approved by the local Ethical Committees (ID 4687) and was carried out according to the Declaration of Helsinki. All patients included signed a written informed consent. Patients underwent a clinical evaluation in the best ON-medication state by a neurologist expert in movement disorders. Motor impairment and the presence of L-Dopa-induced dyskinesia were assessed by means of the MDS -UPDRS parts III-IV and UDysRS, respectively. Also, a minimal dataset of clinical-demographical data was collected (age, age at disease onset, disease duration, ongoing dopaminergic therapy, sex and, if available, DaTSCANⓇ uptake values). Radiotracer uptake values in the bilateral caudate and putamen were calculated using DaTQUANTTM software (GE Healthcare).
Then, patients underwent blood sample collection by means of venipuncture of the median cubital vein after overnight fasting, using commercial collection tubes (BD Vacutainer®). Serum separation was obtained after 30 min of clotting at room temperature and subsequent centrifugation at 1000 × g for 15 min at 4 °C. The upper clear fraction (serum) was collected in 0.5 mL aliquots. The aliquots were stored at −80 °C until analysis. A subgroup of patients underwent lumbar puncture, according to clinical standards, to collect 10–12 mL of CSF in sterile polypropylene tubes. They were then centrifuged at room temperature for 10 min (2000 × g) and aliquots 93 (0.5 mL) were frozen at −80 °C. The levels of total-alpha-syn (t-α-syn) were measured with commercially available ELISA kits in both serum and CSF.
Neurophysiological examination of patients
A subgroup of the enrolled patients in ongoing therapy with L-Dopa also underwent neurophysiological examination. Patients with cognitive impairment (Mini-Mental Status Examination, MMSE ≤ 24), history of epilepsy, implanted medical devices (neurostimulator, cardiac pacemaker, infusion pump), or women who were pregnant or with possible intention/potential, were not eligible for the neurophysiological examination. Before the neurophysiological evaluation, the UDysRS was assessed in the best ON-medication state. All patients were naive to the effects of TMS and unaware of the differences between TMS protocols.
Non-dyskinetic and dyskinetic patients were evaluated in a on motor state 2 h after taking half of the first dose of L-Dopa in the morning (to avoid any dyskinesia that could affect the results) in two different sessions of TBS over M1 (Suppl. Fig. 1). Two stimulation paradigms already validated and described in the past were used12,60: these paradigms allow to induce LTP- and depotentiation-like effects in the motor cortex of the patients and therefore to subsequently evaluate the presence and validity of the impairment of these synaptic mechanisms. In the first paradigm (“potentiation session”), a train of cTBS lasting 20 s (300 stimuli) was delivered to the motor cortex of the patients, followed by the 1 min voluntary contraction of the target muscle (cTBSc0): this procedure induces a significant increase of motor evoked potential (MEP) amplitude recorded by the target muscle for about 20 min. In the second paradigm (“depotentiation session”), the procedure of potentiation described above (cTBSc0) was followed by the delivery of a second train of cTBS 150 stimuli lasting 10 s (cTBS150): this stimulation sequence induces a significant decrease of MEP amplitude recorded from the target muscle for about 20 min. Each experimental session was performed at least 1 week apart from each other in a pseudo-randomized order. TMS procedures were performed in accordance with the guidelines for the safe use of non-invasive brain stimulation61.
Statistical analysis
For electrophysiological results, a Student’s t-test was used to compare pre-and post-HFS/LFS EPSP amplitudes. Behavioral data were analyzed by Student’s t-test and two-way ANOVA (Interaction and Treatment), followed by Bonferroni’s post-hoc test. Immunostaining data were analyzed with a two-tailed Student’s t-test and are expressed as mean ± SEM. Results are presented as mean ± SEM. For all analyses, a p < 0.05 was considered statistically significant. All statistical analysis was performed with GraphPad Prism 6.
Continuous clinical and demographic features of the cohort considered in this study are summarized as mean ± SD and categorical features as count. Comparisons between the groups of dyskinetic and non-dyskinetic patients were performed by unpaired two-tailed Student’s t-test for continuous variables and Chi2-test for categorical variables. Logistic regression was employed to assess relationships between categorical and continuous variables in the total sample of patients and in the subgroups who underwent DaTSCANⓇ-SPECT and those who underwent neurophysiological examination. In this latter subgroup, MEP amplitudes were normalized to the pre-TBS baseline amplitude of each session. Two-way ANOVA (Time and Pattern) was used to examine the effects of the cTBS150 intervention on MEP amplitude in both the groups of dyskinetic and non-dyskinetic patients. Spearman correlation was performed to correlate the amount of depotentiation and UDysRS III. The averaged per cent change of normalized MEP amplitudes between cTBSc0 and depotentiation sessions [(MEPcTBSc0 − MEPDepotentiation)/MEPcTBSc0] at time points T2, T3, T4, T5 after cTBSc0 was calculated as the amount of depotentiation. A p-value <0.05 was deemed statistically significant to reject the null hypothesis. All the analyses were performed with SPSS version 26 for MAC.
References
Poewe, W. et al. Parkinson disease. Nat. Rev. Dis. Primers 3, 17013 (2017).
Spillantini, M. G. et al. Alpha-synuclein in Lewy bodies. Nature 388, 839–840 (1997).
Calabresi, P., Di Lazzaro, G., Marino, G., Campanelli, F. & Ghiglieri, V. Advances in understanding of the function of alpha-synuclein: implications for Parkinson’s disease. Brain 146, 3587–3597 (2023).
Bastide, M. F. et al. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog. Neurobio.l 132, 96–168 (2015).
Ahlskog, J. E. & Muenter, M. D. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov. Disord. 16, 448–458 (2001).
Fahn, S. et al. Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med. 351, 2498–2508 (2004).
Calabresi, P., Di Filippo, M., Ghiglieri, V., Tambasco, N. & Picconi, B. Levodopa-induced dyskinesias in patients with Parkinson’s disease: filling the bench-to-bedside gap. Lancet Neurol. 9, 1106–1117 (2010).
Espay, A. J. et al. Levodopa-induced dyskinesia in Parkinson disease: current and evolving concepts. Ann. Neurol. 84, 797–811 (2018).
Bove, F. & Calabresi, P. Plasticity, genetics, and epigenetics in l-dopa-induced dyskinesias. Handb. Clin. Neurol. 184, 167–184 (2022).
Calabresi, P. et al. Hyperkinetic disorders and loss of synaptic downscaling. Nat. Neurosci. 19, 868–875 (2016).
Picconi, B. et al. Loss of bidirectional striatal synaptic plasticity in L-DOPA-induced dyskinesia. Nat. Neurosci. 6, 501–506 (2003).
Huang, Y. Z., Rothwell, J. C., Lu, C. S., Chuang, W. L. & Chen, R. S. Abnormal bidirectional plasticity-like effects in Parkinson’s disease. Brain 134, 2312–2320 (2011).
Prescott, I. A. et al. Lack of depotentiation at basal ganglia output neurons in PD patients with levodopa-induced dyskinesia. Neurobiol. Dis. 71, 24–33 (2014).
Singh, A. et al. Human striatal recordings reveal abnormal discharge of projection neurons in Parkinson’s disease. Proc. Natl Acad. Sci. USA 113, 9629–9634 (2016).
Campanelli, F., Natale, G., Marino, G., Ghiglieri, V. & Calabresi, P. Striatal glutamatergic hyperactivity in Parkinson’s disease. Neurobiol. Dis. 168, 105697 (2022).
Kishore, A., Joseph, T., Velayudhan, B., Popa, T. & Meunier, S. Early, severe and bilateral loss of LTP and LTD-like plasticity in motor cortex (M1) in de novo Parkinson’s disease. Clin. Neurophysiol. 123, 822–828 (2012).
Morgante, F., Espay, A. J., Gunraj, C., Lang, A. E. & Chen, R. Motor cortex plasticity in Parkinson’s disease and levodopa-induced dyskinesias. Brain 129, 1059–1069 (2006).
Suppa, A. et al. Lack of LTP-like plasticity in primary motor cortex in Parkinson’s disease. Exp. Neurol. 227, 296–301 (2011).
Eusebi, P. et al. Risk factors of levodopa-induced dyskinesia in Parkinson’s disease: results from the PPMI cohort. NPJ Parkinsons Dis. 4, 33 (2018).
Lee, C. S. et al. In vivo positron emission tomographic evidence for compensatory changes in presynaptic dopaminergic nerve terminals in Parkinson’s disease. Ann. Neurol. 47, 493–503 (2000).
Paille, V. et al. Distinct levels of dopamine denervation differentially alter striatal synaptic plasticity and NMDA receptor subunit composition. J. Neurosci. 30, 14182–14193 (2010).
Calabrese, V. et al. Rapamycin, by inhibiting mTORC1 signaling, prevents the loss of striatal bidirectional synaptic plasticity in a rat model of L-DOPA-induced Dyskinesia. Front Aging. Neurosci. 12, 230 (2020).
Cenci, M. A. & Lundblad, M. Ratings of L-DOPA-induced dyskinesia in the unilateral 6-OHDA lesion model of Parkinson’s disease in rats and mice. Curr. Protoc. Neurosci. Chapter 9, Unit9.25 (2007).
Natale, G. et al. Transcranial magnetic stimulation exerts “rejuvenation” effects on corticostriatal synapses after partial dopamine depletion. Mov. Disord. 36, 2254–2263 (2021).
Tozzi, A. et al. Dopamine-dependent early synaptic and motor dysfunctions induced by α-synuclein in the nigrostriatal circuit. Brain 144, 3477–3491 (2021).
Negrini, M. et al. Sequential or simultaneous injection of preformed fibrils and AAV overexpression of alpha-synuclein are equipotent in producing relevant pathology and behavioral Deficits. J. Parkinsons Dis. 12, 1133–1153 (2022).
Ravina, B. et al. Dopamine transporter imaging is associated with long-term outcomes in Parkinson’s disease. Mov. Disord. 27, 1392–1397 (2012).
Parnetti, L. et al. CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol. 18, 573–586 (2019).
Vucic, S. et al. Clinical diagnostic utility of transcranial magnetic stimulation in neurological disorders. Updated report of an IFCN committee. Clin. Neurophysiol. 150, 131–175 (2023).
Alcacer, C. et al. Chemogenetic stimulation of striatal projection neurons modulates responses to Parkinson’s disease therapy. J. Clin. Invest. 127, 720–734 (2017).
Picconi, B. et al. l-DOPA dosage is critically involved in dyskinesia via loss of synaptic depotentiation. Neurobiol. Dis. 29, 327–35 (2008).
Katzenschlager, R. et al. Fourteen-year final report of the randomized PDRG-UK trial comparing three initial treatments in PD. Neurology 71, 474–480 (2008).
Cilia, R. et al. The modern pre-levodopa era of Parkinson’s disease: insights into motor complications from sub-Saharan Africa. Brain 137, 2731–2742 (2014).
Nutt, J. G., Chung, K. A. & Holford, N. H. Dyskinesia and the antiparkinsonian response always temporally coincide: a retrospective study. Neurology 74, 1191–1197 (2010).
Dehay, B. et al. Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol. 14, 855–866 (2015).
Calabresi, P. et al. Alpha-synuclein in Parkinson’s disease and other synucleinopathies: from overt neurodegeneration back to early synaptic dysfunction. Cell Death Dis. 14, 176 (2023).
Mellone, M. et al. NMDA receptor GluN2A/GluN2B subunit ratio as synaptic trait of levodopa-induced dyskinesias: from experimental models to patients. Front Cell Neurosci. 9, 245 (2015).
Durante, V. et al. Alpha-synuclein targets GluN2A NMDA receptor subunit causing striatal synaptic dysfunction and visuospatial memory alteration. Brain 142, 1365–1385 (2019).
Papa, S. M. & Chase, T. N. Levodopa-induced dyskinesias improved by a glutamate antagonist in Parkinsonian monkeys. Ann Neurol 39, 574–578 (1996).
Hadj Tahar, A. et al. Effect of a selective glutamate antagonist on L-dopa-induced dyskinesias in drug-naive parkinsonian monkeys. Neurobiol. Dis. 15, 171–176 (2004).
Wessell, R. H. et al. NR2B selective NMDA receptor antagonist CP-101,606 prevents levodopa-induced motor response alterations in hemi-parkinsonian rats. Neuropharmacology 47, 184–194 (2004).
Chase, T. N. & Oh, J. D. Striatal dopamine- and glutamate-mediated dysregulation in experimental parkinsonism. Trends Neurosci. 23, S86–91 (2000).
Hallett, P. J. & Standaert, D. G. Rationale for and use of NMDA receptor antagonists in Parkinson’s disease. Pharmacol. Ther. 102, 155–174 (2004).
Klockgether, T. & Turski, L. Toward an understanding of the role of glutamate in experimental parkinsonism: agonist-sensitive sites in the basal ganglia. Ann Neurol. 34, 585–593 (1993).
Ahmed, I. et al. Glutamate NMDA receptor dysregulation in Parkinson’s disease with dyskinesias. Brain 134, 979–986 (2011).
Bellomo, G. et al. α-Synuclein seed amplification assays for diagnosing synucleinopathies: the way forward. Neurology 99, 195–205 (2022).
Calabresi, P., Picconi, B., Tozzi, A. & Di Filippo, M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 30, 211–219 (2007).
Calabresi, P., Picconi, B., Tozzi, A., Ghiglieri, V. & Di Filippo, M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nat. Neurosci. 17, 1022–1030 (2014).
Turrigiano, G. G. The dialectic of Hebb and homeostasis. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 372, 20160258 (2017).
Huang, Y. Z., Edwards, M. J., Rounis, E., Bhatia, K. P. & Rothwell, J. C. Theta burst stimulation of the human motor cortex. Neuron 45, 201–206 (2005).
Kojovic, M. et al. Functional reorganization of sensorimotor cortex in early Parkinson disease. Neurology 78, 1441–1448 (2012).
Paxinos, G., Watson, C., Pennisi, M. & Topple, A. Bregma, lambda and the interaural midpoint in stereotaxic surgery with rats of different sex, strain and weight. J. Neurosci. Methods 13, 139–143 (1985).
Mineo, D. et al. Dopamine drives binge-like consumption of a palatable food in experimental Parkinsonism. Mov. Disord. 34, 821–831 (2019).
Marino, G. et al. Intensive exercise ameliorates motor and cognitive symptoms in experimental Parkinson’s disease restoring striatal synaptic plasticity. Sci. Adv. 9, eadh1403 (2023).
Giordano, N. et al. Motor learning and metaplasticity in striatal neurons: relevance for Parkinson’s disease. Brain 141, 505–520 (2018).
Cenci, M. A., Lee, C. S. & Björklund, A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur. J. Neurosci. 10, 2694–2706 (1998).
Guatteo, E. et al. Functional alterations of the dopaminergic and glutamatergic systems in spontaneous α-synuclein overexpressing rats. Exp. Neurol. 287, 21–33 (2017).
Ledonne, A. et al. Morpho-functional changes of nigral dopamine neurons in an α-synuclein model of Parkinson’s disease. Mov. Disord. 38, 256–266 (2023).
Postuma, R. B. et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 30, 1591–1601 (2015).
Huang, Y. Z., Rothwell, J. C., Edwards, M. J. & Chen, R. S. Effect of physiological activity on an NMDA-dependent form of cortical plasticity in human. Cereb Cortex 18, 563–570 (2008).
Rossi, S. et al. Safety and recommendations for TMS use in healthy subjects and patient populations, with updates on training, ethical and regulatory issues: expert guidelines. Clin. Neurophysiol. 132, 269–306 (2021).
Acknowledgements
We thank all patients who participated in the study. This work was supported by grants from Italian Ministry of Education, University and Research (MIUR)- PRIN (Bando 2017, Prot. 2017ENN4FY, P.C.), Italian Ministry of Health, Ricerca Corrente (P.C.), NIH (NS045962, P.C. in collaboration with Prof. Stella Papa of Emory University, Atlanta, GA), Italian Ministry of University and Research (MUR), PNRR project MNESYS (PE0000006).
Author information
Authors and Affiliations
Contributions
P.C. conceived the project. P.C., V.G., and M.T.V. organized the experiments and designed the statistical analysis. F.C., G.N., G.M., A.L., and E.P. prepared the animal models. F.C., G.N., and G.M. carried out electrophysiological and behavioral experiments. F.S., M.D.C., and M.M.C. conducted the morphological experiments. P.C. and G.D.L. organized the clinical protocols. G.D.L., F.B., R.D.I., B.A., and A.T.C. enrolled patients and collected data. A.P., L.P., and G.B. analyzed the patient’s samples. F.B., R.D.I. and B.A. carried out the neurophysiology protocol in PD patients. D.D.G. performed the SPECT imaging in PD patients. F.C., G.M., G.N. and G.D.L. analyzed the data. F.S., M.D.C., G.D.L., V.G., and P.C. wrote the first draft of the manuscript. F.C., G.N., G.M., G.D.L., and F.B. prepared the current figures. P.C., V.G., M.T.V., G.D.L., L.P., N.B.M., B.P., A.R.B., and G.B. revised the article for critical discussion and content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Servillo, F., De Carluccio, M., Di Lazzaro, G. et al. Molecular and cellular determinants of L-Dopa-induced dyskinesia in Parkinson’s Disease. npj Parkinsons Dis. 10, 228 (2024). https://doi.org/10.1038/s41531-024-00836-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41531-024-00836-6
This article is cited by
-
Backtracking α-synuclein pathology: social deficits precede motor symptoms in Parkinson’s disease
npj Parkinson's Disease (2025)
-
CSF phospho-tau levels at Parkinson’s disease onset predict the risk for development of motor complications
Journal of Neurology (2025)
