
Abstract
Bi-allelic pathogenic GBA1 variants cause Gaucher disease (GD), whereas certain heterozygous missense variants increase the risk of Parkinson’s disease (PD), although the underlying mechanisms are unclear. Here, we classified GBA1 missense variants using predictive and structural scores, and analysed their associations with enzyme activity, Saposin C (SapC) interaction and PD progression in 639 patients with heterozygous GBA1 variants from five cohorts. Principal component analysis (PCA) identified two components: PC1, associated with reduced β-glucocerebosidase activity, the GD clinical severity classification, younger age at PD diagnosis, and faster cognitive and motor decline; and PC2, associated with surface-exposed, flexible regions involved in SapC interactions, younger age at PD diagnosis, and slightly with motor decline. These findings highlight that impaired SapC interactions, in addition to reduced activity, may contribute to PD severity in GBA1 variant carriers. This is relevant for therapeutic approaches aimed at stabilizing β-glucocerebosidase or enhancing its enzymatic activity in PD.
Similar content being viewed by others
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder mainly characterized by the pathological aggregation and spreading of alpha-synuclein (enriched in the so-called Lewy bodies), and the loss of dopaminergic and non-dopaminergic neurons1. This leads to the onset and progression of a constellation of motor (akinesia, resting tremor, and rigidity) and non-motor signs and symptoms (cognitive impairment, dysautonomia, sleep issues)2.
β-Glucocerebrosidase (GCase), a lysosomal enzyme encoded by GBA1, is involved in the catabolism of glycosphingolipids such as glucosylceramide (GlcCer) and glucosylsphingosine, and appears to play a role in the regulation of alpha-synuclein aggregation3. The activity and stability of GCase are modulated by Saposin C (SapC), a lysosomal cofactor that facilitates substrate access and protects GCase from proteolytic degradation4. Biallelic GBA1 pathogenic variants cause Gaucher disease (GD), an autosomal recessive lysosomal disorder with a variable phenotype, ranging from asymptomatic cases to severe systemic involvement5. Most of these pathogenic variants are missense variants, such as N370S and L444P. Heterozygous GBA1 variants have been identified as genetic risk factors for developing PD and earlier cognitive impairment6,7. More recently, non-GD-causing GBA1 missense variants, such as E326K and T369M have also been identified as risk factor for PD8,9.
GBA1 variants were originally classified according to the phenotype of patients with GD, with “mild” variants causing non-neuronopathic GD (type 1 GD) and “severe” variants causing neuronopathic GD (types 2 and 3 GD)10,11. In GD, the phenotype is linked to the loss of function of GCase, and severe pathogenic variants are indeed associated with a lower GCase activity than mild pathogenic variants. Several studies have demonstrated that the classification of GBA1 variants into ‘mild’ or ‘severe’ based on GD correlates with PD risk, age at onset, and disease progression. Patients carrying severe variants, such as L444P, exhibit a higher PD risk and earlier onset than those with mild variants or risk factors such as E326K7,12,13. However, the mechanisms by which heterozygous GBA1 variants cause PD and the relationship with GCase activity remain unclear. Therefore, GD clinical classification applied to heterozygous pathogenic variants raises questions about their relevance in terms of association with PD progression, the severity of non-GD-causative missense variants, and the effect of numerous unclassified GBA1 variants14. Standard in silico prediction scores used in clinical laboratories have been developed to assess the pathogenicity of missense variants, such as differences in enzymatic activity8. Other structural scores, such as solvent accessibility, provide information on the effect of missense variants on protein surface exposure, which has been shown to be an important feature for pathogenicity15,16. In the context of GBA1, these in silico scores may allow for the inference of the mechanisms associated with PD progression and gain a better understanding of the PD pathophysiology in patients with heterozygous GBA1 variants.
In this work, we first aimed to disentangle the effects of GBA1 missense variants using in silico tools and then determine whether these components were associated with age at PD diagnosis, motor, and cognitive progression in a large cohort of GBA1 PD carriers.
Results
Population
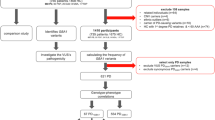
Of the 732 patients with identified GBA1 variants, we excluded 93 patients, including 20 patients with homozygous variants, 30 with compound heterozygous variants and 43 with other variants (recombination, nonsense GBA1 variants and variants in other PD genes) (Fig. 1). After exclusion, the study population included a total of 639 PD patients with heterozygous GBA1 variants, distributed across several cohorts. Specifically, 280 patients were from the AMPPD study (6.9% of the cohort), 204 patients from the NGC cohort (4.8%), 16 patients from the ICEBERG study (9.5%), 61 patients from the NSPARK cohort (7%), and 78 patients from the PREDISTIM study (9.4%). Patients carried 46 different heterozygous missense GBA1 variants, with 33% (15/46) of GBA1 variants classified in the unknown GD category (Table S1). The patients included 195 E326K carriers, 58 L444P carriers, 185 N370S carriers, 95 T369M carriers and 106 patients with other variants (Table 1). The sex ratio between variant types was balanced between groups. Patients with severe and unknown variant categories had a lower median age at diagnosis compared to the other categories (respectively 51.0 y IQR: 11.9 y and 53.0 y IQR: 21.1 y), whereas patients with mild variant had a later median age at diagnosis (59.0 y IQR: 14.9 y) (Table 1).

Flow chart.
Variant analysis
We retained 2906 GBA1 variants from 495 residues with available VEP and structural scores. Following standardization, we performed a PCA on these variants. The first principal component (PC1) accounted for 53% of the variance, while the second principal component (PC2) explained 12% (Fig. 2A, B). The contributions of the various scores to PC1 and PC2 are detailed in Table S2A, and scores are available in Table S1.

A Percentage of explained variance by the principal component analysis. B Variable plot of the first two principal components. DDG free energy difference, RSA relative solvent accessibility, DSA delta of solvent accessibility, LOF Loss of function.
Variants located within the catalytic cavity exhibited significantly higher PC1 scores compared to other residues, while those located in the SapC contact region showed elevated PC2 scores (Fig. 3A). Similarly, variants positioned in flexible loop regions displayed significantly higher PC2 scores compared to non-loop residues, while no significant difference in PC1 was observed (Fig. 3B). in silico modeling of the GCase–SapC interface predicted that variants destabilizing the protein–protein interaction had significantly higher PC2 scores, with no association observed for PC1 (Fig. 3C). In in vitro SapC activation assays, variants classified as non-responsive to SapC-mediated activation exhibited significantly higher PC2 values compared to responsive variants (Fig. 3D). PC1 scores were strongly associated with reduced recombinant GCase enzymatic activity (P < 0.001), whereas PC2 showed no significant relationship with enzymatic activity (Fig. 3E). Protease resistance assays based on Cathepsin D digestion revealed no significant differences in either PC1 or PC2 scores between protease-resistant and protease-sensitive variants (Fig. 3F). While PC1 was significantly different across GD classifications, no significant difference was observed for PC2 (Fig. 4).

A Distributions of PC1 and PC2 scores for GBA1 missense variants stratified by structural spatial context: non-catalytic cavity/non–Saposin C contact residues (other residues, blue, n = 2659), catalytic cavity residues (purple, n = 96), and Saposin C contact residues (orange, n = 151)61. B Comparison of PC1 and PC2 distributions between loop residues (red, n = 220) and non-loop residues (blue, n = 2686)61. Loop regions are typically associated with increased flexibility and surface exposure. C in silico analysis of the GCase–Saposin C interface using AlphaFold3 predictions. Variants were classified as stabilizing (blue, n = 511) or destabilizing (red, n = 2395) based on predicted effects on protein–protein interactions. D In vitro Saposin C activation response among variants for which recombinant β-glucocerebosidase activity was measured in the presence or absence of Saposin C53. Variants were grouped as responsive (blue, n = 11) or non-responsive (red, n = 14). E Relationship between PC scores and recombinant β-glucocerebosidase activity (n = 28)54. PC1 and PC2 scores were plotted against in vitro activity values. F Protease resistance analysis based on Cathepsin D digestion assays53. Variants were classified as protease-resistant (blue, n = 18) or protease-sensitive (red, n = 9), reflecting differences in protein folding stability. Violin plots display the distribution density, with overlaid boxplots showing the median and interquartile range. Statistical significance was assessed using Wilcoxon rank-sum; ns = not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Spearman’s correlation coefficient (R) is shown, with non-linear loess smoothing used to visualize trends. Shaded regions represent 95% confidence intervals.

A Gaucher disease classification according to PC1 and PC2, respectively. Variants are classified as risk variants (N = 3), mild (N = 55) or severe (N = 74). Statistical significance was assessed using Wilcoxon rank-sum; ns not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. B Plot of GBA1 missense variants according to PC1 and PC. Color key: Blue = Risk variant, Orange = Mild, Red = Severe.
In the Partial Least Squares (PLS) regression model using enzymatic activity as the response variable, the lowest prediction error was achieved with a single latent component—indicating that PLS Component 1 (PLS1) captures the most relevant signal predictive of GCase activity (Fig. S1A, B). When applied across the full set of GBA1 variants, inverted PLS1 scores were strongly and positively correlated with PC1 (Pearson’s R = 0.99, P < 0.001) with similar feature loadings observed between the two components (Table S2B), whereas a weaker inverse correlation was observed with PC2 (Pearson’s R = –0.10, P < 0.001) (Fig. S1C, D).
GBA1 variant and enzymatic activity ex vivo
Blood GCase activity was available for 127 visits from 43 patients with GBA1 variants, 17 patients with E326K, 10 patients with T369M, 7 with N370S, 3 with L444P and 6 with other variants. GD classification was significantly associated with GCase enzymatic activity (P = 0.021). In the post hoc analysis, the only significant association was a lower GCase activity in severe GBA1 carriers compared to risk variant carriers (P = 0.036) (Fig. 5A). Patients with higher PC1 had a significantly lower blood GCase activity (P = 0.014) while PC2 was not significantly associated with lower GCase activity (Fig. 5B, C). These results were similar when excluding L444P carriers (Table S3).

These results are derived from linear mixed-effects models, where β-glucocerebrosidase activity is the dependent variable. A Estimated marginal means of β-glucocerebosidase activity (μmol/L/h) across Gaucher disease classification (risk variant, mild or severe). Pairwise comparisons of estimated means are shown with associated p values. B Relationship between β-glucocerebosidase activity and principal component 1 (PC1). Each dot represents an individual sample; the red line indicates the model-predicted slope with a 95% confidence interval. C Relationship between β-glucocerebosidase activity and principal component 2 (PC2). No significant association was observed. Gcase β-glucocerebosidase.
GBA1 classification and age at diagnosis
Among the 639 patients, GD classification was significantly associated with age at diagnosis (P < 0.001). Patients with severe variants had lower age at diagnosis compared to patients with risk variants (P = 0.013) and patients with mild variants (P < 0.001) (Fig. 6A). Higher PC1 and PC2 values were significantly associated with younger age at diagnosis (P = 0.010 and P = 0.015, respectively) (Fig. 6B, C). These associations remained significant in the sensitivity analysis, excluding L444P carriers (Table S4).

These results are derived from linear mixed-effects models. A Estimated marginal means of age at diagnosis (in years) across Gaucher disease classification (risk variant, mild or severe). Pairwise comparisons of estimated means are shown with corresponding p values. B Relationship between age at diagnosis and principal component 1 (PC1). Each dot represents an individual; the red line shows the fitted slope with 95% confidence interval. C Relationship between age at diagnosis and principal component 2 (PC2), also showing model-estimated slope and confidence interval.
GBA1 classification and disease progression
In the longitudinal analysis, 384 patients were included, corresponding to 1558 visits, with a median of two visits per patient (IQR 5), and a median follow-up of one year (IQR 3.6 years). Sex ratios, variant proportions and age at diagnosis were similar to the whole cohort (Table 1). The median duration between the diagnosis and baseline visit was 3.8 years (IQR: 6.7 years), with no significant differences between GD classification categories. Education was significantly different between GD categories, with more patients with education below twelve years in the severe and unknown categories (Table 1).
In the cognitive analysis using linear mixed-effects model (LMM), GD classification was significantly associated with cognitive decline during follow-up (P = 0.002) (Table S5). In post hoc analysis at ten years, severe variant carriers had a higher cognitive decline compared to risk variant (P = 0.002) and mild variants (P = 0.040) carriers (Table S5A, Fig. 7A). Higher PC1 values were associated with higher cognitive decline (P < 0.001). The predicted decrease of MoCA over 10-year follow-up was 0.1 ± 1.1 point for patients with low PC1 tercile, 2.8 ± 1.1 points for medium PC1 tercile and 5.7 ± 1.5 points for high PC1 tercile. PC2 was not significantly associated with cognitive decline during follow-up. In the sensitivity analysis using generalized estimating equations (GEE), the results were consistent for the PC-based classification, which remained significantly associated with cognitive decline. However, the association with GD classification was no longer statistically significant. The results remain similar in the sensitivity analysis excluding L444P carriers (Table S5).

These results are derived from linear mixed-effects models. The estimated marginal means represent predicted slopes of cognitive decline (MoCA score). A Estimated marginal mean trajectories of MoCA scores over time by Gaucher disease classification (risk variant, mild or severe). A significant difference in cognitive decline slopes is observed between groups. (Color key: Blue = Risk variant, Orange = Mild, Red = Severe). B Predicted MoCA score change by PC1. Individuals with higher PC1 scores show significantly faster cognitive decline. (Color key: Green = Low tercile PC score, Blue = Medium tercile, Red = High tercile). C Predicted MoCA score change by PC2. No significant differences were detected. (Color key: Green = Low tercile PC score, Blue = Medium tercile, Red = High tercile). Shaded areas represent 95% confidence intervals. The estimated slopes reflect adjusted marginal means derived from the fitted LMMs.
For the motor analysis using LMM, GD classification was significantly associated with motor progression (P = 0.031). In the post hoc analysis at 10 years, only severe variants showed significantly higher motor progression compared with mild variants (P = 0.034) (Fig. 8A, Table S6A). The association was no longer significant in the sensitivity analysis after excluding L444P carriers. Higher PC1 values were significantly associated with faster motor progression (P = 0.003) (Fig. 8B, Table S6B). The predicted increase of MDS-UPDRS III ON over 10-year follow-up was 4.6 ± 3.5 points for patients with low PC1 tercile, 12.8 ± 3.5 points for medium PC1 tercile, and 21.9 ± 5.5 points for high PC1 tercile. There was a trend toward higher motor progression for higher PC2 (P = 0.084) (Fig. 8C, Table S6B). This association was significant after excluding L444P carriers. In contrast, the sensitivity analysis using GEE yielded non-significant results for both GD classification and PC-based classifications (Table S6).

These results are derived from linear mixed-effects models. The estimated marginal means represent predicted slopes of motor symptom progression (MDS-UPDRS III ON condition). A Predicted trajectories of MDS-UPDRS III scores over time by Gaucher disease classification (risk variant, mild or severe). A significant difference in motor progression slopes is observed across groups. (Color key: Blue = Risk variant, Orange = Mild, Red = Severe). B Predicted MDS-UPDRS III score change by PC1. Individuals with higher PC1 scores show significantly faster motor progression. (Color key: Green = Low tercile PC score, Blue = Medium tercile, Red = High tercile). C Predicted MDS-UPDRS III score change by PC2. Individuals with higher PC2 scores show a trend toward faster motor progression. (Color key: Green = Low tercile PC score, Blue = Medium tercile, Red = High tercile). Shaded areas represent 95% confidence intervals. The estimated slopes reflect adjusted marginal means derived from the fitted LMMs.
Discussion
Our analysis identified distinct disease progression profiles in PD patients with GBA1 variants based on two components of in silico scores. Higher PC1 values were linked to lower GCase activity and faster motor and cognitive decline, while higher PC2 values were associated with younger age at diagnosis, and a trend toward faster motor progression, but not with GCase activity or cognitive decline. Additionally, in this large dataset, we confirmed that severe GBA1 variants, according to the classic clinical GD classification, were associated with lower GCase activity, earlier onset, and faster progression of both cognitive and motor symptoms. We demonstrate that VEP scores, which are designed to estimate the impact of variants on protein function, were mostly represented in PC1, and reflect GCase enzyme activity, as further confirmed by PLS regression. In PD, the enzymatic activity was shown to be reduced for mild and severe variants compared to risk variant carriers17, and this GD classification has been shown to influence age at onset and disease progression7,12,13. We consistently show that PC1 was associated with reduced enzymatic activity of GCase and cognitive progression in PD GBA1 patients. The main representative of the mild form, the N370S variant in the glycosyl hydrolase TIM domain, results in a stable GCase with reduced enzyme activity18. Experimental evidence suggests that reduced GCase activity is associated with the propagation of the synucleinopathy19. In cell culture models, GCase deficiency has been demonstrated to exacerbate existing alpha-synuclein pathology and facilitate the spread of alpha-synuclein fibrils20,21,22. Consistently, in vivo studies have shown that reduced GCase activity significantly increases the dissemination of alpha-synuclein pathology in mice21,23. The reduction in GCase enzymatic activity leading to increased propagation of synuclein may provide a mechanistic explanation for its association with accelerated cognitive decline.
Structural scores were mostly represented in PC2, which was associated with residue surface exposure, local flexibility, and proximity to the SapC interaction interface. These features suggest that PC2 reflects a distinct axis of structural perturbation—less related to catalytic impairment. Consistently, in silico modeling indicated that variants predicted to destabilize the GCase–SapC interface were also enriched for higher PC2 values, and in vitro data showed that variants with altered response to SapC activation exhibited higher PC2 scores. Notably, the risk variant E326K—which is associated with PD but not GD—has been reported to impair GCase–SapC binding24,25. This observation supports the hypothesis that disruption of cofactor interactions, as captured by PC2, may contribute to PD pathogenesis. Clinically, higher PC2 scores were associated with earlier onset of motor symptoms and showed a trend toward faster motor progression, while no significant association was observed with cognitive decline. This pattern aligns with growing evidence that GCase dysfunction may promote synucleinopathy through mechanisms beyond enzymatic loss. SapC normally binds to GCase, enhancing its enzymatic activity and protecting it from proteolysis4. Impaired binding to SapC may destabilize GCase, reducing its functional activation and increasing its vulnerability to degradation. Furthermore, impaired GCase–SapC interaction may destabilize GCase and compromise chaperone-mediated autophagy, promoting α-synuclein accumulation—particularly in dopaminergic neurons26,27. This may explain why higher PC2 scores are selectively associated with motor progression but not cognitive decline. Although PC2 correlates with structural metrics such as B-factor and solvent accessibility—features known to influence protein folding and stability28,29—we did not observe significant differences in protease susceptibility. This suggests that PC2 may reflect a structural flexibility that, while not inherently destabilizing, could predispose to misfolding under stress or during biogenesis. Clinical laboratories commonly use tools such as CADD and REVEL for variant classification, while structural prediction tools are less commonly used, despite their potential relevance. Such tools offer complementary insights by assessing the impact of variants on protein interaction interfaces, which could be particularly valuable in understanding the variant pathogenicity beyond what is captured by traditional scores.
This study presents a well-characterized cohort of PD patients with GBA1 variants, with a particular focus on heterozygous carriers and detailed clinical profiling. However, this study has several limitations. We used data from different cohorts, which may have introduced heterogeneity in the studied population, and differences in genetic screening methods across cohorts could have introduced bias. To address these issues, we accounted for the study effect in the LMM and excluded post-DBS visits in the PREDISTIM cohort to avoid potential bias in cognitive and motor progression assessments30. Although our models adjusted for key clinical covariates and cohort-level effects, we acknowledge that other potential confounders—such as polygenic risk scores, environmental exposures, and comorbidities—were not systematically accounted for and may also influence disease trajectories. The frequency of GBA1 missense variants was similar to previous reports, with E326K, T369M, N370S, and L444P accounting for more than 80% of PD cases7,31,32. Due to this overrepresentation, the weight of frequent variants was particularly high, potentially driving the observed differences, especially for severe variants. The robustness of our results was tested by excluding carriers of the severe L444P variant in sensitivity analyses, which yielded similar findings. To further evaluate the robustness of our findings, we used GEE to assess population-averaged effects of GBA1 variants on longitudinal changes in cognitive and motor scores. The results from the GEE analysis were consistent with our main findings for cognitive progression, thereby reinforcing the robustness of the association. However, for motor progression, GEE results were less stable and non-significant, likely reflecting the greater inter-individual variability in motor symptoms, which are influenced by heterogeneous disease trajectories and treatment effects. Unlike GEE, LMMs incorporate random effects, allowing for the modeling of subject-specific slopes and intercepts; hence, they are better suited for capturing individual-level variability in motor progression33. Studies with larger sample sizes are needed to confirm our findings, particularly for rare variants. Given that our study population primarily includes individuals of European ancestry, caution is warranted when generalizing these findings to non-European populations. However, the proposed framework may help to prioritize and functionally contextualize rare or population-specific variants. Establishing the link between heterozygous variants and biological mechanisms is challenging, and our data on GCase activity are limited. Although a significant difference in ex vivo GCase activity was observed between severe and risk variant carriers, other comparisons (e.g., risk vs. mild, mild vs. severe) did not reach statistical significance, likely reflecting the limited sample size in this subset (n = 43). Similarly, the lack of a statistically significant association between PC2 and ex vivo GCase activity should be interpreted with caution, as the observed trend suggests a potential biological signal that may become significant with larger cohorts. Future studies incorporating high-throughput functional assays, such as Multiplex Assays for Variant Effects (MAVEs), will be essential to systematically characterize the effects of GBA1 missense variants on enzymatic function and cofactor interactions, thereby refining our understanding of their contribution to disease risk and progression34. Integration of multimodal biomarkers—such as neuroimaging, fluid-based measures, and transcriptomic profiles—may further enhance the predictive power of variant classification by capturing complementary dimensions of disease biology and individual heterogeneity.
In conclusion, we show that the PC1, predominantly represented by VEP scores, aligns with GD classification and is associated with reduced enzymatic activity, as well as motor and cognitive decline. In contrast, PC2, predominantly represented by structural scores, was associated with age at diagnosis and progression of motor symptoms. These results suggest that, beyond diminished GCase enzymatic activity, pathogenicity in PD among GBA1 variant carriers may also be driven by impaired SapC interaction. Our PCA-based classification provides a scalable and biologically informed tool for prognostic assessment and future stratification of PD patients with heterozygous GBA1 variants, particularly in the context of clinical trials evaluating targeted therapies, such as chaperone molecules aimed at stabilizing GCase.
Methods
Study design
Data from five cohorts were analyzed: Accelerating Medicine Partnership – Parkinson’s Disease (AMPPD) cohort, from eight North American cohorts (N = 4038)35; ICEBERG a French monocentric cohort, recruiting patients between 2014 and 2022 (N = 168) (NCT02305147); the “Noyaux Gris Centraux” (NGC) cohort, a French multicentric cohort recruiting patients between 1990 and 2021, enriched for patients with familial and early-onset PD (N = 4210); NSPARK, a French real-life multicentric prospective cohort, recruiting patients between 2021 and 2024 (N = 867)36; and PREDISTIM, a multicentric French cohort, recruiting patients between 2013 and 2019 if eligible and planning to undergo deep brain stimulation (DBS) (N = 833)37.
All clinically diagnosed PD patients38 with complete demographic data (sex, date of birth, date of diagnosis, date of visit) and carrying a heterozygous pathogenic missense variant of GBA1 were included. Pathogenic missense variants of GBA1 were selected if they were defined as pathogenic or likely pathogenic by the American College of Medical Genetics (ACMG)39 or were identified in the literature as a risk factor for PD. For consistency, patients with nonsense variants, homozygous and compound heterozygous missense variants, and structural variants of GBA1 were excluded. Patients with mutations identified in other PD genes (LRRK2, PRKN, VPS35, PARK7, SNCA and PINK1) were also excluded. For the longitudinal analysis, only patients with available motor and cognitive data were retained. Patients with deep brain stimulation (DBS) were not included because of its potential effect on cognitive and motor function.
All participants provided written informed consent. The genetic and clinical studies were approved by local ethics committees: PREDISTIM (RCB: 2013-A00193-42), ICEBERG (RCB: 2014-A00725-42), and NSPARK (CPPIDF1-2020-ND58). Studies involving the NGC were reviewed and approved by the CCPPRB (Comité Consultatif de Protection des Personnes dans la Recherche Biomédicale) of the Groupe Hospitalier Pitié-Salpêtrière, Paris, France. AMPPD data were accessed under a data use agreement; all contributing cohorts obtained informed consent and ethics approval at their respective sites.
Data collection
Sex and age at PD diagnosis were collected in all five cohorts. The baseline visit was defined as the first visit reported in each cohort. In the AMPPD, NSPARK, ICEBERG and PREDISTIM cohorts, level of education (above and below twelve years), Montreal Cognitive Assessment (MoCA), Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) part III ON state were collected at baseline and follow-up visits.
For the GCase activity, we used previously published data available in the AMPPD cohort using dried blood spots prepared from blood40. GCase enzyme activity was measured using a multiplex assay. The enzyme activity of each sample was calculated from the ratio of the ion abundance of the product to that of the internal standard measured by mass spectrometry. Activity was expressed as micromoles of product per litre of whole blood per hour (µmol/L/h).
Genetic analysis
Patients were screened for pathogenic variants in PD-associated genes using a combination of sequencing and targeted molecular techniques. In the NGC, NSPARK, ICEBERG and PREDISTIM cohorts, patients were screened for GBA1 variants using long-range PCR (LR-PCR) with two overlapping fragments covering all exons and gene-specific primers to avoid GBA1 pseudogene (GBA1LP)41, followed by sequencing on MiSeq 2000 (Illumina)42 or Sanger sequencing as previously described43. In addition, the most common autosomal dominant PD mutations were assessed using the same method: LRRK2 Ex41, VPS35 Ex15, and SNCA Ex3. Patients were also screened for the LRRK2 Gly2019Ser variant using the TaqMan allelic discrimination Assay-By-Design method. PRKN or SNCA rearrangements were evaluated using Multiplex Ligation-dependent Probe Amplification (MLPA) with MRC-Holland Salsa MLPA P051/P052 Parkinson kits, following the manufacturer’s protocol. Furthermore, PD patients with age at onset below 45 years old or with an autosomal recessive form underwent WES to look for rare variants in genes linked to PD or Parkinsonian syndromes.
Genetic data from AMPPD subjects for the 7 PD genes (GBA1, LRRK2, VPS35, SNCA, PRKN, PARK7 and PINK1) were obtained by analyzing DNA microarray data, WES, whole genome sequencing (WGS), Sanger sequencing of variants in the GBA1 gene and RNA sequencing. For the E326K and T369M GBA1 variants, rs2230288 and rs75548401 were extracted from WGS data.
Variant analysis
GBA1 variants were classified according to GD–based severity as “risk factor”, “mild”, or “severe”, following the classifications provided by the GBA1 Browser11. For each variant, multiple variant effect predictors (VEPs) were gathered, including two widely used scores (CADD44 and REVEL45), two newer ones (PrimateAI46 and AlphaMissense47), and two scores focused on loss-of-function (LOF) effects (DeMask48 and LOF score49) (Table S7). We also collected structural scores, including free energy difference calculated by FoldX (version 5.0), relative solvent accessibility (RSA), delta solvent accessibility (DSA), and B-factor available in SIGMA15 and Envision16 (Table S7). Structural annotations of residues were derived from a previously published reference50, categorizing residues into catalytic cavity (n = 96) and SapC contact sites (n = 151). Loop residues (n = 220) were similarly defined. in silico modeling of the GCase–SapC interface was performed using AlphaFold3-predicted structures51, and the impact of missense variants on the interaction was assessed using the SAAMBE prediction tool52. Variants were classified as “stabilizing” (n = 511) or “destabilizing” (n = 2395) based on predicted effects on the GCase–SapC binding energy. We collected in vitro data from previously published sources53,54. GCase enzymatic activity was measured as the mean of three independent replicates of GBA1 missense variants expressed in HEK293T cells, reported in nmol/mg/min54. SapC activation status was determined based on recombinant GCase activation profiles relative to wild-type (WT) GCase in the presence of SapC53. Variants were classified as SapC-responsive (n = 11) or non-responsive (n = 14) depending on whether their mean enzymatic activity after SapC exposure, plus one standard deviation, was higher than the WT mean activation level. Protease sensitivity was assessed based on susceptibility to Cathepsin D digestion assays reported in the same study53. Variants labeled as “highly unstable” or “dead enzyme” were considered protease-sensitive (n = 9), while all other variants were classified as protease-resistant.
All missense variants with available scores were included in the variant analysis. We performed a principal component analysis (PCA) on the different scores, retaining the first two principal components (PC1 and PC2) for each variant. For the first two PCs, we performed a Spearman correlation with in vitro GCase activity. Group comparisons were performed using Wilcoxon rank-sum tests. To investigate the relationship between in silico scores of GBA1 missense variants and GCase enzymatic activity, we also performed a partial least squares (PLS) regression analysis55. We selected the 28 variants for which in vitro enzymatic activity measurements were available. Structural and functional annotations for each variant were scaled and used as predictors, while the corresponding normalized enzymatic activity served as the response variable. A PLS model was trained using leave-one-out cross-validation, and model performance was evaluated using the root mean squared error of prediction (RMSEP)56. The optimal number of components was selected based on the lowest RMSEP. The trained model was then applied to the full dataset to compute PLS scores for all annotated variants. To facilitate interpretation, the direction of Component 1 scores (PLS1) was inverted, and the resulting scores were used for downstream correlation analyses and visualization. We classified GBA1 variants according to i) GD classification categories: risk variants (non-GD causing but risk variants for PD), mild (causing type I GD) and severe (causing type II and III GD as previously described11) and ii) PC classification (PC1 and PC2).
Statistical analyses
Quantitative variables were described using median and interquartile range (IQR), and categorical variables were described using counts and percentages (%). Comparisons of demographic characteristics between the variant categories and the GD classification categories were made using the Kruskal–Wallis test for numerical variables and the Chi-2 test for categorical variables.
To model the enzymatic activity in patients, we used the available blood enzymatic activity of GCase in the AMPPD cohort. We analyzed enzymatic activity according to the GD classifications and PCA classification through an LMM, with enzymatic activity as the dependent variable, patient groups with the two covariates sex and age at visit as fixed effects and subject identifiers as random effects. We analyzed age at diagnosis according to the GD classification and PCA classification through an LMM for each classification with age at diagnosis as the dependent variable, including sex as a covariate and a random effect on the cohorts. A sensitivity analysis was used to assess the robustness of the model after excluding the L444P carriers.
We used data from the AMPPD, NSPARK, ICEBERG and PREDISTIM cohorts to model the effect of GBA1 variant classifications on motor and cognitive progression. For motor decline, we performed LMM with MDS-UPDRS III total score in ON condition as the dependent variable and subject identifiers and cohort as random effects. The fixed effects of the model were sex, age at diagnosis, disease duration between diagnosis and baseline, GBA1 variant classifications and interaction of each of these variables with duration since baseline.
To model cognitive decline, we performed LMM with MoCA score as the dependent variable, and with subject identifiers and cohort as random effects. The fixed effects were sex, age at diagnosis, education (below 12 years), disease duration between diagnosis and baseline, GBA1 variant classifications, and interaction of each of these variables with duration since baseline during the study. Sensitivity analyses were performed by excluding the L444P carriers.
Missing data were not imputed: only patients with complete demographic and clinical data were included in each analysis. Analyses were performed with R software version 4.0.3 (version 4.0.3, R Core Team 2020; https://www.R-project.org/). All models were obtained with the restricted maximum likelihood estimation method using the ‘lme4’ R package (v1.1-32)50. Model assumptions and fit were checked by a visual inspection of the diagnostic residual plots generated with the ‘ggResidpanel’ R package (v0.3.0)57.
Based on the models, the relationships of enzyme activity, age of onset, cognitive and motor progression with the GBA1 variant classifications were reported in terms of p value. P values were calculated using Type II Wald Chi-square tests from the ‘car’ R package (v3.1–2)58. To examine the effect of the interaction between GBA1 classification and time to diagnosis on the dependent variable, the estimated marginal means were calculated using the ‘emmeans’ R package (v1.8.9)59. The significance level for statistical tests was set at 0.05 (two-tailed). Whenever the GD classification was significant, we conducted a post hoc analysis using Tukey’s method from ‘emmeans’ to identify pairwise differences between categories for GCase activity, age at diagnosis, and at the 10-year mark from inclusion for the longitudinal analysis.
To assess the robustness of our findings on clinical progression, we conducted a sensitivity analysis using GEE, modeling the population-averaged effects of GBA1 variant classifications on both motor and cognitive scores over time. GEE models were specified with an exchangeable correlation structure to account for within-subject dependencies and included the same fixed effects as in the LMMs: sex, age at diagnosis, education (for MoCA), disease duration between diagnosis and baseline, GBA1 variant classification, and interactions of these variables with follow-up time. Analyses were performed using the geepack R package (v1.3–2)60, with robust sandwich estimators for standard errors. A sensitivity analysis excluding L444P carriers was also conducted within the GEE models.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author upon request. The genotype and clinical data for the AMP PD cohorts are available through the Accelerating Medicine Partnership® (AMP®) Parkinson’s Disease (AMP PD) Knowledge Platform. For up-to-date information on the study, visit https://www.amp-pd.org. Clinical longitudinal data and genotyping data for the other cohorts included are accessible through appropriate data-sharing agreements that protect participant privacy with the institutions that conducted or are conducting study consents and clinical assessments under local institutional review board approvals.
Change history
21 November 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41531-025-01204-8
References
Morris, H. R., Spillantini, M. G., Sue, C. M. & Williams-Gray, C. H. The pathogenesis of Parkinson’s disease. Lancet 403, 293–304 (2024).
Kalia, L. V. & Lang, A. E. Parkinson’s disease. Lancet 386, 896–912 (2015).
Aflaki, E., Westbroek, W. & Sidransky, E. The complicated relationship between Gaucher disease and Parkinsonism: insights from a rare disease. Neuron. 93, 737–746 (2017).
Liou, B. et al. Combination of acid β-glucosidase mutation and Saposin C deficiency in mice reveals Gba1 mutation dependent and tissue-specific disease phenotype. Sci. Rep. 9, 5571 (2019).
Platt, F. M., d’Azzo, A., Davidson, B. L., Neufeld, E. F. & Tifft, C. J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 4, 1–25 (2018).
Blandini, F. et al. Glucocerebrosidase mutations and synucleinopathies: Toward a model of precision medicine. Mov. Disord. 34, 9–21 (2019).
Liu, G. et al. Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann. Neurol. 80, 674–685 (2016).
Mallett, V. et al. GBA p.T369M substitution in Parkinson disease: polymorphism or association? A meta-analysis. Neurol. Genet. 2, e104 (2016).
Huang, Y., Deng, L., Zhong, Y. & Yi, M. The Association between E326K of GBA and the Risk of Parkinson’s Disease. Parkinsons Dis. 2018, 1048084 (2018).
Beutler, E., Gelbart, T. & Scott, C. R. Hematologically important mutations: Gaucher disease. Blood Cells Mol. Dis. 35, 355–364 (2005).
Parlar S. C., Grenn F. P., Kim J. J., Baluwendraat C., Gan-Or Z. Classification of GBA1 variants in Parkinson’s disease; the GBA1-PD browser. Mov. Disord. 38, 489–495 (2023).
Gan-Or, Z. et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 84, 880–887 (2015).
Cilia, R. et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 80, 662–673 (2016).
Zhou, Y. et al. Mutational spectrum and clinical features of GBA1 variants in a Chinese cohort with Parkinson’s disease. NPJ Parkinsons Dis. 9, 129 (2023).
Zhao, H. et al. SIGMA leverages protein structural information to predict the pathogenicity of missense variants. Cell Rep. Methods 4, 100687 (2024).
Gray, V. E., Hause, R. J., Luebeck, J., Shendure, J. & Fowler, D. M. Quantitative missense variant effect prediction using large-scale mutagenesis data. Cell Syst. 6, 116–124.e3 (2018).
Huh, Y. E. et al. β-Glucocerebrosidase activity in GBA-linked Parkinson disease. Neurology 95, e685–e696 (2020).
Wei, R. R. et al. X-ray and biochemical analysis of N370S mutant human acid β-glucosidase. J. Biol. Chem. 286, 299–308 (2011).
Menozzi, E., Toffoli, M. & Schapira, A. H. V. Targeting the GBA1 pathway to slow Parkinson disease: Insights into clinical aspects, pathogenic mechanisms and new therapeutic avenues. Pharmacol. Ther. 246, 108419 (2023).
Gegg, M. E., Verona, G. & Schapira, A. H. V. Glucocerebrosidase deficiency promotes release of α-synuclein fibrils from cultured neurons. Hum. Mol. Genet. 29, 1716–1728 (2020).
Henderson, M. X. et al. Glucocerebrosidase activity modulates neuronal susceptibility to pathological α-synuclein insult. Neuron. 105, 822–836.e7 (2020).
Bae, E. J. et al. Glucocerebrosidase depletion enhances cell-to-cell transmission of α-synuclein. Nat. Commun. 5, 4755 (2014).
Migdalska-Richards, A. et al. The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain 140, 2706–2721 (2017).
Liou, B. & Grabowski, G. A. Is E326K glucocerebrosidase a polymorphic or pathological variant?. Mol. Genet Metab. 105, 528–529 (2012).
Pietrafesa, D. et al. Investigating the Impact of the Parkinson’s-associated GBA1 E326K mutation on β-glucocerebrosidase dimerization and interactome dynamics through an in silico approach. Int. J. Mol. Sci. 25, 11443 (2024).
Kuo, S. H. et al. Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci. Adv. 8, eabm6393 (2022).
Kuo, S. H., Tasset, I., Cuervo, A. M. & Sulzer, D. Misfolded GBA/β-glucocerebrosidase impairs ER-quality control by chaperone-mediated autophagy in Parkinson disease. Autophagy 18, 3050–3052 (2022).
Zhang, H. et al. On the relation between residue flexibility and local solvent accessibility in proteins. Proteins 76, 617–636 (2009).
Mishra, A., Ranganathan, S., Jayaram, B. & Sattar, A. Role of solvent accessibility for aggregation-prone patches in protein folding. Sci. Rep. 8, 12896 (2018).
Mangone, G. et al. Early cognitive decline after bilateral subthalamic deep brain stimulation in Parkinson’s disease patients with GBA mutations. Parkinsonism Relat. Disord. 76, 56–62 (2020).
Malek, N. et al. Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J. Neurol. Neurosurg. Psychiatry 89, 702–709 (2018).
Straniero, L. et al. The SPID-GBA study: sex distribution, Penetrance, Incidence, and Dementia in GBA-PD. Neurol. Genet. 6, e523 (2020).
Alencar, A. P., Singer, J. M. & Rocha, F. M. M. Competing regression models for longitudinal data. Biom. J. 54, 214–229 (2012).
Gasperini, M., Starita, L. & Shendure, J. The power of multiplexed functional analysis of genetic variants. Nat. Protoc. 11, 1782–1787 (2016).
Iwaki, H. et al. Accelerating medicines partnership: Parkinson’s disease. genetic resource. Mov. Disord. 36, 1795–1804 (2021).
Mariani, L. L. et al. Descriptive analysis of the French NS-Park registry: Towards a nation-wide Parkinson’s disease cohort?. Parkinsonism Relat. Disord. 64, 226–234 (2019).
Barbosa, R. P. et al. The impact of subthalamic deep-brain stimulation in restoring motor symmetry in Parkinson’s disease patients: a prospective study. J. Neurol. 271, 2582–2595 (2024).
Clarke C. E. et al. UK Parkinson’s Disease Society Brain Bank Diagnostic Criteria [Internet]. Clinical effectiveness and cost-effectiveness of physiotherapy and occupational therapy versus no therapy in mild to moderate Parkinson’s disease: a large pragmatic randomised controlled trial (PD REHAB). NIHR Journals Library. Available at: https://www.ncbi.nlm.nih.gov/books/NBK379754/ (2016).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 17, 405–424 (2015).
Alcalay, R. N. et al. Longitudinal measurements of glucocerebrosidase activity in Parkinson’s patients. Ann. Clin. Transl. Neurol. 7, 1816–1830 (2020).
Miocić, S. et al. Identification and functional characterization of five novel mutant alleles in 58 Italian patients with Gaucher disease type 1. Hum. Mutat.25, 100 (2005).
Zampieri, S., Cattarossi, S., Bembi, B. & Dardis, A. GBA analysis in next-generation era: pitfalls, challenges, and possible solutions. J. Mol. Diagn. 19, 733–741 (2017).
Lesage, S. et al. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. 20, 202–210 (2011).
Schubach, M., Maass, T., Nazaretyan, L., Röner, S. & Kircher, M. CADD v1.7: using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. 52, D1143–D1154 (2024).
Ioannidis, N. M. et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99, 877–885 (2016).
Sundaram, L. et al. Predicting the clinical impact of human mutation with deep neural networks. Nat. Genet. 50, 1161–1170 (2018).
Cheng, J. et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381, eadg7492 (2023).
Munro, D. & Singh, M. DeMaSk: a deep mutational scanning substitution matrix and its use for variant impact prediction. Bioinformatics 36, 5322–5329 (2021).
Stein, D. et al. Genome-wide prediction of pathogenic gain- and loss-of-function variants from ensemble learning of a diverse feature set. Genome Med. 15, 103 (2023).
Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 67, 1–48 (2015).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Pahari, S. et al. SAAMBE-3D: predicting effect of mutations on protein–protein interactions. Int. J. Mol. Sci. 21, 2563 (2020).
Liou, B. et al. Analyses of variant acid beta-glucosidases: effects of Gaucher disease mutations. J. Biol. Chem. 281, 4242–4253 (2006).
Pokorna, S. et al. Design of a stable human acid-β-glucosidase: towards improved Gaucher disease therapy and mutation classification. FEBS J. 290, 3383–3399 (2023).
Liland K. H., Mevik B. H., Wehrens R., Hiemstra P. pls: partial least squares and principal component regression. Available at: https://cran.r-project.org/web/packages/pls/index.html (2024).
Liland K. H., Mevik B. H., Wehrens R., Hiemstra P. pls: Partial Least Squares and Principal Component Regression [Internet]. Available at: https://cran.r-project.org/web/packages/pls/index.html (2024).
Panels and Interactive Versions of Diagnostic Plots using ggplot2 [Internet] Available at: https://goodekat.github.io/ggResidpanel/ (2024).
Fox J. et al. car: companion to applied regression [Internet]. Available at: https://cran.r-project.org/web/packages/car/index.html (2023).
Lenth R. V. et al. emmeans: estimated marginal means, aka least-squares means [Internet]. Available at: https://cran.r-project.org/web/packages/emmeans/index.html (2023).
Højsgaard S., Halekoh U., Yan J., Ekstrøm C. T. geepack: generalized estimating equation package. Available at: https://cran.r-project.org/web/packages/geepack/index.html (2023).
Romero, R. et al. Mechanism of glucocerebrosidase activation and dysfunction in Gaucher disease unraveled by molecular dynamics and deep learning. Proc. Natl. Acad. Sci. 116, 5086–5095 (2019).
Acknowledgements
The authors gratefully acknowledge all participants and technicians who participated and helped in the studies. Data used in the preparation of this article were obtained from the ICEBERG, NGC, PREDISTIM, NSPark cohorts and the Accelerating Medicine Partnership® (AMP®) Parkinson’s Disease (AMP PD) Knowledge Platform. ICEBERG cohort was supported by grants from DHOS-Inserm, France Parkinson, Ecole des NeuroSciences de Paris (ENP), Fondation pour la Recherche Médicale (FRM), and the French State “Investissements d'avenir” (ANR-10-JAIHU-06), ANR-11-INBS-0006, Fondation d’Entreprise EDF, BIOGEN Inc., Fondation Thérèse and René Planiol, Fondation Saint-Michel, ANR-21-JPW2-000 (JPND Control-PD). The NGC cohort has received funding from the program “Investissements d’avenir” ANR-10-IAIHU-06. NSPARK cohort has received funding from the program “Investissements d’avenir” ANR-10- IAIHU-06, French National Institute of Health & Medical Research (INSERM), Ministère des Solidarités et de la Santé - DGOS, Paris, France and Agence nationale de la recherche, PIA 1 “INBS”, F-CRIN, Paris, France. The AMPPD program is a public-private partnership managed by the Foundation for the National Institutes of Health and funded by the National Institute of Neurological Disorders and Stroke (NINDS) in partnership with the Aligning Science Across Parkinson's (ASAP) initiative; Celgene Corporation, a subsidiary of Bristol-Myers Squibb Company; GlaxoSmithKline plc (GSK); The Michael J. Fox Foundation for Parkinson's Research; Pfizer Inc.; AbbVie Inc.; Sanofi US Services Inc.; and Verily Life Sciences. Clinical data and biosamples used in preparation of this article from AMPPD were obtained from the (i) Michael J. Fox Foundation for Parkinson’s Research (MJFF) and National Institutes of Neurological Disorders and Stroke (NINDS) BioFIND study, (ii) Harvard Biomarkers Study (HBS) and the Stephen & Denise Adams Center for Parkinson’s Disease Research of Yale School of Medicine (CPDR-Y), (iii) National Institute on Aging (NIA) International Lewy Body Dementia Genetics Consortium Genome Sequencing in Lewy Body Dementia Case-control Cohort (LBD), (iv) MJFF LRRK2 Cohort Consortium (LCC), (v) NINDS Parkinson's Disease Biomarkers Program (PDBP), (vi) MJFF Parkinson’s Progression Markers Initiative (PPMI), and (vii) NINDS Study of Isradipine as a Disease-modifying Agent in Subjects With Early Parkinson Disease, Phase 3 (STEADY-PD3) and (viii) the NINDS Study of Urate Elevation in Parkinson’s Disease, Phase 3 (SURE-PD3).
Author information
Authors and Affiliations
Consortia
Contributions
A.L. undertook the statistical analysis and wrote the first draft of the manuscript, and C.T. contributed to the genetic analysis and the study protocol. A.B and F-X. Contributed to the statistical analysis and the study protocol. L-L.M., S.L., A.B. and J-C.C. assisted with the protocol design and the finalization of the manuscript. All other authors, including G.C., G.M., S.S., N.B., M.A., I.A., S.A., J-P.B., C.B-C, L.D., S.D., A.E., M.F., C.G., E.H., S.L., A.M., C.M., E.M., F.O., A-S.R., S.T., M.V. and D.D., reviewed and provided corrections to the manuscript. All authors contributed to and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
Elena Moro was the Associate Publisher of npj Parkinson’s Disease at the time of publication and was not involved in the peer review or editorial decision for this article. All other authors declare no competing interests..
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lanore, A., Tesson, C., Basset, A. et al. Classification of GBA1 variants and their impact on Parkinson’s disease: an in silico score analysis. npj Parkinsons Dis. 11, 226 (2025). https://doi.org/10.1038/s41531-025-01060-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41531-025-01060-6
This article is cited by
-
Very late-onset Krabbe disease with concomitant dementia: case description and a critical review of the literature
Neurological Sciences (2026)
