
Abstract
Epithelial ovarian cancers are largely comprised of immunogenic tumor sub-types with the degree of CD8+ T cell infiltration being prognostic of clinical outcome. Tumor antigen-specific T cells are identified among these infiltrating T cell populations which has spurred a decade of development towards antigen-specific immunotherapies. Despite these efforts, the success of such immunotherapies has shown to be limited. In this study, we used state-of-the art immunopeptidomics approach and a novel proteogenomic profiling method to identify potential immunogenic human leukocyte antigen class I-presented peptides from patient-derived high-grade serous ovarian cancer. From 11 patients’ tumors, we identified promising candidates for their therapeutic potential. Of these, we selected the best 13 candidates and validated their immunogenicity in both healthy donors and cancer patients.
Similar content being viewed by others


Introduction
Epithelial ovarian cancer is the fifth most common cancer affecting women, comprising nearly a quarter-million cases worldwide each year. Seventy to 80% of cases are high-grade serous ovarian cancer (HGSC) which arises from the surface of the ovary or from the distal fallopian tube1. Complete surgical tumor removal has been the only significant curative treatment for early-stage, non-metastatic ovarian cancer. However, in most cases, surgery is performed at advanced stages of cancer development. In addition to surgery and chemotherapy, which constitute the standard of care for ovarian cancer patients, targeted anticancer agents, such as poly ADP-ribose polymerase inhibitors (PARPi) and anti-angiogenic drugs, have shown promising clinical results2. Despite these advances, the prognosis for ovarian cancer remains poor due to high recurrence rates and drug resistance, highlighting the urgent need for new therapies to improve patient outcomes3. While immune checkpoint blockade (ICB) agents have significantly improved survival in other solid tumor types, monospecific ICB antibodies exhibit minimal efficacy in HGSC4.
HGSC infiltration by lymphocytes is associated with a higher survival rate5. A subset of these infiltrating lymphocytes may target tumor antigens, providing protection against the tumor. Consequently, immunotherapy approaches, such as therapeutic cancer vaccination designed to generate and/or enhance tumor-reactive T lymphocytes, represent promising therapeutic strategies. Therapeutic cancer vaccines are designed to elicit an immune response against known tumor associate or cancer germline antigens, and the full capitalization of this immunotherapy is dependent on the choice of the optimal antigen target6,7. Currently few well-established vaccine targets are undergoing clinical trial evaluation for ovarian cancer, including Mesothelin, NY-ESO-1, WT-1, and MUC-1. Recent studies have also identified novel putatively antigenic peptides derived from ovarian tumors8,9. These studies leverage the direct identification of tumor antigens through immunopeptidomics and primary patient tumor material.
In this study, we present a comprehensive approach for discovering candidate ovarian tumor antigens, with a specific focus on tumor peptides that have broad applicability to the HGSC patients. Ideal tumor antigens should exhibit high tumor-selective expression, be presented by HLA alleles prevalent in the population, and demonstrate strong immunogenicity. To capture the full spectrum of shared tumor antigen, we expanded our immunopeptidome search beyond canonical exons and employed a proteogenomic discovery strategy using a personalized reference database. Our candidate antigens selection process incorporates several factors to assess therapeutic potential, such as the dysregulation of source genes in ovarian cancer, the population frequency of target HLA alleles, and the similarity of peptides to human pathogen antigens10,11,12,13.
Results
Proteogenomic approach discovers shared tumor antigens in 11 ovarian cancer patients
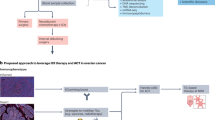
To investigate the tumor antigen landscape of HGSC, we collected tumor samples from 11 patients during surgical resection. We initially evaluated the HLA class I-presented peptides using a direct immunopeptidomics pipeline. HLA-I binding peptides were enriched using state-of-the-art immunoaffinity purification methods. The eluted peptides were characterized by LC-MS/MS, and the spectra were resolved to peptide sequences using the human canonical UniProt proteome as a reference database. Additionally, to broaden our antigen discovery for HLA-I -presented peptides originating outside of the canonical proteome we performed a second search of the MS/MS spectra to a custom reference database. This custom database was constructed from patient tumor RNA sequencing reads using the StringTie assembler, representing the personalized transcriptomes of the 11 ovarian tumor patients (Fig. 1).

Samples are collected, identified by pathology, and biobanked for characterization. Tumor specimens are split to undergo RNA sequencing and HLA-I immunoaffinity purification. The assembled RNA-Seq reads generated by StringTie are utilized to construct a de novo transcriptome and to identify HLA alleles. Peptides purified through immunoaffinity are analyzed via LC-MS/MS and compared against both Uniprot proteins and the custom StringTie database. Identified peptides are then ranked based on their binding potential using MHCFlurry and differential expression in ovarian cancer. Further filtering steps eliminate rare HLA types and prioritize peptides shared by multiple patients and binding to more prevalent HLAs. Finally, candidate peptides are evaluated for immunogenicity in healthy donors and PBMCs derived from ovarian cancer patients. Infographic was created using Infographic was created using BioRender.com.
Using the reference UniProt database, each immunopeptidomics run gathered on average 1633 peptides (range 503–2553). On the other hand, using this proteogenomic approach to matching peptide spectra resolved around half of the number of peptides, on average 750 peptides per run (range 337–1386) (Fig. 2A). However, we observed that around 90% of the peptides, regardless of whether they were identified using UniProt or StringTie corresponded to the interval of 8 to 12 amino acids in length (Fig. 2B) with around 40% of them corresponding to 9mers (Fig. 2C).

A Number of eluted peptides of all lengths identified in each immunopeptidomics run, using either the UniProt (black) or StringTie (magenta) databases. Each dot represents a single immunopeptidomics run. B Percentage of total peptides with lengths between 8 and 12 amino acids. C Percentage of total peptides that are 9 amino acids long. D Distribution of peptide lengths, represented by the number of eluted peptides for each dataset. E The percentage of 9mers peptides predicted to be binders in each immunopeptidomics run. Binding peptide classification was performed using the MHCflurry tool and the corresponding set of HLA-I alleles for each sample. Peptides were classified as HLA-I ligands (binders) when their predicted “affinity percentile” was lower than 2. F Evaluation of the overall overlap between datasets of all 9mers derived by matching against UniProt (dark gray) or StringTie database (magenta). G Percentage of 9mers peptides found in the IEDB repository. H Comparison of the predicted HLA binding affinity profiles between peptides which were found in the IEDB repository or not, for both the peptide dataset obtained from UniProt or StringTie. The vertical gray dotted lines represent, from left to right, the affinity thresholds for “strong binders” (50 nM) and “weak binders” (500 nM), respectively.
Both searches methods gave the standard expected peak at 9 amino acids in length as expected from HLA-I eluted peptides (Fig. 2D). We evaluated the performance of our HLA-I peptide elution in two ways: first, by assessing the sequence motifs of the eluted peptides, and second, by assessing the fraction of peptides considered binders for the specific HLA alleles corresponding to each analyzed sample. Peptide motif deconvolution, performed using Gibbs Clustering tool14, showed that the motifs of the eluted peptides largely overlapped with the expected motifs for the HLA alleles of each subject (Supplementary Fig. 1). However, as previously noted15, this method has some limitations in specificity, which may arise from either a limited number of peptides or the high complexity of the expected haplotypes. Such complexity occurs when two or more HLA alleles in the haplotype exhibit overlapping or similar presentation motifs.
To address peptide specificity more thoroughly, we used the machine learning-based method MHCflurry16. On average, 90% of the 9mers were predicted as strong binders to their respective HLA alleles across all samples (Fig. 2E). Machine learning-based peptide-MHC specificity deconvolution showed consistent pattern of presentation among peptides identified using Uniprot or StringTie (Supplementary Fig. 2).
Additionally, we observed that over 95% of 9mers identified using StringTie were identified using the UniProt dataset, while only the 38.48% of the 9mers in the UniProt dataset were found in the search using the StringTie assembled transcripts database (Fig. 2F).
Next, we focused on comparing individual tumor samples. Overall, peptide overlap analysis revealed the formation of 4 to 5 clusters, with the highest overlap occurring—as expected—between UniProt- and StringTie-derived datasets from the same samples or biological replicates (Supplementary Fig. 3A).
Since the dataset sizes varied, with StringTie-derived peptide sets being approximately half the size of the UniProt-derived sets, we further examined directional overlaps to account for this imbalance. This analysis revealed more clearly defined clusters—specifically, four—and showed that StringTie-derived peptides were largely contained within the corresponding UniProt-derived datasets (Supplementary Fig. 3B).
To have a better understanding of the similarity among immunopeptidomic profiles of different individual regardless of the source database, we performed an overlap search separately on the samples derived by the two searches. We observed that patents’ immunopeptidomics overlapped in consistent pattern regardless of the method (Supplementary Fig. 4A, B). Based on the nature of the immunopeptidomics, we hypothesized that such overlap was a reflection of the similarity in HLA haplotypes among different subjects. Therefore, we have performed of one-hot encoding of each patient using HLA-allele name with a 2-digit specificity and we performed hierarchical clustering, and we observed that we obtained the same clusters indeed underlining that the peptides that we observed are heavily influenced by the MHC composition of each patient (Supplementary Fig. 4C, D). Therefore, the more HLAs molecules subjects have in common the more overlap we will be able to observe. This further underline the need to have a cohort which is large enough to have a sufficient coverage of frequent HLAs.
Interestingly, when considering the 9mers of each datasets, over 90% had already been previously identified as HLA-I ligands and reported in the IEDB database (Fig. 2G), further strengthening confidence of our ovarian cancer dataset. The main difference between peptides found in or absent from the IEDB repository was their differential HLA-binding affinity. Peptides absent from IEDB exhibited weaker binding affinity profiles compared to those previously annotated (Fig. 2H).
These findings collectively support the reliability of the discovered peptides in our ovarian cancer dataset.
Identification of abundant overexpressed genes in ovarian tumor
To further prioritize relevant candidate targets for the development of therapeutically effective vaccines, we sought to identify genes overexpressed in ovarian tumor tissue compared to matching healthy tissue controls. To achieve this, we conducted differential gene expression (DGE) analysis using RNAseq data from healthy ovarian tissues in the GTEx repository and ovarian tumor samples from the TCGA database. To account for potential batch effects that could render the results uninterpretable, we specifically utilized the UCSC Xena Toil Recompute results17. This dataset presents a batch normalization which brings data from both tumor and healthy subjects into a comparable numerical space. Additionally, we processed our ovarian tumor RNAseq dataset utilizing the same quantification and normalization methods. We performed principal component analysis (PCA) of RSEM-normalized transcript counts of the eleven ovarian cancer subjects reported here, the Cancer Genome Atlas HGSC (TCGA-OV) and the GTEx healthy ovarian tissue database (GTEX-OV). We observed that ovarian cancer patients involved in this study clustered with those in the TCGA-OV dataset, while remaining distinct from the GTEX-OV cohort (Fig. 3A). These results support the tumorigenic identity of the collected ovarian tumor samples and demonstrate that meaningful gene expression differences can be obtained when proper precautions are taken to avoid over-interpretation (Fig. 3A).

A Principal component analysis (PCA) plot shows that the gene expression patterns of the ovarian cancer patients subjects exhibit greater similarity to those of the TCGA-OV subjects than to the GTEX-OV subjects. B Scatter plot displays significantly upregulated genes in ovarian cancer patients tumor samples compared to healthy tissue (GTEX). In gray, the genes having base mean >50, s value < 0.05, Log2 Fold Change >1, while, highlighted in red are 65 genes with the following characteristics: base mean >500, s value < 0.001, Log2 Fold Change ≥4.
Next, we performed differential gene expression analysis (DGE) to explore overexpressed genes in tumor tissues compared to the heathy controls. Using Ape-GLM shrinkage, we identified 8311 differentially expressed genes in total. Of these, 4433 were found to be down-regulated in tumor and 3878 were up-regulated (Fig. 3B). To prioritize candidates for subsequent immunogenicity screening, we focused on transcripts which were both strongly over-expressed in tumor and were abundant (i.e. had high baseline gene expression). Applying very stringent criteria (s-value < 0.001, log2(shrunken FC) ≥ 4, and baseline-mean expression ≥ 500) we identified 65 genes. This set included, several known ovarian cancer tumor-associated genes such as MUC1, MUC16, MSLN, CLDN1, further validating the robustness of our approach.
Peptide selected using additional immunological criteria show promising immunogenicity profile
To further prioritize the remaining peptides for experimental immunogenicity testing and to account for multiple criteria related to therapeutic potential, we applied several complementary approaches in parallel rather than sequentially (Fig. 4A and Supplementary Figs. 5, 6), followed by manual curation. We used MHCflurry, a state-of-the-art HLA-binding affinity predictor, to evaluate the allele-specificity of each eluted peptide and prioritize strong binders over weak or non-binders (potential contaminant peptides). Second, we adopted differential expression data to prioritize peptides derived from genes overexpressed in ovarian cancer. Third, we favored peptides empirically observed in multiple subjects in this study over those that appeared to be more “private”. Fourth, we prioritized peptides with binding specificity to highly frequent HLA alleles. We fetched the alleles with a frequency higher than 1/100 of the “USA NMDP European Caucasian” population deposited at the Allele frequency net database (AFND)18. Fifth, we prioritized peptides with expression support from both StringTie and UniProt searches or from StringTie alone. Finally, we used bioinformatic approach (HEX)13 to identify tumor peptides with similarity to pathogen-derived sequences. Additionally, we disfavored peptides identified in the HLA Ligand-Atlas19, as they were considered to be presented in healthy tissues, reducing the probability of being immunogenic.

A Schematic listing criteria used for peptide prioritization. B Interferon-gamma (IFN-γ) secretion by PBMCs from healthy donors (HDs) stimulated with selected peptides. Each dot represents an individual peptide (average of technical duplicates). C IFN-γ secretion by PBMCs from ovarian cancer (OVA) patients stimulated with the same peptides; each dot represents a single peptide. In graphs B and C, each dot represents a single peptide (average of technical duplicates). D Interferon gamma secretion upon activation of HD PBMCs stimulated with the selected peptides. Response is deconvoluted at peptide level and each dot represent the response of a different HD. E Interferon gamma secretion upon activation of OVA patients’ PBMCs stimulated with the selected peptides. Response is deconvoluted at peptide level and each dot represent the response of a different patient. F Percent response rate deconvoluted by peptides for both HDs (black) and OVA patients (magenta). Dotted line represents 25% response rate as reference. G Focus on assessment of cancer specific response for peptides with tumor-specific response (4, 8 and 13). Data from peptide 4 and 13 were analyzed using Mann–Whitney test, while data from peptide 8 were analyzed using Wilcoxon signed-rank test (*p value < 0.05) as all HD responses were 0. In all the graphs showing ELISpot data, the dotted line on the y axis represents a response threshold which correspond to 23.3 spots/1*106 PBMCs as from ref. 12.
Combining all the above-described methods, we identified 13 candidate peptides for further immunological characterization (Table 1).
Next, to assess the immunogenicity of the selected peptides and evaluate their therapeutic potential, we synthesized the peptides at high purity and tested their ability to activate T cells derived from PBMCs obtained from healthy donors or ovarian cancer patients. PBMCs from healthy donors were selected based on their HLA haplotypes, allowing us to test each peptide multiple times. Upon stimulation of PBMCs from healthy donors with the selected peptides, we observed that our selected peptides were able to produce T cell activation. Interestingly, on average, we observed that our peptides were less prone to activate PBMCs derived from male donors (Fig. 4B). Additionally, 4 out of 9 ovarian cancer patients exhibited T cell responses to one or more of the selected peptides (Fig. 4C). When analyzing the responses at the individual peptide level, we found that some peptides did not induce T cell activation in any of the subjects, while others elicited stronger responses in PBMCs from either healthy donors or patients (Fig. 4D, E). Peptides 5, 6, 7, and 10 triggered responses in more than 25% in both healthy donors’ and ovarian cancer patients PBMCs (Fig. 4F). However, only peptide 8 elicited a significantly stronger response in ovarian cancer patients compared to healthy donors, suggesting a tumor-specific activation profile (Fig. 4G).
Overall, our results demonstrate that several peptides identified through immunopeptidomics exhibit promising immunogenicity profiles, suggesting their potential for use in developing therapeutic cancer vaccines for ovarian cancer.
Discussion
Despite initial response to current therapies, most ovarian cancer patients experience recurrence and need further treatment. The development of chemotherapy-resistant disease poses a significant challenge, highlighting the necessity for new therapeutic interventions, such as immunotherapy20,21.
Indeed, ovarian cancer present variable levels of T cell infiltration which generally considered to be quite low22,23. Overall, the presence of tumor-infiltrating lymphocytes (TILs) has been associated with increased progression-free survival (PFS) and overall survival (OS)24,25,26. Ovarian cancer represents no exception, and the presence of TILs, especially CD8+ T cells, is indicative of a good prognosis in patients, regardless of tumor stage24,27. Consequently, ovarian cancer is regarded as a promising target for immunotherapeutic approaches, including cancer vaccines.
Numerous clinical trials over the past three decades have investigated peptide vaccines for ovarian cancer treatment, yet with limited efficacy28.
Several unmet challenges hinder the development of effective peptide vaccines and require careful consideration in their design. One critical issue identified in previous trials, which have potentially hindered their results, was the use of “classical” tumor antigens with poor tumor expression or limited evidence of presentation in the immunopeptidome29.
To address this, it is essential to identify peptides directly presented on HLA molecules at the surface of cancer cells30. Advanced techniques such as immunoaffinity purification of the HLA complex, followed by elution and the identification of naturally presented HLA ligands using liquid chromatography and tandem mass spectrometry, represents the current state-of-the-art method to uncover the immunopeptidome landscape of cancer cells31.
Integrating novel sequencing data with large public databases can present challenges, particularly due to batch effects that complicate the interpretation of results32. To overcome this issue, we opted to utilize the Toil dataset developed by Vivian et al., which is one of the most extensive and consistently analyzed repositories of human RNA-seq expression data17. This dataset integrates both tumor and healthy samples and normalized and brought into a comparable numerical space, ensuring more reliable and meaningful comparisons.
The low tumor mutation burden (TMB)22,23,33,34 in ovarian cancer makes it inconvenient to target tumor-specific antigens (TSAs) resulting from mutations for vaccine development. In contrast, tumor-associated antigens (TAAs), originating from aberrantly expressed proteins, are less immunogenic but more commonly shared among patients, enhancing their therapeutic potential.
To conduct our analysis, we used transcripts assembled with StringTie. While the vast majority of peptides identified using the StringTie dataset were also found in the UniProt search, only about 48% of the peptides identified in the UniProt search were present in the StringTie-derived dataset. We believe this discrepancy is primarily due to technical rather than biological reasons. Specifically, this might be due to the larger nature of the proteogenomic search space in comparison to the use of UniProt as a reference. Indeed the StringTie database is produced by a six-frame translation by PEAKS, substantially increasing the search space. As a result, the number of peptide identifications decreases, which is consistent with findings reported by Chong et al.35.
In future analyses, the search space could be refined by avoiding six-frame translation, for example, by using open reading frame (ORF) prediction tools such as TransDecoder (https://github.com/TransDecoder/TransDecoder), as implemented in other pipelines36.
In our study we identified high-frequency candidate target epitopes presented across multiple ovarian cancer specimens supporting their suitability for vaccine development. We are not disheartened to learn that many of our highly ranked peptide candidates are already identified and patented for this specific purpose. For example, we identified the “ALKARTVTF” peptide, derived from the CCR5 gene, which is overexpressed 16-fold in ovarian tumors. Additionally, this peptide was predicted to have strong binding, presentation, and was empirically observed in more than half of our subjects’ tumors. Interestingly, CCR5 has low expression level in multiple healthy tissues (Supplementary Fig. 5A, B) while having elevated expression in multiple cancer types. Indeed, in addition to ovarian cancer, CCR5 has high tumor-specific expression in breast, pancreas, renal, testis and skin cancers (Supplementary Fig. 6)37. CCR5 has already been chosen as a target in multiple clinical trials and is well known as a hallmark of cancer progression38. Indeed, it was patented for the treatment of ovarian cancer in 2017 (WO2018138257A1). However, while we observed increased T cell activation toward the CCR5 epitope in ovarian cancer patients compared to healthy donors, this difference was not statistically significant.
Another noteworthy target that supports the validity of our pipeline is peptide 9, “TYSEKTTLF,” an HLA-A24-restricted epitope derived from MUC16. MUC16 is largely undetectable in normal ovarian tissues and in many other healthy tissues (Supplementary Fig. 5A, B), yet it is highly expressed in over 80% of ovarian cancer samples (Supplementary Fig. 6)39. Despite this strong tumor-associated expression, we unexpectedly observed no difference in T cell reactivity to this epitope between ovarian cancer patients and healthy donors.
Additionally, we tested two splice-variant aberrations variants: DRYLLVSQF from DDX42 and KYLTIYLQK from TRMT10B. DDX42 has been associated with tumor progression in epithelial ovarian cancers and poor prognosis in HGSC40,41, whereas no specific link has been established between TRMT10B and ovarian cancer. DDX42 exhibited moderately consistent expression across multiple healthy tissues (Supplementary Fig. 5A, B), while TRMT10B showed low expression levels in normal tissues. Notably, both genes were expressed at higher levels in normal tissues compared to tumors (Supplementary Fig. 6), suggesting that, based on conventional gene expression profiles, they may not be optimal candidates. However, the peptides selected from these genes originate from cancer-specific aberrant splice variants, which may confer tumor specificity not apparent from gene-level expression alone.
Interestingly, overexpression of TRMT10C, a related family member, has been linked to poorer survival in gynecologic cancers42. In contrast to TRMT10B, TRMT10C shows higher expression in multiple cancer types (Supplementary Fig. 6), highlighting a divergent expression pattern within this gene family.
Ultimately, among all the tested peptides, only VVHLIKNAY from ITGB2 was able to produce significantly different activation of PBMCs from cancer patients compared to healthy donors, who showed no reactivity. Interestingly, the peptide was identified only in the search using the StringTie assembled database, and was overlooked by the search using the UniProt database highlighting the added value of our customized approach. The role of ITGB2 in ovarian cancer appears controversial. A comprehensive analysis conducted by Li et al. demonstrated a close association between ITGB2 upregulation and ovarian tumorigenesis43. Additionally, it has been observed that increased ITGB2 expression correlated with an unfavorable clinical outcome, heightened immune cell infiltration, including M2 macrophages, and moderate correlation with CD4+/CD8+ T cells and B cells44. On the other hand, expression profiling revealed that ITGB2 is also moderately elevated in healthy lung tissue and highly expressed in the spleen (Supplementary Fig. 5A, B). Although ITGB2 is overexpressed in several tumor types (Supplementary Fig. 6), this signal is largely overshadowed by its high expression in healthy blood. While this didn’t seem to hinder its immunogenicity in ovarian cancer patients, this pattern raises concerns about potential toxicity and off-target immune responses.
Further research is required to determine whether the peptide-specific immune responses elicited by our vaccine candidates translate into effective tumor cell killing. The detection of immune activation in response to tumor-associated antigens (TAAs)—which are typically considered poorly immunogenic—in a cohort of immunotherapy-naïve subjects is already a notable finding. While we have demonstrated the immunogenic potential of these peptides, we have not yet shown that the immune cells recognizing these peptides can functionally eliminate tumor cells presenting them. This represents a key limitation of our current study and a critical focus for future investigations.
One additional limitation of the vaccination approach in ovarian cancer is the immunosuppressive tumor microenvironment (TME) which hampers the efficacy of T cells in killing and controlling malignant cells45. Indeed, OC have been observed to express high level of PD-L1 and TILs in these tumors frequently exhibit elevated levels of LAG333,46. To overcome this challenge, future investigations should explore combining cancer vaccine strategies with immune checkpoint blockade therapies to counteract TME-induced immunosuppression.
Methods
Patient samples
Hospital sampling was performed from 28 ovarian cancer patients from mid-2020 until mid-2021. Samples were collected according to the declaration of Helsinki. All patients signed informed consent prior to samples’ collection. The study was approved by the Research Ethics Committee of the Northern Savo Hospital District (approval number 350/2020).
Ovarian tumor samples were dissected from primary ovary tumors (pelvic location) and metastatic nodes which were spread through the abdomen (omental and splenic locations). Fatty, necrotic, and bloody areas were removed and the remaining, cleaned tissues snap-frozen in liquid nitrogen prior to HLA-bound peptide enrichment. A second, pathologically similar portion of the same tumor specimen was stored in RNAprotect Tissue Reagent (Qiagen, # 76106) prior to RNA extraction. Although 28 subjects were enrolled in the clinical study, only eleven were given HGSC diagnoses, had sufficient metastatic omental tissues collected and could be included in further analysis (Supplementary Table 1).
Purification of HLA class-I complexes
HLA-I –peptide complexes were immunoaffinity purified from ovarian tumor samples using HLA-I antibody (anti-human HLA-A, HLA-B, HLA-C, clone W6/32, InVivoMab) via the method described by Bassani-Sternberg47 with minor modifications. Frozen tumor tissue was cut into 1 × 1–3 × 3 mm3 fragments and further mechanically dissociated using a gentleMACS™ Dissociator (without any enzymatic treatment). Dissociated tissue was lysed with 0.25% sodium deoxycholate, 0.2 mM iodoacetamide, 1 mM EDTA, 1 mM PMSF (phenylmethylsulfonyl fluoride), and 1% octyl-β-D glucopyranoside in the presence of protease inhibitors in PBS at 4 °C for 2 h. The lysate was precleared (2000 × g, 5 min at 4 °C) and cleared by centrifugation at 20,000 × g, 30 min at 4 °C prior to loading to the immunoaffinity column (AminoLink, Pierce) with covalently linked antibody. Following binding (o/n at 4 °C) the affinity column was washed using 10 column volumes of each buffer (150 mM NaCl, 20 mM Tris-HCl; 400 mM NaCl, 20 mM Tris-HCl; 150 mM NaCl, 20 mM Tris-HCl and 20 mM TrisHCl, pH 8.0) and bound complexes were eluted in 10% acetic acid.
Eluted peptide-HLA-I complexes were desalted using SepPak-C18 cartridges (Waters). The cartridge was prewashed with 80% acetonitrile in 0.1% trifluoro acetic acid (TFA) and with 0.1% TFA prior to loading of the samples. Samples were washed with 0.1% TFA and peptides were eluted and in 30% acetonitrile in 0.1% TFA prior to drying using vacuum centrifugation (Eppendorf).
LC-MS/MS analysis of HLA-I peptides and proteomics database search
LC-MS/MS and peptide identification were performed on a fee-for-service basis by the CIC bioGune in Bilbao, Spain.
Samples (200 ng) were loaded in a timsTOF Pro with PASEF (Bruker Daltonics) coupled online to either an Evosep ONE (Evosep) or a NanoElute (Bruker) liquid chromatograph. The 30 samples-per-day protocol (44-min gradients) was used with the Evosep ONE, whereas a custom 30-min gradient was used for the NanoElute runs. The runs were performed for each of the samples: (1) a preliminary load of 1/20 of the sample in order to check sample load, (2) an adjusted sample load where only z = 1 ions are analyzed, and (3) an adjusted sample load where z > 1 ions are analyzed. Protein identification and quantification was carried out using the PEAKS X software (Bioinformatics solutions). All three loads for each sample were summed and considered as a single sample. Searches were carried out against a database consisting of Homo sapiens (Uniprot/Swissprot) or a multi-sample, merged Stringtie-generated Expressed Sequence Tag (EST) ad hoc database (described more in depth later), with precursor and fragment tolerances of 20 parts per million (ppm) and 0.05 Da respectively. HLA class I-presented peptides with a mass range between 400 and 650 m/z, and with charge states <4+ were considered for further analysis in line with what has been done elsewere48,49,50.
Total RNA extraction, RNA sequencing and comparative analysis with GTEX and TCGA database
As previously mentioned, the tumor specimen’s portion to be dedicated to RNA-seq was stored in tumor specimen was stored in RNAprotect Tissue Reagent (Qiagen, # 76106) prior RNA extraction. Total RNA was extracted from ~30 mg of patient tumor tissues by RNAeasy Protect Cell Mini Kit (Qiagen, # 74624) according to the manufacturer’s instruction. RNA sequencing was performed by Eurofins Genomics to a minimum depth of 30 M reads on Illumina NovaSeq 6000 instrument with 2x150bp paired-end, strand-specific sequencing procedure. Raw data from healthy omentum, and metastatic omentum were analyzed for using UCSC Toil RSEM gene expression pipeline (hg38, gencode v23)17, and merged with the GTEx (Genotype-Tissue Expression), TARGET, and TCGA (The Cancer Genome Atlas) Toil Recompute archive [github, link]. This particular pipeline and reference pairing was selected in order to maintain bioinformatic methods comparability between the new clinical samples and the well-known healthy-tissue efforts available in public databases.
RNA-sequencing of ovarian tumor tissues and AS identification
Each patient’s transcriptome was processed separately, and subsequently merged into a “pan-transcriptome” using Stringtie in “—merge” mode. The pan-transcriptome’s splice junctions were annotated by merging with their overlapping canonical isoforms (Gencode v23) using the “gffread” utility, and repeats were masked using “gffcompare”. For compatibility with the MS spectral identification program (PEAKS X), as reported in the software's user manual (Section 6.1), splice junctions were converted into mature transcripts in EST format, using the gffread, ‘-w’ flag from the hg38 reference genome coordinates.
To maintain congruent comparisons, Stringtie was not run using a HISAT2 re-alignment (per best practices), but rather run using the same aligned reads matrix from above (“Total RNA extraction, RNA sequencing and comparative analysis with GTEX and TCGA database”).
HLA-typing
HLA typing was inferred from RNA-Seq data using the ArcasHLA pipeline51. The ArcasHLA suite contains an explicit nucleotide sequence database which was constructed from the implicit HLA allele differences as described by The International ImMunoGeneTics database (IMGT) consortium. ArcasHLA performs typing inference by extracting aligned reads from chromosome 6 and then uses the Kallisto pseudoaligner to rapidly bin reads against each reference sequence. The sequence, or pair of sequences which matches the most reads is inferred to be the correct set.
A handful of subjects had multiple sequenced biological specimens. Specimens were typed independently and the few conflicts that were observed were resolved in favor of the type for which binding affinity data was available in MHCFlurry.
Because our peptide immunoaffinity methods only targeted class-I HLA molecules, only class I matches were retained.
European ancestry was selected as a prior expectation for the purposes of identifying rare alleles.
To further maintain congruent comparisons, ArcasHLA was run on the same aligned reads matrix from above (“Total RNA extraction, RNA sequencing and comparative analysis with GTEX and TCGA database”). The table of patients’ HLA-typing is showed in Supplementary Table 2.
RNA-seq differential expression
RSEM values were imported from either the Xena precalculated RSEM isoform values, or from the Toil pipeline “rsem_isoforms.results” files. Transcript-specific counts were imported by the “tximport” function in “scaledTPM” mode. Counts were normalized by DESeq2 for differences in sequencing effort. PCA biplots were created from the variance-stabilized values. Differential expression was calculated using DESeq2 between the 11 new tumor samples and the GTEX healthy ovarian tissue.
Shrunken Log2-fold changes were calculated using the “apeglm” model using an expected “lfcThreshold” value of 1. Genes were considered differentially expressed if their shrunken log-fold change was greater than 1, their base-mean values were greater than 40.
Peptide overlap calculation
Pairwise overall overlap was recursively calculated as the number of peptides common to both sets (i.e., the intersection) divided by the total number of unique peptides identified in either set (i.e., the union).
On the other hand, left overlap was recursively calculated as the number of peptides common to both sets divided by the total number of peptides in one of the two sets being compared.
Class I MHC binding affinity prediction
Peptides derived from MS were filtered for those which were 8–15 amino acid residues in length. Each peptide MHC specificity was annotated using a predicted class I MHC binding affinity (nM), presentation probability score, and processing probability score as calculated by MHCflurry16.
HEX ranking
Putative neo-antigens between 8 and 12 amino acid residues in length were ranked using the HEX pipeline. HEX is a proprietary solution owned by Helsinki University which depends on a neural network to rank binding affinity (NetMHCpan 4.1b52), and uses a proprietary custom position-specific weight-matrix (PWM) to assess a peptide’s similarity to pathogens which humans have immuno-historically encountered.
The binding domain of the HLA molecule is approximately 9 residues in width, and therefore we chose to assess viral similarity with strong preference towards the central portion. This algorithmic weighting is achieved using HEX’s custom PWM.
HEX also includes a preformatted pathogen database which provides the “hit” sequences against which putative neoantigens are compared. HEX uses the NCBI BLASTp to perform this search. Although the HEX database can be easily substituted to support other pathogens, this work uses only the 17705-record Uniprot “Viruses [10239]” database by default. Candidate peptides are matched with potential viral orthologs by BLASTp (BLOSUM62)53, and then further refined by pairwise positional-weighted alignment using PMBEC54. Peptides with any viral ortholog then have their binding affinity predicted by NetMHCpan for each supplied HLA allele. Peptide specificity was considered as the highest binding affinity for a given MHC. Finally, an aggregate score is calculated to rank the predicted probability of a peptide for eliciting an immune response in the originating subject. When comparing tumor- and virus-derived pair of peptides the average of the binding affinity was computed for display, and pair of peptides were considered good candidates only when IC50 for both tumor and viral peptides was below 500 nM. The specific mathematics of the HEX viral similarity assessment are detailed briefly as follows. Similarity to viral sequences was further refined by pairwise positional-weighted alignment using PMBEC (link below) substitution matrix favoring the similarity in the central portion of the peptides as follows.
Where :
Final similarity score was normalized according to the following formula:
Where “a” is the similarity score calculated between each tumor and its cognate viral peptide, “b” and “c” are the similarity scores calculated by self-aligning the tumor peptide and the viral peptide respectively.
Epitope selection for immunological analysis
Identified peptides eluted from ovarian cancer specimens were searched into the HLA Ligand Atlas database19 which is the gold standard repository for ligand peptides commonly expressed in healthy tissues. Peptides identified in the HLA Ligand Atlas were considered to have lower chances to be immunogenic as they were presented in healthy tissues and probably covered by central tolerance.
Additionally, peptides were searched within the IEDB T Cell database55. Presence of a given peptide in this database was only used to gather information about its prior investigations. The database can be obtained at the following link.
Identified peptides MHC allele-specificity information were further annotated with the seroprevalence data available for the US and European population using information provided by the Allele Frequency Net Database (AFND) available at allelefrequencies.net18. Annotations were made using the “USA NMDP European Caucasian” (n = 1,242,890) dataset as retrieved in December 2020. Before adding the information to the peptide results, HLA types were filtered to only those with a population frequency greater than 1 in 100. Peptides with higher allele seroprevalence were favored over others specific to more rare alleles. When peptides presented multiple specificities, cumulative sum of the specific alleles’ frequencies were used.
PBMCs purification
Ten to thirty mL of blood was collected from patients immediately prior to the surgical tumor resection in heparin salts coated tubes. PBMC were purified using Leucosep™ tubes according to manufacturer’s instruction. PBMCs were then counted and cryopreserved in Human AB serum (Sigma) 10% DMSO (Sigma).
Healthy donors PBMCs
Healthy donors PBMCs were purchased from CTL (Bonn, Germany). The subjects were selected based on their HLA types to optimize the peptide immunogenicity testing.
Peptide synthesis
Selected peptides to be tested for immunogenicity (listed in Table 1) were synthetized by Genscript (Netherlands) at 4 mg, 90% purity.
IFN-γ ELISpot assay
T cell epitope-specific activation was detected using commercially available IFN-γ ELISpot reagents (ImmunoSpot, Bonn, Germany), accordingly to the manufacturer’s instructions. According to their HLA haplotype, each subject was tested only for the peptides that had corresponding HLA specificity. Each subject’s PBMCs were seeded at the maximum density possible (depending on the number of stimuli) but always below 600 K cells/well. Seeded PBMCs were stimulated in vitro with 2 ug/well of each peptide at 37 °C for 72 h. After 3 days of stimulation, the number of cytokine-producing, antigen-specific T cells was evaluated using an ELISpot reader system (ImmunoSpot) and all the spots’ counts were normalized for 1 × 106 seeded PBMCs. A response was deemed positive if the IFNγ ELISpot count exceeded a minimum threshold of 23.3 spots per 1*106 PBMCs as shown elsewhere12.
Data availability
RNA-seq count data and a summary table of eluted peptides are provided in the Supplementary Materials. Raw datasets generated during this study are not publicly available due to patient data privacy reasons but are available from the corresponding author upon reasonable request.
References
Karnezis, A. N. & Cho, K. R. Preclinical models of ovarian cancer: pathogenesis, problems, and implications for prevention. Clin. Obstet. Gynecol. 60, 789–800 (2017).
Kuroki, L. & Guntupalli, S. R. Treatment of epithelial ovarian cancer. BMJ 371, m3773 (2020).
Peng, Z. et al. PD-1/PD-L1 immune checkpoint blockade in ovarian cancer: dilemmas and opportunities. Drug Discov. Today 28, 103666 (2023).
Wan, C. et al. Enhanced efficacy of simultaneous PD-1 and PD-L1 immune checkpoint blockade in high-grade serous ovarian cancer. Cancer Res. 81, 158–173 (2021).
Curiel, T. J. et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 10, 942–949 (2004).
Feola, S. et al. Uncovering the tumor antigen landscape: what to know about the discovery process. Cancers 12, 1660 (2020).
Feola, S., Chiaro, J. & Cerullo, V. Integrating immunopeptidome analysis for the design and development of cancer vaccines. Semin. Immunol. 67, 101750 (2023).
Schuster, H. et al. The immunopeptidomic landscape of ovarian carcinomas. Proc. Natl. Acad. Sci. USA 114, E9942–e9951 (2017).
Zhao, Q. et al. Proteogenomics uncovers a vast repertoire of shared tumor-specific antigens in ovarian cancer. Cancer Immunol. Res. 8, 544–555 (2020).
Balachandran, V. P. et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551, 512–516 (2017).
Łuksza, M. et al. Neoantigen quality predicts immunoediting in survivors of pancreatic cancer. Nature 606, 389–395 (2022).
Rojas, L. A. et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 618, 144–150 (2023).
Chiaro, J. et al. Viral molecular mimicry influences the antitumor immune response in murine and human melanoma. Cancer Immunol. Res. 9, 981–993 (2021).
Andreatta, M., Lund, O. & Nielsen, M. Simultaneous alignment and clustering of peptide data using a Gibbs sampling approach. Bioinformatics 29, 8–14 (2013).
Chiaro, J. et al. Development of mesothelioma-specific oncolytic immunotherapy enabled by immunopeptidomics of murine and human mesothelioma tumors. Nat. Commun. 14, 7056 (2023).
O’Donnell, T. J., Rubinsteyn, A. & Laserson, U. MHCflurry 2.0: improved pan-allele prediction of MHC class I-presented peptides by incorporating antigen processing. Cell Syst. 11, 42–48.e7 (2020).
Vivian, J. et al. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 35, 314–316 (2017).
Gonzalez-Galarza, F. F. et al. Allele frequency net database (AFND) 2020 update: gold-standard data classification, open access genotype data and new query tools. Nucleic Acids Res. 48, D783–d788 (2020).
Marcu, A. et al. HLA ligand Atlas: a benign reference of HLA-presented peptides to improve T-cell-based cancer immunotherapy. J. Immunother. Cancer 9, e002071 (2021).
Lheureux, S. et al. Epithelial ovarian cancer. Lancet 393, 1240–1253 (2019).
Schaar, B. et al. Cell-based immunotherapy in gynecologic malignancies. Curr. Opin. Obstet. Gynecol. 31, 43–48 (2019).
Cristescu, R. et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362, eaar3593 (2018).
Park, J., Lee, J. Y. & Kim, S. How to use immune checkpoint inhibitor in ovarian cancer? J. Gynecol. Oncol. 30, e105 (2019).
Clarke, B. et al. Intraepithelial T cells and prognosis in ovarian carcinoma: novel associations with stage, tumor type, and BRCA1 loss. Mod. Pathol. 22, 393–402 (2009).
Zhang, L. et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 348, 203–213 (2003).
Mantia-Smaldone, G. M., Corr, B. & Chu, C. S. Immunotherapy in ovarian cancer. Hum. Vaccin. Immunother. 8, 1179–1191 (2012).
Hwang, W. T. et al. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol. Oncol. 124, 192–198 (2012).
Chow, S., Berek, J. S. & Dorigo, O. Development of therapeutic vaccines for ovarian cancer. Vaccines 8, 657 (2020).
Nelde, A. et al. Immunopeptidomics-guided warehouse design for peptide-based immunotherapy in chronic lymphocytic leukemia. Front. Immunol. 12, 705974 (2021).
Di Marco, M., Peper, J. K. & Rammensee, H. G. Identification of immunogenic epitopes by MS/MS. Cancer J. 23, 102–107 (2017).
Falk, K. et al. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature 351, 290–296 (1991).
Wang, Q. et al. Unifying cancer and normal RNA sequencing data from different sources. Sci. Data 5, 180061 (2018).
Morand, S. et al. Ovarian cancer immunotherapy and personalized medicine. Int. J. Mol. Sci. 22 (2021).
Vareki, S. M. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 6, 157 (2018).
Chong, C. et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat. Commun. 11, 1293 (2020).
Fallon, T. R. et al. transXpress: a Snakemake pipeline for streamlined de novo transcriptome assembly and annotation. BMC Bioinforma. 24, 133 (2023).
Aldinucci, D., Borghese, C. & Casagrande, N. The CCL5/CCR5 axis in cancer progression. Cancers 12, 1765 (2020).
Jiao, X. et al. Recent advances targeting CCR5 for cancer and its role in immuno-oncology. Cancer Res. 79, 4801–4807 (2019).
Zhai, Y. et al. MUC16 affects the biological functions of ovarian cancer cells and induces an antitumor immune response by activating dendritic cells. Ann. Transl. Med. 8, 1494 (2020).
D’Oronzo, S. et al. DEAD-Box Helicase 4 (Ddx4)(+) stem cells sustain tumor progression in non-serous ovarian cancers. Int. J. Mol. Sci. 21, 6096 (2020).
Hashimoto, H. et al. Germ cell specific protein VASA is over-expressed in epithelial ovarian cancer and disrupts DNA damage-induced G2 checkpoint. Gynecol. Oncol. 111, 312–319 (2008).
Zhao, S. et al. The potential regulatory role of RNA methylation in ovarian cancer. RNA Biol. 20, 207–218 (2023).
Li, C. et al. Identifying ITGB2 as a potential prognostic biomarker in ovarian cancer. Diagnostics 13, 1169 (2023).
Li, C. et al. Identifying ITGB2 as a potential prognostic biomarker in ovarian cancer. Diagnostics 13, (2023).
Baci, D. et al. The ovarian cancer tumor immune microenvironment (TIME) as target for therapy: a focus on innate immunity cells as therapeutic effectors. Int. J. Mol. Sci. 21, (2020).
Westergaard, M. C. W. et al. Tumour-reactive T cell subsets in the microenvironment of ovarian cancer. Br. J. Cancer 120, 424–434 (2019).
Bassani-Sternberg, M. Mass spectrometry based immunopeptidomics for the discovery of cancer neoantigens. Methods Mol. Biol. 1719, 209–221 (2018).
Hoenisch Gravel, N. et al. TOF(IMS) mass spectrometry-based immunopeptidomics refines tumor antigen identification. Nat. Commun. 14, 7472 (2023).
Marcu, A. A.-O. et al. HLA Ligand Atlas: a benign reference of HLA-presented peptides to improve T-cell-based cancer immunotherapy. LID https://doi.org/10.1136/jitc-2020-002071 (2021).
Bichmann, L. et al. MHCquant: automated and reproducible data analysis for immunopeptidomics. J. Proteome Res. 18, 3876–3884 (2019).
Orenbuch, R. et al. arcasHLA: high-resolution HLA typing from RNAseq. Bioinformatics 36, 33–40 (2020).
Reynisson, B. et al. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48, W449–W454 (2020).
Altschul, S. F. et al. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Kim, Y. et al. Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinforma. 10, 394 (2009).
Vita, R. et al. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 47, D339–D343 (2019).
Acknowledgements
We would like to thank the staff of Kuopio University Hospital for their assistance with sample collection during surgeries, and Satu Kaipainen and Vita Albers-Skirdenko for their help with sample processing, PBMC purification, and preservation. V.C. acknowledges the support of the European Research Council (ERC) under the Horizon 2020 framework programme (Grant Agreement No. 681219), the Magnus Ehrnrooth Foundation (Project No. 4706235), the Jane and Aatos Erkko Foundation (Project No. 4705796), the Finnish Cancer Foundation (Project No. 4706116), the Helsinki Institute of Life Science (HiLIFE) (Project No. 797011004), and the Digital Precision Cancer Medicine Flagship iCAN. V.C. and S.F. also acknowledge support from the GeneCellNano Flagship. J.C. acknowledges the support of the Doctoral Programme in Drug Research (DPDR) at the University of Helsinki and the Finnish Cultural Foundation. K.P. acknowledges support from the K. Albin Johansson Foundation.
Author information
Authors and Affiliations
Contributions
Conceptualization: J.C., K.P., O.G., T.K., V.C. Investigation: J.C., K.P., S.F., M.E., S.W., K.Õ., S.R., O.G., M.A., F.E. Data curation: J.C., A.B., O.G. Formal analysis: J.C., K.P., M.Az., F.E., S.R., A.B., O.G. Software: J.C., A.B.; Visualization: J.C., A.B. Project administration: O.G., T.K., V.C. Writing - original draft: J.C., K.P., A.B., M.E., O.G. Writing - Review & Editing: all authors. Resources: H.S., M.An. Funding acquisition: T.K., V.C.
Corresponding author
Ethics declarations
Competing interests
V.C. is co-founder and shareholder of Valo Therapeutics Oy. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chiaro, J., Peltonen, K., Õunap, K. et al. Proteogenomic approach to immunopeptidomics of ovarian tumors identifies shared peptide vaccine candidates. npj Vaccines 10, 195 (2025). https://doi.org/10.1038/s41541-025-01234-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41541-025-01234-6
