
Abstract
Ampicillin (AMP) poses a significant environmental hazard to aquatic ecosystems, and previous research has been inadequate in addressing the ecological toxicity of its byproducts. A novel photocatalyst, WO3-BiFeO3/digestate biochar (DSB), denoted WBD, was synthesized for efficient photocatalytic degradation of AMP. In our study, 150 mg WBD achieved 99.93% AMP degradation in 3 h at a concentration of 100 mg/L. WBD maintained a stable degradation performance under various environmental stressors, including pH, and the presence of Cl−, NO3−, and HA. •O2−, h+, •OH, and 1O2 were identified as primary active oxygen species. WBD effectively targeted the -NH2, -SH, and β-lactam ring-forming -COOH of AMP, facilitating its transformation into low-toxic or non-toxic degradation products, and establishing a detoxification pathway. Overall, this study introduces a novel environmentally material that demonstrates a high degradation efficiency, long-lasting effectiveness, and low product toxicity, offering a promising approach for the effective management of emerging pollutants.
Similar content being viewed by others
Introduction
Antibiotics have become a significant global environmental concern, drawing the attention of numerous countries, where they have been identified as critical targets for pollution control1,2,3. In 2021, China alone produced 231,000 tons of antibiotics, representing ~15% of the world’s total production, yet its per capita consumption accounted for 50% of global antibiotic use. During the same period, the United States and the European Union produced 143,000 and 49,300 tons, respectively3,4. However, the excessive use of antibiotics results in their accumulation in aquatic environments and, subsequently, in organisms, thereby posing risks to human, animal, and plant health5. Ampicillin (AMP), a β-lactam antibiotic that is widely used in clinical medicine for treating infections, is classified by the World Health Organization as an essential drug for children. However, a significant portion (50–70%) of administered AMP is excreted unchanged into the environment, leading to pseudo-persistence at ng/L to mg/L levels in water bodies6,7,8,9. Notably, AMP undergoes hydrolysis in aquatic environments under varying conditions, with a half-life that ranges from 5 to 27 d. Although the degradation of AMP has been attempted through environmental self-purification processes, the potential hazards of the resulting degradation products are often overlooked10,11. Upon photoexcitation, AMP undergoes carbonyl addition and dehydrogenation, generating products that are known for their ecological toxicity. More specifically, these products are suspected to inhibit algal growth by disturbing hormone levels, thereby disrupting aquatic food chains and potentially promoting the expression of antibiotic resistance genes (ARGs) and the proliferation of resistant bacteria12,13,14. Therefore, the effective management of AMP should not only prioritize its efficient removal, but should also focus on minimizing the toxicity of its degradation products.
Photocatalysis is a promising approach for the treatment of persistent organic pollutants due to its robust catalytic oxidation ability, high degradation efficiency, stable performance, and environmentally friendly outcomes. Previous studies have found that photocatalytic materials such as C3N4-MoS2/3DG and CNTs/AG/Ti demonstrate excellent AMP degradation efficiencies. However, the toxicities of the resulting by-products tend to be higher than that of the parent AMP, thereby posing a persistent environmental hazards15,16. Consequently, there is an urgent requirement to explore new degradation materials capable of efficiently decomposing AMP, while mitigating their toxic impacts on biological organisms.
Bismuth-based materials, including Bi2O3, Bi2WO6, BiVO4, and BiOX (X = halogen), represent the forefront of photocatalysis research17. BiFeO3 is known to exhibit ferroelectric and antiferromagnetic properties. This material generates depolarization fields through internal charge movement at room temperature, suppressing the recombination of photogenerated holes and electrons, thereby enhancing its photocatalytic efficiency18. In addition, WO3 is an n-type semiconductor metal oxide with wide energy band gap (2.5–2.7 eV) and a hole diffusion strength of 150 nm19. Although this material is stable, inexpensive, and less polluting to the environment20, its photocatalytic ability is limited owing to the fast rate of electron–hole complexation, which can be effectively solved through heterostructure construction. Previous studies have successfully co-doped BiFeO3 with WO3 to create a type-II heterojunction, significantly boosting the photocatalytic capability of the material, particularly in the degradation of the organic dye methylene blue21. However, a low photocatalytic efficiency, limited reuse cycling characteristics, and a poor magnetic recyclability have impeded the further development of such materials22. It is therefore desirable to identify suitable electron transfer mediators to form a three-component dual Z-scheme heterojunction photocatalyst to provide highly reducing electrons (e−) and highly oxidizing holes (h+) to enhance the catalytic efficiency and lifetime23.
In the treatment of kitchen waste, anaerobic digestion generates large amounts of organic digestate, which is mainly treated by incineration, and results in a waste of resources. Biochar (BC) derived from agriculture, forestry, animal husbandry, and urban sludge, has been extensively applied in water treatment processes, offering an effective pathway for the resource utilization of organic solid waste24. BC possesses a variety of defect structures that enhance charge separation efficiency. Its composite with metal oxides can further improve intrinsic properties, such as narrowing the bandgap width and extending the lifetime of photocarriers, making it a promising substrate material for photocatalysis25,26. The combination of BC and photocatalysts was demonstrated to generate stable Z-scheme heterojunctions that produced less-toxic products; however, the pathways associated with their production remain relatively unknown27. Therefore, the study of low-toxicity pathways of biochar-based photocatalysts in AMP degradation is not only a waste-to-resource management method, but also an innovative endeavor.
This study therefore aims to develop an environmentally friendly photocatalytic material for the degradation of AMP in water. For this purpose, BC derived from the anaerobic digestion of kitchen waste is used as a substrate and is modified by metal oxide doping to enhance its photocatalytic properties. Microfluidics is employed to prepare WO3-BiFeO3/digestate biochar (DSB) magnetic gel balls. The photocatalytic oxidation capability of the material is investigated, along with the detoxification pathways of its degradation products. The material ratios, dosage, pH, anions, and natural organic matter (NOM) are systematically evaluated to optimize the photocatalytic performance. A range of characterization techniques are used to analyze the surface morphology and structural composition of the catalyst, and to assess its photoresponse, charge transfer efficiency, and redox capability. Computational simulations using the density functional theory (DFT) approach, ecological structure–activity relationships (ECOSAR), and a toxicity estimation software tool (T.E.S.T) are performed to predict the ecological toxicities of the intermediate products generated during AMP degradation.
Results
Characterization
The X-ray diffraction (XRD) results are depicted in Fig. 1a–d, wherein Fig. 1a–c shows the crystal phase structures of the three raw materials. It can be seen that WO3 (PDF#89-8052) showed peaks at 2θ values of 24.12, 49.63, and 52.58° corresponding to the (110), (104), and (114) crystal planes, respectively. In addition, BiFeO3 (PDF#71-2494) exhibited peaks at 31.65, 45.72, and 56.24°, corresponding to the (104), (024), and (018) crystal planes, respectively. Simultaneously, peaks corresponding to the (110), (002), (400), and (402) crystal planes of Bi24Fe2O39 (PDF#42-0201), a secondary phase of BiFeO3, were also observed due to the instability of the BiFeO3 prepared by the sol-gel method28. For the DSB specimen, sharp peaks were observed at 29.26, 47.39, and 48.42°, which correspond to the (104), (018), and (116) crystal planes, respectively, of aragonite calcium carbonate (PDF#86-2334). Figure 1d displays the corresponding XRD results for the composite material (i.e., WO3-BiFeO3/DSB, denoted WBD), wherein the distribution of all three crystal types was confirmed. The slightly reduced peak intensities were attributed to potential interference from the amorphous carbon formed by organic compounds29.

XRD patterns of a WO3, b BiFeO3, c DSB, d W-D, W-B, B-D, and WBD. e FTIR spectra, f BET analyses, and g pore size distributions of the synthesized photocatalysts.
Figure 1e shows the Fourier transform infrared (FTIR) spectra recorded for the various samples, illustrating the vibrational modes of the functional groups present on the material surfaces. For all samples, the sharp peaks at 1597 and 1348 cm−1 correspond to the in-plane bending vibrations of the H-O-H and -OH moieties, respectively30. In addition, the peak at 3524 cm−1 in the WO3 spectrum arises from the W-OH stretching vibration resulting from the adsorption of water molecules by W, while the peak at 935 cm−1 corresponds to the stretching vibration of the W-O-W bonds31,32. The weaker peaks at 635 and 547 cm−1 in the BiFeO3 spectrum correspond to the stretching vibrations of the Bi-O and Fe-O bonds, respectively33. Furthermore, the sharp peak at 1114 cm−1 in the WBD spectrum corresponds to the C-N deformation vibration of the chitosan acetylamine groups, while those at 2819 and 2719 cm−1 originate from the -CH stretching vibrations of the benzene ring and the aldehyde groups, respectively, thereby confirming successful biochar incorporation34,35. A distinct band was also observed at 875–940 cm−1 for the W-B and WBD samples, providing information regarding the Bi-O, Fe-O, and W-O bonds. This observation of this band suggests the formation of a heterojunction between BiFeO3 and WO3, which enhances the charge transfer and photoresponse capabilities of the catalyst36.
The Brunauer–Emmett–Teller (BET) method was subsequently employed to analyze the pore structures and adsorption mechanisms of the materials. Since the photocatalytic degradation efficiency is closely related to the catalyst active sites and adsorption capacity, N2 adsorption/desorption isotherms were recorded for the samples, as shown in Fig. 1f. It was found that WO3, BiFeO3, and W-B exhibited Type I adsorption isotherms with H3 hysteresis loops. In particular, W-B showed a decreased adsorption capacity and specific surface area (SSA) after composite formation. The presence of Type I adsorption isotherms demonstrates that the unmodified photocatalysts exhibit microporous structures. In addition, the H3 hysteresis loops indicate that the unmodified photocatalytic materials possessed fewer or incomplete pore structures, resulting in reduced adsorption performances37. n the high-pressure region, WBD exhibits a distinct enhancement in its adsorption capability, reaching a maximum N2 adsorption capacity of 107.68 cm3/g STP. This corresponds to a Type IV adsorption isotherm with a H3 hysteresis loops, indicating that WBD formed a well-defined mesoporous structure, which enhanced its adsorption capabilities38.
Barrett–Joiner–Halenda pore size analysis was employed to evaluate the roles of the metal salts in regulating the pore size. As illustrated in Fig. 1g and Table 1, WBD exhibits the most prominent porous structure, with a mesopore volume accounting for 48.03% of the total pore volume. This indicates that the metal salts provide additional catalytic active sites, and when co-doped with biomass carbon, they effectively prevent internal pore collapse, ensuring a higher porosity39.
The morphological characteristics of the samples were subsequently investigated using scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HRTEM). Supplementary Fig. 1 depicts the surface microstructures of the unmodified WO3, BiFeO3, and W-B materials, which exhibit square flake-like crystals bearing dense irregular spherical structures. The HRTEM images of WBD shown in Fig. 2a–f show a high degree of crystallinity, wherein the spacing distances of the (110) crystal plane of CaCO3, the (102) crystal plane of WO3, and the (110) crystal plane of BiFeO3 were 0.236, 0.324, and 0.260 nm, respectively.

a–c HRTEM images of the WBD specimen. d, e SEM-EDS images of WBD (5,000× magnification). f Elemental mapping results for WBD.
The morphology of the WBD specimen is illustrated in Fig. 2a–c, wherein it can be seen that the internal structure exhibits a regular porous, layered arrangement. Compared to BiFeO3, WO3, and DSB, the WBD sample exhibited a looser three-dimensional structure and a more pronounced pore architecture. In addition, the EDS surface scans shown in Fig. 2d, e reveal the predominance of C, and the even dispersion of Ca, Bi, W, Fe, and O throughout the interior and surface of the photocatalyst. This suggests that the biochar framework incorporates WO3, BiFeO3, and CaCO3 aggregates through intermolecular forces, such as π-π stacking and hydrophobic interactions40. Despite the significant decrease in the O/C ratio of WBD after multiple high-temperature calcinations, which is unfavorable for capturing organic molecules, the open-pore structure increased the contact probability between the photocatalyst and the AMP molecules, potentially enhancing the catalytic efficiency41.
Full X-ray photoelectron spectroscopy (XPS) scanning was performed for WBD before and after the reaction with AMP. As shown in Fig. 3a, the elemental content did not change significantly after the reaction, indicating a good stability, even after five cycles. The corresponding Bi, C, W, O, and Fe spectra presented in Fig. 3b–g were subsequently examined to observe changes in their oxidation states.

a Full survey spectrum, and the corresponding b Bi 4f, c C 1s, d W 4f, e O 1s spectra. f NLLSS fitting and g peak fitting for the Fe 2p component.
More specifically, Bi 4f (Fig. 3b) exhibits characteristic peaks at 158.99 and 164.38 eV, which correspond to Bi 4f5/2 and Bi 4f7/2, respectively, and indicate that Bi primarily exists in the form of Bi3+ 42. In addition, in the C 1 s spectrum (Fig. 3c), the peaks at 284.8, 286.68, and 288.38 eV, which were attributed to the C-C, C-O, and C=O species, respectively. The satellite peak at 292.18 eV corresponds to the π-π bonds in the benzene ring, confirming the successful incorporation of DSB43. In Fig. 3d, the strong peaks at 35.16 and 37.28 eV correlate with the W 4f7/2 and W 4f5/2 components, respectively, while the broader peak at 40.62 eV is the WO3 energy loss peak44. Notably, the shapes of the Bi, C, and W peaks did not change significantly after five cycles, indicating a good material stability. Furthermore, from the O 1s spectrum (Fig. 3e), three peaks corresponding to lattice oxygen (OL, 529.83 eV), oxygen vacancies (OV, 531.18 eV), and surface-adsorbed oxygen (OC, 532.78 eV) were detected. Importantly, such oxygen vacancies can adjust the bandgap and enhance the photogenerated charge-carrier separation efficiency. They also act as active sites for catalytic reactions and play crucial roles in the degradation of AMP45. After five cycles, the amount of surface-adsorbed oxygen decreased, and the oxygen vacancies became occupied.
Nonlinear least-squares fitting (NLLSF) and peak decomposition were applied to the Fe 2p spectrum to investigate the oxidation states and abundance of Fe in the WBD specimen. The NLLSF results shown in Fig. 3f indicate that the Fe3+ : Fe2+ ratio was close to 3:1 in WBD. Fe3+ was involved in the construction of BiFeO3, whereas Fe2+ provided binding sites for oxygen vacancies, facilitating photocarrier transfer. After five cycles of photocatalysis, the Fe 2p peak underwent a chemical shift with an increased binding energy, suggesting the oxidation of Fe2+ to Fe3+. Figure 3g shows that, for Fe3+, strong peaks are observed at 710.28 eV (Fe3+ 2p3/2) and 724.14 eV (Fe3+ 2p1/2), with satellite peaks at 717.88 eV and 731.69 eV, respectively. Fe2+ exhibits peaks at 708.53 eV (Fe2+ 2p3/2) and 717.88 eV (Fe2+ 2p1/2), with satellite peaks at 713.38 eV and 722.54 eV46. In conclusion, e- from the surface of BiFeO3 are released outward during the catalytic reaction in WBD, participating in the reaction of active species.
Photocatalytic kinetics
Kinetic experiments were subsequently performed to reveal the photocatalytic degradation efficiency of WBD under varying conditions. The kinetic reaction constants are presented in Supplementary Fig. 2, while Fig. 4a presents the degradation capabilities of the various materials, revealing distinct patterns. The initial findings showed that unaltered materials generally display a limited ability to degrade AMP. Notably, chitosan exhibited minimal AMP degradation, suggesting that its role in the WBD surface gel mainly involves confining domains and providing structural support. In contrast, WBD demonstrated a 26.40% enhancement in the AMP degradation efficiency compared to the W-B heterojunction photocatalyst. As presented in Fig. 4b, the optimal WO3/BiFeO3:DSB ratio was determined to be 3:1, giving the highest degree of degradation (56.44%) and a reaction rate of 0.265 min−1, identify it as the benchmark catalyst for subsequent experiments. It was also found that increasing the dosage enhanced the adsorption and photocatalytic capabilities of the WBD catalyst (Fig. 4c). This was attributed to the fact that an increased dosage significantly enhanced the abundance of catalytically active sites in the reaction system, resulting in the production of more oxidizing species47. However, beyond a catalyst dosage of 150 mg, a plateau was reached, giving a maximum adsorption amount of 34.67% in dark conditions. Notably, an excess of the photocatalyst can lead to particle aggregation, thereby reducing the catalytic efficiency48. Thus, using a WBD loading of 150 mg, 99.93% AMP removal was achieved consistently over a period of 2–2.5 h. In addition, upon variation in the pH (Fig. 4d) it was found that the WBD adsorption efficiency improved under alkaline conditions, particularly at pH 10, wherein the adsorption amount increased significantly from 32.78 to 51.74%. The stability of oxygen species is strongly influenced by pH. Alkaline conditions can promote the production of most oxygen species49. Under acidic conditions, •O2− easily combines with H+ to form H2O250, however, the production of •OH in the system will be increased by Fe2+51. When pH < 2, WBD’s photocatalytic capability intensifies, possibly due to the crystalline structure of DSB acting as a buffer, releasing CO32− to suppress H+ and enhance •OH generation52. Overall, the highest reaction rate of 2.728 min−1 was achieved at pH 10. Moreover, as shown in Fig. 4e, at low AMP concentrations (10–20 mg/L), WBD effectively adsorbs AMP. Upon increasing the AMP concentration to 50 mg/L, adsorption saturation begins to occur. At this time, the photocatalytic degradation of AMP by WBD reached completion within 1.5 h. However, above 100 mg/L AMP, the photocatalytic efficiency decreased due to active site occupation, and complete degradation within 3 h was unattainable.

a catalyst material (pH 7, C0 = 100 mg L−1, T = 25 ± 1 °C, t = 3 h, m/v = 0.5 g L−1); b material ratio (pH 7, C0 = 100 mg L−1, T = 25 ± 1 °C, t = 3 h, m/v = 0.5 g L−1); c catalyst dosage (pH 7, C0 = 100 mg L−1, T = 25 ± 1 °C, t = 3 h); d pH (C0 = 100 mg L−1, T = 25 ± 1 °C, t = 3 h, m/v = 1.0 g L−1); and e AMP concentration (pH 7, T = 25 ± 1 °C, t = 3 h, m/v = 1.0 g L−1).
As shown in Fig. 5a–d, the photocatalytic efficiency of WBD was inhibited to varying degrees in the presence of different anions and NOM. More specifically, increasing concentrations of all four anions and humic acid (HA) inhibited the photocatalytic efficiency of WBD. HA is a class of NOM that is commonly found in aquatic ecosystems. Cationic antibiotics can inhibit its degradation binding to the negatively charged HA molecules through electrostatic interactions53. In addition, Cl− and NO3− can influence the photocatalytic performance of a catalyst, acting as scavengers of photogenerated radicals54. Furthermore, H2PO4− can significantly impact the photocatalytic performance through the release of H+ into water, which inhibits the generation of hydroxyl radicals55. Thus, in addition to competing with AMP for adsorption, these species inhibit selective degradation by •OH and h+, ultimately the catalytic performance. Moreover, the results of reusability experiments results indicated that WBD maintained a good catalytic stability over five cycles. In a previous report, Bargozideh et al. used MoS2 to modify BiFeO3. In their photocatalytic degradation experiments with Rhodamine B, the degree of pollutant degradation decreased to ~70% after three cycles due to the occupation of oxygen vacancies during the reaction, which reduced to catalytic ability56. In the current study, the degree of degradation dropped to 72.48% after five cycles, indicating its superior cycling stability.

Anti-interference experiment (pH 7, T = 25 ± 1 °C, t = 3 h, m/v = 1.0 g L−1) a 2, 5, and 10 mM KCl; b 2, 5, and 10 mM KNO3; c 1, 5, and 10 mg/L HA; and d 2, 5, and 10 mM KH2PO4. e Variation in the reaction rate constant; f Reusability experiments.
Optoelectronic properties
As shown in Fig. 6a, the photocatalysts were subjected to diffuse reflectance spectroscopy (DRS) in the 200–800 nm range to assess their optical responses. BiFeO3 exhibits a broad absorption band from 200 to 500 nm, which is attributed to charge transfer between Bi and Fe under optical excitation. Additionally, the weak peak at 650 nm arises from d-d transitions from the valence band (VB) to the conduction band (CB); these transitions involve the Fe 3d or Bi 6p orbitals, and are guided by the O 2p orbitals57. The absorption band of WO3 is relatively narrow compared with that of BiFeO3, resulting in peaks at 300 and 450 nm arising from the d-d transitions. Upon co-doping BiFeO3 with WO3, the absorption spectrum of W-B underwent a red shift due to the formation of defect levels, which reduces the bandgap and enhances the photocatalytic activity58. After doping with DSB, the absorbance of WBD in the 600–800 nm range was enhanced, thereby broadening the optical response range of the material.

(a) UV-vis DRS results, (b) Tauc plots, (c) Mott–Schottky plots, and (d) schematic diagram of the WBD band gap.
The forbidden bandwidths of the samples were estimated using Tauc plots, as shown in Fig. 6b. BiFeO3 and WO3 possess band gaps of 2.16 and 2.78 eV, respectively, and after doping, W-B exhibits a band gap of 2.02 eV, whereas WBD has a narrower band gap of 1.99 eV. This narrow bandgap enhances photocarrier generation, boosting the catalytic redox capability59.
Subsequently, the flat-band potentials of the catalysts were determined using Mott–Schottky plots. As shown in Fig. 6c, the slopes of all curves are positive, suggesting that both WO3 and BiFeO3 are n-type semiconductors; thus, W-B and WBD formed n–n heterojunctions after co-doping60. Simultaneously, Fig. 6c shows that the flat-band potential of WBD is −0.26 eV vs. NHE. For n-type semiconductors, the conduction band energy (Ec) is typically higher than the flat-band potential (Ef) by 0.1–0.3 eV. In this study, a value of 0.2 eV was employed, placing the conduction band energy at −0.46 eV61. The valence band energy (EV) of WBD was calculated using the equation EV = EC + Eg, where Eg is the forbidden bandwidth, giving EV = 1.53 eV62. These results are depicted in Fig. 6d, wherein visible light excitation promotes e− transitions from the VB to the CB, leaving behind h+. The DSB acts as an electron acceptor and donor at the interface, facilitating charge transfer, and representing a Z-scheme heterojunction. This structure effectively reduces the bandgap of a photocatalytic material, thereby enhancing the photocarrier separation efficiency63,64.
Linear sweep voltammetry (LSV) was employed to assess the current density response capabilities of the WBD, W-B, BiFeO3, and WO3 catalysts. As shown in Fig. 7a, WBD demonstrates a faster current response rate than the other catalysts, indicating that the incorporation of DSB provides abundant surface active sites and enhances the electron–hole separation efficiency through vacancy defect electron capture65.

a LSV results; b EIS results; and c i–t curves.
The electrochemical impedance spectroscopy (EIS) results presented in Fig. 7b indicate that WBD exhibits a smaller impedance arc than the other materials, demonstrating that DSB doping enhanced the charge-transfer efficiency of the Z-scheme heterojunction. The porous structure of the biochar facilitates the formation of an interfacial electric field between WO3 and BiFeO3, thereby promoting the separation of electron–hole pairs66.
Furthermore, the i–t curves (Fig. 7c) show that the photocurrent densities of the samples decreased in the order: WBD > W-B > BiFeO3 > WO3. The maximum value of 8.71 μA/cm2 obtained for WBD demonstrates its superior current response capability. Compared to the study by Zhang et al., wherein BiFeO3 was modified using ZnIn2S4, this represented an 5-fold increase in the photocurrent density67.
These results clearly demonstrate that DSB doping enhanced the charge-transfer efficiency of WBD due to its ability to act as a bridge for carrier exchange, effectively reducing the bandgap between the CB and the VB. This reduction facilitates excitation and electron–hole separation.
Photoluminescence (PL) spectroscopy is commonly used to characterize the separation and migration efficiencies of photogenerated carriers. In photocatalytic reactions, a weaker PL intensity indicates superior electron–hole separation, leading to a higher catalytic efficiency68. The PL spectra of the four catalysts recorded at an incident wavelength of 340 nm are depicted in Fig. 8a. The prominent peak at 432 nm in all samples arose from the excitation of metal–oxygen bonds. Additionally, WO3, W-B, and WBD exhibited a broad peak at 611 nm, which was attributed to the defect structures and vacancy oxygen effects within the catalysts69. The PL intensity of W-B was significantly lower than those of the pure BiFeO3 and WO3 species, indicating that heterojunction formation enhanced the photocatalytic activity. The weakest PL intensity was observed for WBD due to its larger SSA enhancing the effective surface oxygen-exchange reaction area, and promoting electron–hole separation70.

a PL; b FLT; c saturation magnetization; and d remnant magnetization and coercivity results.
The fluorescence lifetime (FLT) spectra of the four materials are presented in Fig. 8b, wherein it can be seen that within a 50 ns timeframe, WBD exhibits a longer fluorescence decay time, with an average FLT of 1.95 ns. This indicates that the use of biochar as a photocatalyst carrier prolongs the lifetime of photoexcited carriers and suppresses rapid charge recombination71.
Furthermore, Fig. 8c, d show that the magnetic susceptibilities of W-B and WBD range from 10−4 to 10−1, indicative antiferromagnetic behavior. In addition, the observed narrow hysteresis loops, low coercivity, and low spontaneous magnetization were attributed to structural distortion in the BiFeO3 structure72,73. The saturation magnetization of WBD was determined to be 8.86 emu/g, which is 2.43-fold higher than that of W-B and almost 9-fold higher than that of the BiFeO3/Bi-modified g-C3N4 heterojunction photocatalyst prepared by Ammar et al.74. This high saturation magnetization was attributed to the sensitivity of the BiFeO3 crystal structure and dimensions, wherein its loose, porous microstructure enhances the anisotropy, suppresses spiral spin structures, and increases the magnetization intensity75,76. In summary, WBD belongs to soft magnetic materials characterized by high permeability, low magnetic energy loss, excellent stability, and able to be reused multiple times.
Mechanism and ecotoxicology
As presented in Fig. 9a, the initial total organic content (TOC) degradation efficiency in wastewater was 49.14 mg/L, and after 3 h of photocatalytic degradation, 53.48% degradation was achieved. This indicates that WBD does not completely degrade AMP, suggesting that the photocatalyst primarily contributes to detoxification rather than the complete mineralization of persistent organic pollutants. As shown in Fig. 9b, c, the catalytic efficiencies decreased to 57.03%, 54.47%, 76.00%, and 56.07% (c.f., the control) upon the addition of four different quenchers. The reaction rate constants also decreased from 0.829 min−1 to 0.322, 0.295, 0.514, and 0.315 min−1, respectively. To further reveal the contributions of different ROS and elucidate the reaction mechanism, the electron paramagnetic resonance (EPR) spectroscopy was performed. As shown in Fig. 9d, the response peaks of the hydroxyl radicals before and after illumination were compared for the WBD system77. Consequently, the •O2−, h+1, O2, and •OH species were identified as the main active oxygen species contributing to the catalytic reaction, with their the photocatalytic effects decreasing in the order of •OH > 1O2 > •O2− > h+.

(a) the TOC degradation capability, (b) the AMP degradation rate, and (c) the first-order rate constant. (d) EPR spectra of the reactive oxygen species in the WBD photocatalytic system.
By combining free-radical capture experiments, the pathways for the generation of several key radicals were defined Under visible light excitation, the electrons released from BiFeO3 can react with ambient O2 to produce O2− 78. In addition, the presence of h+ on WO3 facilitates the conversion of OH− and H2O to •OH. In the absence of additional active intermediates1,O2 is primarily formed via the conventional Type I mechanism, wherein •O2− combines with h+. As a supplementary catalytic species1,O2 exhibits a stronger resistance to environmental interference and a greater persistence than •OH, enabling its participation in degradation reactions through non-radical pathways79,80. The generation of active species proceeds according to the following reaction equations:
AMP possesses two dissociation constants (pKa1 2.5, PKa2 7.2) at which the amino and carboxyl groups are ionized. At pH > 7.2, the carboxyl group is deprotonated to generate AMP−, at pH < 2.5, the amino group is protonated to give AMP+, and at pH 2.5–7.2, both forms coexist to give the zwitterionic form (AMP0)8,81.
HOMO/LUMO depict the outer electron layer information of the four structural forms of AMP. As depicted in Fig. 10a, the three dissociation forms of AMP in water (Pka1 = 2.5, Pka2 = 7.2) exhibit similar charge orbital distributions. Moreover, the energy band gaps between HOMO and LUMO remain around 5 eV, indicating a reluctance for charge transfer to higher or lower states. This suggests that changes in pH do not easily disrupt the original structure of AMP, highlighting its low chemical reactivity and the need for higher energy excitation for reactions82. HOMO is concentrated around the benzene ring, -NH2 at C3, and -C=O at C1, which act as electron acceptors and facilitate reduction reactions; in contrast, LUMO is concentrated around -COOH at C13, -C=O at C11, and atoms C12, C14, N17, and S19, where electrons are prone to delocalization and loss. According to the electrostatic potential (ESP) calculations in Fig. 10b, AMP molecules primarily accumulate electrons around the benzene ring, N7, O8, O18, and O21, making them susceptible to electron loss reactions and decomposition. These findings indicate that O8, O18, O21, N7, and the benzene ring in AMP exhibit high electron charge densities, making them favorable sites for electrophilic radical attack. Due to steric hindrance effects, N17 and S19 are difficult binding sites for reactions, thus maintaining the fundamental structure of the β-lactam and thiazole rings of AMP. However, the ecological toxicity of degradation products primarily arises from the exposure of -COOH and thiol groups (-SH) due to the opening of the aforementioned structures83,84. Therefore, the ring-opening of the β-lactam and thiazole rings within the AMP molecule is a crucial step in its decomposition process.

a HOMO/LUMO plots; b ESP of AMP; c f−, d f+, e f0, and f CDD isosurfaces of AMP; g selectivity of radical attack on AMP (blue circles: sites susceptible to electrophilic attack; green circles: sites susceptible to nucleophilic attack).
Fukui function is employed to predict the selectivity of nucleophilic attack (f+), electrophilic attack (f-), and radical attack (f0) on AMP. Computational results are presented in Fig. 10c–e and Supplementary Table 2. As results, -COOH connected to C13, -C=O) at C11 and C1, are susceptible to nucleophilic attack (like •O2−). -NH2 attached to C3, as well as S19, N7, and N9, are prone to electrophilic attack (like h+). Free radicals (like •OH) primarily affect the structures of S19, N7, O8, O21, O18, and the benzene ring. As shown in Fig. 10f, Condensed Dual Descriptor (CDD) further analyze the propensity of nucleophilic or electrophilic attacks on specific sites of AMP. The results indicate that -COOH connected to C13 and -C=O at C11 exhibit nucleophilicity. -NH2 attached to C3 and O8 display electrophilicity. On the benzene ring, C33, C35, C36, and C40 show nucleophilicity, while C34 and C38 exhibit electrophilicity. Within carbon framework, all non-carbon elements demonstrate nucleophilicity, whereas carbon elements display electrophilicity. Combining these findings, a schematic representation of radical attacks on AMP is depicted in Fig. 10g. This illustration underscores that non-carbon groups such as -NH2, -C=O, -N, and -S are significantly affected by electrophilic radicals, thereby contributing primarily to the ecological toxicity associated with AMP.
Based on the above analyses, the possible AMP degradation pathways was predicted under WBD catalysis conditions. As depicted in Fig. 11, following the photocatalytic reactions, AMP degradation yielded 10 intermediate products (Supplementary Fig. 3), suggesting the occurrence of three distinct pathways. More specifically, pathway I involves opening of the benzene ring and the loss of carbonyl and amine groups from the peptide bond to generate intermediates P1 and P2. Furthermore, cleavage of the cyclobutene carbonyl group generates P3, while the loss of a carboxyl and an amino group forms intermediate P4. Pathway II begins with the detachment of two methyl groups from the thiazole ring, along with the detachment of carbonyl and amine groups from the peptide bond, producing intermediates P5 and P6. Further breakdown of the benzene and β-lactam rings generates intermediates P7 and P8. Moreover, pathway III involves the dissociation of carbonyl, amine, thiol groups, producing intermediates P9 and P10. The relatively high detected abundances of P1 and P7 suggest that pathways I and II are the primary degradation routes, with pathway II representing more thorough degradation than pathway I.

Proposed AMP degradation pathways.
Finally, the toxicity levels of AMP and its degradation products were predicted in aquatic organisms using ECOSAR 2.2. As shown in Table S3, AMP exhibits chronic toxicity in green algae and water fleas. Additionally, P4 and P9 showed slightly higher toxicities compared to the parent compound, while P1 and P2 demonstrated ecological toxicity levels comparable to those of the parent compound. The degradation products from pathway II (P5–P8) were less toxic than those from pathway I, while P9 was found to be more ecologically toxic than P10. These results suggest that the detachment of amine, carboxyl, and thiol groups is crucial in the detoxification of AMP85.
T.E.S.T Result is depicted in Supplementary Table 4. Owing to potential bioaccumulation through the food chain, the bioconcentration factor (BCF), chronic toxicity, and mutagenicity characteristics of the degradation products were assessed using quantitative structure–activity relationship (QSAR) models. According to the TEST results (Supplementary Table 4), the BCF values of P2, P3, P4, P6, P8, P9, and P10 were >1, indicating that these structures are prone to bioaccumulation in organisms. However, since P3, P4, P8, and P10 possess chronic toxicity indices >0.5, developmental toxicity was possible; fortunately, these intermediates are not abundant in the degradation products. No mutagenic structures were identified, and it was deduced that the developmental toxicities of the Pathway II degradation products were notably lower than those of the other pathways86. This was attributed to the fact that these degradation products are primarily formed through the attack of non-nucleophilic free radicals, which evidently contribute positively to AMP detoxification.
In summary, the photocatalytic degradation products of AMP by WBD show low ecological toxicity, indicating a superior detoxification effect compared to the study by Liu et al.16. It’s primarily attributed to WBD’s ability to generate a greater quantity of •OH, h+, and other non-nucleophilic pathways such as 1O2. These radicals continuously cleave -NH2, -SH, and -COOH groups on the β-lactam ring of AMP, achieving the objective of reducing the toxicity of the degradation products. However, this computer-simulated degradation pathway has uncertainties, the precise regulation of the degradation pathway of organic pollutants with low toxicity has not been thoroughly studied.
Discussion
In this study, a novel photocatalyst, namely WO3-BiFeO3/digestate-derived biochar (DSB), denoted WBD, was synthesized for use in the degradation of AMP into low-toxicity or non-toxic degradation products. The use of DSB to prepare this catalyst significantly enhanced the number of reactive sites, the SSA, and the mesopore volume. A Z-scheme heterojunction was formed inside WBD, boosting the photogenerated charge-carrier concentration and enhancing the charge-transfer efficiency. Under the optimized conditions (150 mg WBD, 100 mg/L AMP, 3 h), 99.93% degradation was achieved. In addition, over 5 catalytic cycles, WBD exhibited a robust photocatalytic degradation capability and maintained a high efficiency, demonstrating a strong resilience against environmental interference. During degradation, primary reactive oxygen species (i.e., •O2−, h+,•OH, and 1O2) effectively target he recalcitrant functional groups (i.e., -NH2, -SH, and -COOH) generated by β-lactam ring opening. This process yielded low-toxicity or non-toxic degradation products, and a clear detoxification pathway was established. Overall, this economical and environmentally functional material (WBD) exhibits a superior degradation efficiency and stability compared to state-of-the-art photocatalysts, and provides excellent prospects for lowering the toxicity profiles of antibiotics in water.
In future studies, the catalytic degradation efficiencies of Bi-based photocatalysts will be evaluated under diverse antibiotic stress conditions, and modulation of the energy band structure will be performed for further investigation of the degradation pathways and by-products. Concurrently, a pilot-scale photocatalytic reactor system will be constructed for the continuous treatment of antibiotic-laden wastewater to facilitate material recycling, ensure catalytic stability, and safeguard environmental integrity. These endeavors are poised to establish a robust foundation for the industrial-scale implementation of such photocatalytic technologies.
Methods
Raw materials
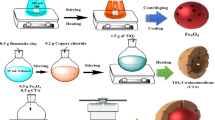
AMP (CAS: 69-53-4 98%) was purchased from Macklin Biochemical Technology Co., Ltd. (Shanghai, China) and stored at 2–8 °C. Digestate (DS) was obtained from a kitchen waste treatment plant in Ningbo. Its physicochemical properties are detailed in Table 2. DS as a byproduct of kitchen waste fermentation, contains abundant cellulose, hemicellulose, lignin, and other substances. Its high C/N ratio and rich organic content provide excellent carbonization potential. Na2WO4•2H2O, Fe(NO3)3•9H2O, Bi(NO3)3•5H2O, chitosan, (C2H2OH)2, polyethylene glycol (PEG), and CH3CN are all of analytical reagent (AR) purity, purchased from Macklin Biochemical Technology Co., Ltd. (Shanghai, China) and stored dry at room temperature (Scheme 1).

a Preparation of WBD, b the photocatalytic mechanism, and c investigation of the low toxicity degradation pathway.
Preparation
DSB was produced via slow pyrolysis in a muffle furnace. WO3, BiFeO3, and WO3-BiFeO3 composites were synthesized using the sol-gel method87,88, and WO3-BiFeO3/DSB magnetic gel balls were prepared through droplet microfluidics89,90. Specific details of preparation processes can be found in following text. The schematic diagram of WO3-BiFeO3/DSB magnetic gel balls (WBD) preparation is shown in Scheme 2.

Schematic representation showing preparation of WBD magnetic gel balls.
Preparation of digested- derived biochar
The digested sludge was taken from the fermentation residue of a food waste treatment plant in Ningbo, air-dried for 48 h, then underwent processing involving crushing, grinding, and drying until a constant weight was achieved. Subsequently, the material was sieved through a 100-mesh screen and transferred to a drying dish. The powder was then subjected to pyrolysis in a muffle furnace at 600 °C, with a heating rate of 10 °C/min over a period of 2 h. Following pyrolysis, the resultant biochar was washed three times with distilled water and subsequently dried. The final product was designated as DSB.
Preparation of WO3
5.280 g Na2WO4•2H2O were added to 300 mL of 33.3% HCl solution. The mixture was stirred at 300 r/min for 36 h, resulting in the formation of a yellow solid. The precipitate was separated by centrifugation and washed by ultrapure water. Then it was dried at 80 °C for 8 h to obtain crude WO3. The product was subsequently added to 100 mL of ultrapure water and subjected to ultrasonication for 3 h to yield a light-yellow suspension. After centrifugation at 4000 r/min for 15 min and vacuum drying, the final WO3 product was obtained.
Preparation of BiFeO3
7.760 g Bi(NO3)3•5H2O and 6.464 g Fe(NO3)3•9H2O were added to 50 mL of ethylene glycol. The resulting solution was stirred at 300 r/min for 2 h until a stable reddish-brown sol was formed. Next, the sol was dried in a vacuum oven at 100 °C. The dried light-yellow gel was then calcined in a muffle furnace at 450 °C for 1.0 h to synthesize BiFeO3.
Preparation of WO3-BiFeO3
7.760 g Bi(NO3)3•5H2O and 6.464 g Fe(NO3)3•9H2O were dissolved in 100 mL of 50% ethylene glycol, followed by the addition of 3 mL 69% HNO3. To this solution, 1.856 g WO3 prepared from 2.2.2 was added. The mixture was stirred at 300 r/min at 115 °C for 2 h until it transformed into a brownish dry gel solid. Obtained gel was dried in an oven at 150 °C to yield a dry brown powder. Subsequently, this powder was calcined in a muffle furnace at 550 °C for 2 h to produce a composite powder of WO3 and BiFeO3, named W-B.
Preparation of WO3-DSB and BiFeO3-DSB
In a 200 mL beaker, 100 mL 1% acetic acid solution was added. 2 g chitosan were then introduced into the acetic acid solution and dissolved with magnetic stirring at 300 r/min. Subsequently, 2 g WO3 and 2 g DSB were added to the solution, and the mixture was stirred vigorously to achieve homogeneity. Following this, 10 mL 1% polyethylene glycol (PEG) was added, and the solution was stirred at 50 °C until a gel precipitated. The gel was allowed to mature by standing at 25 °C for 12 h. Then, 45 mL 1% NaOH solution was added to the gel to adjust the pH to 9.0. The gel was heated in a water bath at 50 °C for 2 h and then treated with 10 mL of acetone. Finally, the precipitate was washed and the supernatant removed until neutral. The precipitate was dried, ground, and processed to obtain WO3-DSB, designated as W-D. The same preparation method was used to prepare BiFeO3-DSB except that WO3 was replaced by BiFeO3. The products were named as B-D.
Preparation of WO3-BiFeO3/DSB magnetic gel balls
DSB and W-B were combined in various mass ratios (1:1, 1:2, 1:3, 1:4) and heated in a muffle furnace at 300 °C for 1.5 h. At 25 °C, 6 g calcined product were added to 100 mL 2% chitosan-acetic acid solution and stirred magnetically at 300 r/min until homogeneous, forming a mixed solution. This mixed solution was then slowly dripped into a 1% NaOH solution using a syringe to form chitosan gel beads with uniform particle size. After 4 h of standing, the gel balls were removed from the solution, washed, and transferred into 400 mL of ultrapure water. To this, 5 mL 5% polyethylene glycol was added, and pH was adjusted to 9.0. Subsequently, the mixture was gently stirred at 60 °C for 2 h to induce cross-linking, followed by 20 h resting period. The beads were then removed, washed to neutrality, and naturally aged for 24 h to prepare chitosan-crosslinked WO3-BiFeO3/DSB carbon-based photocatalytic gel magnetic balls, named WBD X: Y(where XY denotes the mass ratio of DSB to W-B).
Characterization
FTIR spectrometer (Thermo Nicolet 6700, USA) for elucidating functional groups in materials; scanning electron microscope (ZEISS Sigma 300, Germany) and transmission electron microscopy (JEOL JEM F200, Japan) for observing surface morphology, lattice spacing, and pore structure of materials (Fitted by DigitalMicrograph software to give), and internal scanning within the WBD for elemental composition analysis; automated surface area and porosity analyzer (Micromeritics ASAP 2460, USA) for analyzing SSA and pore size distribution; X-ray single crystal diffractometer (Bruker D8 Advance, USA) for characterizing internal structure and crystal phase composition of materials, with data analyzed and fitted using Jade 8.7 software; X-ray photoelectron spectrometer (Thermo ESCALAB 250XI, USA) for analyzing surface elemental composition, chemical states, and molecular structure information of materials.
The materials' optical response range was analyzed using UV-visible diffuse reflectance spectrometer (Shimadzu UV-3600i Plus, Japan), and Tauc plot was generated to determine the band gap of WBD. The Mott-Schottky curve of the materials was obtained using an electrochemical workstation (PINE-Wave Driver 100, USA), allowing determination of the positions of the valence and conduction bands, and impedance spectroscopy (EIS) measurements were conducted. LSV and chronoamperometry (i-t) curves were measured using an electrochemical workstation (Zahner PP211, Germany). The three-electrode electrochemical system comprised ITO-coated glass as the working electrode, platinum foil as the counter electrode, and Ag/AgCl as the reference electrode, with 0.5 M HCl serving as the electrolyte.
Transient-state fluorescence spectrometer (Edinburgh FLS1000, UK) was utilized to analyze the optoelectronic properties of the material, the rate of energy transfer, and the fluorescence excited-state lifetimes. Vibrating sample magnetometer (LakeShore 7404, USA) was employed to characterize the magnetic properties and changes in magnetic intensity of DSB before and after W-B doping.
Photocatalytic kinetics
HPLC condition
High-performance liquid chromatography (HPLC) was used to determine the concentration of AMP in the solution. The instrument used was a Waters_e2695 HPLC system equipped with a 2489 UV detector. A GL Sciences WondaSil C18 column (4.6 mm × 150 mm × 5 μm) was employed to separate organic components, with the column temperature maintained at 25 °C. The mobile phase A (1 L) was prepared with a volume ratio of 12% acetic acid solution : 0.2 mol/L potassium dihydrogen phosphate solution : acetonitrile : water = 0.5 : 50 : 50 : 900. Mobile phase B (1 L) was prepared with a volume ratio of 12% acetic acid solution : 0.2 mol/L potassium dihydrogen phosphate solution : acetonitrile : water = 0.5 : 50 : 400 : 550. During operation, an isocratic elution with mobile phase A and mobile phase B (v:v = 85:15) was first performed to wash the column, followed by a linear gradient elution over 1 h according to Supplementary Table 1 immediately after elution of the AMP peak. The flow rate was set to 1 mL/min, the column temperature control at 35 °C with an injection volume of 20 μL per sample. Detection of the separated components was performed at a wavelength of 254 nm using a UV detector, with AMP eluting at a retention time of 5.26 min.
Kinetic experiment
In this study, a 100 mg/L AMP solution was used to simulate wastewater. We conducted a 3 h photocatalytic kinetic experiment, varying materials, material ratios, dosage, and pH as experimental parameters. A 300-W xenon lamp, filtered at 400 nm, served as the light source for the photocatalytic reactions. Materials were in darkness for 45 min to reach adsorption equilibrium. Samples were collected every 15 min. After 45 min, xenon lamps were turned on for photocatalysis, with samples taken every 30 min over the subsequent hour. The AMP content of the samples was determined by HPLC. The details of method are given in Supplementary Material. Degradation rate was used to assess photocatalytic performance, and first-order reaction kinetics were fitted to analyze kinetic constants. We employed KCl, KH2PO4, and KNO3 at concentrations of 2, 5, and 10 mM as anionic sources, and humic acid (HA) at concentrations of 1, 5, and 10 mg as NOM. These environmental factors were investigated to study their influence on photocatalytic kinetics. Degradation rate and first-order reaction rate constants were calculated using the Langmuir Hinshelwood equations:
Where q denotes the degradation rate; t is the reaction time; k represents the first-order reaction rate constant; C signifies the initial reactant concentration and C0 denotes the reactant concentration after degradation.
Quenching experiment and electron paramagnetic resonance (EPR) spectroscopy
Incorporating 1 mM of quenchers such as benzoquinone, ethylenediaminetetraacetic acid, ferulic acid, and tert-butanol into photocatalytic reactions corresponds to quenching agents for superoxide radicals (•O2−), holes (h+), singlet oxygen (1O2), and hydroxyl radicals (•OH), respectively. This approach is used to analyze the contributions of these radicals in photocatalytic reactions. Degradation rate and first-order reaction rate constants were calculated using Eqs. (1–2). Assays were performed on an EPR spectrometer (Bruker EMXplus-6/1) to identify active species and vacancies in the samples. In a 1 mL water/methanol solution, 20 mM 5,5-dimethyl-1-pyrroline N-oxide (DMPO) and 3 mg of catalyst were utilized for the detection of •OH and •O2- radicals; DMPO was replaced with 2,2,6,6-tetramethylpiperidine (TEMP) for 1O2 detection, and with 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) for h+ detection.
Theoretical calculations
DFT calculations were performed using Gaussian 09 W to generate three dissociation models of AMP in water, optimizing their structures with the B3LYP/6-311G basis set, using the DFT-D3 dispersion correction with BJ-damping to correct the weak interaction and improve the calculation accuracy91. Then were using Multiwfn 3.8_dev for analyzed92. Visualizations were exported using Visual Molecular Dynamics to study AMP’s structural features, charge distributions, and potential degradation pathways93. Which including Highest Occupied Molecular Orbital and Lowest Unoccupied Molecular Orbital (HOMO/LUMO), ESP, and Fukui functions.
Toxicity analysis
Using high-performance liquid chromatography-mass spectrometry (Agilent 1290-6550-Qtof, America) to determine the structures of degradation products of WBD photocatalysis and analyzed the degradation pathways. The toxicity of AMP and its derivatives on aquatic organisms, including fish, daphnid, and green algae, was predicted using the US EPA-developed computational toxicology software ECOSAR 2.2, with median lethal concentration (LC50), median effect concentration (EC50), and Chronic Value (ChV) as the assessment criteria. According to the toxicity concentration limits defined by the Guidelines for the Hazard Evaluation of New Chemical Substances (HJ/T154-2004), which categorize substances as highly toxic (<1 mg/L), toxic (1–10 mg/L), harmful (10–100 mg/L), and non-toxic (>100 mg/L)94, we analyzed the toxicity of AMP degradation products on aquatic organisms. Furthermore, utilizing the computational toxicology software T.E.S.T to predicted the developmental toxicity, mutagenicity, and bioaccumulation factor of the degradation products95.
Data processing
The raw data were processed using Origin 2018.
Data availability
No datasets were generated or analyzed during the current study.
Code availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Ministry of Ecology and Environment of the People’s Republic of China, Ministry of Industry and Information Techonology of the People’s Republic of China, Ministry of Agriculture and Rural Affairs of the People’s Republic of China, Ministry of Commerce of the People’s Republic of China, General Administration of Customs of the People’s Republic of China, State Administration for Maket Regulation. List of priority new pollutants for control (2023 edition). https://www.mee.gov.cn/gzk/gz/202212/t20221230_1009192.shtml (2023).
The National Science and Technology Council. National Emerging Contaminants Research Initiative. https://www.whitehouse.gov/wp-content/uploads/2022/08/08-2022-National-Emerging-Contaminants-Research-Initiative.pdf (2022).
European Medicines Agency. Sales of veterinary antimicrobial agents in 31 European countries in 2018. https://www.ema.europa.eu/en/documents/report/sales-veterinary-antimicrobial-agents-31-european-countries-2019-2020-trends-2010-2020-eleventh_en.pdf (2023).
U.S. Food & Drug Administration. Summary Report on antimicrobials sold or distributed for use in food-producing animals. https://www.fda.gov/media/154820/download (2023).
Nouri, N., Khorram, P., Duman, O., Sibel, T. & Hassan, S. Overview of nanosorbents used in solid phase extraction techniques for the monitoring of emerging organic contaminants in water and wastewater samples. Trends Environ. Anal. Chem. 25, e00081 (2020).
Liu, X., Lu, S., Guo, W., Xi, B. & Wang, W. Antibiotics in the aquatic environments: a review of lakes. China. Sci. Total Environ. 627, 1195–1208 (2018).
Grace, L. et al. Global sales of oralantibiotics formulated for children. Bull. World Health Organ. 98, 458–466 (2020).
Ioannou-Ttofa, L., Raj, S., Prakash, H. & Fatta-Kassinos, D. Solar photo-Fenton oxidation for the removal of ampicillin, total cultivable and resistant E. coli and ecotoxicity from secondary-treated wastewater effluents. Chem. Eng. J. 355, 91–102 (2019).
Wang, H., Li, J., Wang, B. & Chen, G. Deciphering pollutants removal mechanisms and genetic responses to ampicillin stress in simultaneous heterotrophic nitrification and aerobic denitrification (SHNAD) process treating seawater-based wastewater. Bioresour. Technol. 315, 123827 (2020).
Liang, D., Hu, Y., Huang, R., Cheng, J. & Chen, Y. Effects of various antibiotics on aerobic nitrogen removal and antibiotic degradation performance: Mechanism, degradation pathways, and microbial community evolution. J. Hazard. Mater. 422, 126818 (2022).
Shannon, M. M., Jeffrey, L. U., Amy, L. T. & Richard, J. W. pH and temperature effects on the hydrolysis of three β-lactam antibiotics: Ampicillin, cefalotin and cefoxitin. Sci. Total Environ. 466-467, 547–555 (2014).
Aydogdu, S. & Hatipoglu, A. Theoretical insights into the reaction mechanism and kinetics of ampicillin degradation with hydroxyl radical. J. Mol. Model. 29, 63 (2023).
Pouretedal, H. R. & Sadegh, N. Effective removal of Amoxicillin, Cephalexin, Tetracycline and Penicillin G from aqueous solutions using activated carbon nanoparticles prepared from vine wood. J. Water Process. Eng. 1, 64–73 (2014).
Zhou, W. et al. The co-effect of ampicillin and multi-walled carbon nanotubes on activated sludge in sequencing batch reactors: microbial status, microbial community structure and ARGs propagation. Environ. Sci. Nano. 8, 204–216 (2021).
Yang, W. & Wang, Y. Enhanced electron and mass transfer flow-through cell with C3N4-MoS2 supported on three-dimensional graphene photoanode for the removal of antibiotic and antibacterial potencies in ampicillin wastewater. Appl. Catal. B 282, 119574 (2021).
Liu, H. et al. Insights into electrocatalytic oxidation of aqueous ampicillin: degradation mechanism and potential toxicity from intermediates. J. Environ. Chem. Eng. 10, 108673 (2022).
Song, Y., Bao, Z. & Gu, Y. Photocatalytic enhancement strategy with the introduction of metallic Bi: a review on Bi/semiconductor photocatalysts. Chem. Rec. 24. https://doi.org/10.1002/tcr.202300307 (2024).
Yan, F. et al. Optimization of polarization and electric field of bismuth ferrite based ceramics for capacitor applications. Chem. Eng. J. 417, 127945 (2021).
Potlako, J. M., Mope, E. M., Francis, O. & Bhekie, B. M. Visible light responsive MoS2/Ag@WO3/EG photoanode with highly stable Z-scheme induced circular electron motion pioneered by Exfoliated graphite for bisphenol a photoelectrodegradation. Chem. Eng. J. 464, 142462 (2023).
Mope, E. M., Bulelwa, N., Potlako, J. M. & Mabuba, N. In-situ synthesis of Tetraphenylporphyrin/Tungsten (VI) Oxide/Reduced Graphene Oxide (TPP/WO3/RGO) nanocomposite for visible light photocatalytic degradation of Acid Blue 25. Chem. Eur. J. 4, 8379–8389 (2019).
Subramanian, Y., Ramasamy, V., Gubendiran, R. K., Srinivasan, R. G. & Arulmozhi, D. Structural, optical, thermal and photocatalytic dye degradation properties of BiFeO3-WO3 nanocomposites. J. Electron. Mater. 47, 7212–7223 (2018).
Pastre, M. M. G., Cunha, D. L. & Marques, M. Design of biomass-based composite photocatalysts for wastewater treatment: a review over the past decade and future prospects. Environ. Sci. Pollut. Res. 30, 9103–9126 (2023).
Javaid, A. et al. Graphitic carbon nitride/bismuth-based Z-scheme heterojunctions for the photocatalytic removal of pharmaceuticals and personal care products — a review. Curr. Opin. Chem. Eng. 46, 101054 (2024).
El-Bestawy, E. A., Gaber, M., Shokry, H. & Samy, M. Effective degradation of atrazine by spinach-derived biochar via persulfate activation system: Process optimization, mechanism, degradation pathway and application in real wastewater. Environ. Res. 229, 115987 (2023).
Gong, J. et al. Highly efficient activation of periodate by a manganese-modified biochar to rapidly degrade methylene blue. Environ. Res. 241, 117657 (2024).
Jiang, H. et al. Multi-phase CdS loaded on biochar for photocatalytic activation of peroxymonosulfate for thiamethoxam degradation: π-conjugation improves PMS adsorption. Sep. Purif. Technol. 326, 124842 (2023).
Qi, K. et al. CQDs/biochar from reed straw modified Z-scheme MgIn2S4/BiOCl with enhanced visible-light photocatalytic performance for carbamazepine degradation in water. Chemosphere 287, 132192 (2022).
Čebela, M. et al. BiFeO3 perovskites: a multidisciplinary approach to multiferroics[J]. Ceram. Int. 43, 1256–1264 (2017).
Jayant, V. & Yusuf, M. Insights into polymorphism and inclusion properties in organic compounds. Discov. Chem. 1, 52 (2024).
Bu, X. et al. Fabrication of novel Z-scheme LaCoO3/activated biochar/Ag3PO4 heterojunctions for intensifying visible-light-catalytic degradation of bisphenol A. Appl. Surf. Sci. 588, 152887 (2022).
Thiyagarajan, K., Muralidharan, M. & Sivakumar, K. Defects induced magnetism in WO3 and reduced graphene oxide-WO3 nanocomposites. J. Supercond. Nov. Magn. 31, 117–125 (2018).
Younas, A., Iqbal, T., Almohammedi, A. & Afsheen, S. Experimental and theoretical comparative analysis of pure WO3 and La doped WO3 for degradation of MB. Opt. Quantum Electron. 55, 1172 (2023).
Remya, K. P. et al. Tailoring the morphology and size of perovskite BiFeO3 nanostructures for enhanced magnetic and electrical properties. Mater. Des. 192, 108694 (2020).
Travlou, N. A., Kyzas, G. Z., Lazaridis, N. K. & Deliyanni, E. A. Functionalization of graphite oxide with magnetic chitosan for the preparation of a nanocomposite dye adsorbent. Langmuir 29, 1657–1668 (2013).
Kalumpha, M., Guyo, U., Zinyama, N. P., Vakira, F. M. & Nyamunda, B. C. Adsorptive potential of Zea mays tassel activated carbon towards the removal of metformin hydrochloride from pharmaceutical effluent. Int. J. Phytorem. 22, 148–156 (2020).
Duan, Q. et al. Synthesis and characterization of morphology-controllable BiFeO3 particles with efficient photocatalytic activity. Mater. Res. Bull. 112, 104–108 (2019).
Liu, G., Lin, Y., Li, S., Shi, C. & Zhang, D. Mechanism and efficiency of photocatalytic triclosan degradation by TiO2/BiFeO3 nanomaterials. Water Sci. Technol. 86, 3133–3152 (2022).
Siara, S., Elvis, C., Harishkumar, R. & Chellam, P. V. ZnAl2O4 supported on lychee-biochar applied to ibuprofen photodegradation. Mater. Res. Bull. 145, 111530 (2022).
Li, R. et al. An overview of carbothermal synthesis of metal-biochar composites for the removal of oxyanion contaminants from aqueous solution. Carbon 129, 674–687 (2018).
Li, Y. et al. Removal of Cr(VI) by 3D TiO2-graphene hydrogel via adsorption enriched with photocatalytic reduction. Appl. Catal. B 199, 412–423 (2016).
Wan, Z. et al. Stoichiometric carbocatalysis via epoxide-like C-S-O configuration on sulfur-doped biochar for environmental remediation. J. Hazard. Mater. 428, 128223 (2022).
Navjot, Tovstolytkin, A. I. & Lotey, G. S. Plasmonic enhanced photocatalytic activity of Ag nanospheres decorated BiFeO3 nanoparticles. Catal. Lett. 147, 1640–1645 (2017).
Wang, X. et al. Porous biochar modified with polyethyleneimine (PEI) for effective enrichment of U(VI) in aqueous solution. Sci. Total Environ. 708, 134575 (2020).
Wang, Y. et al. Mesoporous WO3 modified by Au nanoparticles for enhanced trimethylamine gas sensing properties. Dalton Trans. 50, 970–978 (2021).
Cao, E. et al. WO3-LaFeO3 nanocomposites for highly sensitive detection of acetone vapor at low operating temperatures. Chemosensors 11, 439 (2023).
Bagus, P. S. et al. Comments on the theory of complex XPS spectra: extracting chemical information from the Fe 3p XPS of Fe oxides. Comments Inorg. Chem. 41, 373–397 (2021).
Iqbal, A. et al. Methodological optimization for efficient degradation of Acid Violet 49 using advanced oxidation processes and varied photocatalyst combinations. Sci. Rep. 14, 30339 (2024).
Lv, H. et al. CaO composite improves the conversion efficiency of free radicals in the catalytic activation of permonsulfate by Cu-based oxides. J. Environ. Chem. Eng. 12, 114305 (2024).
Hao, Z., Hou, W., Fang, C., Huang, Y. & Liu, X. Sulfite activation by cobaltosic oxide nanohydrangeas for tetracycline degradation: performance degradation pathways and mechanism. J. Hazard. Mater. 439, 129618 (2022).
Liu, Y., Guo, J., Chen, Y., Tan, N. & Wang, J. High-efficient generation of H2O2 by aluminum-graphite composite through selective oxygen reduction for degradation of organic contaminants. Environ. Sci. Technol. 54, 14085–14095 (2020).
Jiang, Z., Wang, L., Lei, J., Liu, Y. & Zhang, J. Photo-Fenton degradation of phenol by CdS/rGO/Fe2+ at natural pH with in situ-generated H2O2. Appl. Catal. B 241, 367–374 (2019).
Xiao, S. et al. Iron-mediated activation of persulfate and peroxymonosulfate in both homogeneous and heterogeneous ways: a review. Chem. Eur. J. 384, 123265 (2020).
Yang, C. et al. Insight into the humic acid/antibiotic complexation for boosting tetracycline degradation by heterogeneous persulfate activation. J. Environ. Chem. Eng. 12, 111712 (2024).
Ahmadian, A., Ahmadi, S. & Goharrizi, B. A. Roles of reactive species in photocatalysis: effect of scavengers and inorganic ions on dye removal from wastewater. Int. J. Environ. Sci. Technol. 20, 6433–6448 (2023).
Li, S. et al. Fabrication of vessel–like biochar–based heterojunction photocatalyst Bi2S3/BiOBr/BC for diclofenac removal under visible LED light irradiation: mechanistic investigation and intermediates analysis. J. Hazard. Mater. 391, 121407 (2020).
Bargozideh, S., Tasviri, M. & Kianifar, M. Construction of novel magnetic BiFeO3/MoS2composite for enhanced visible-light photocatalytic performance towards purification of dye pollutants. Int. J. Environ. Anal. Chem. 102, 6390–6404 (2022).
Lam, S., Sin, J. & Mohamed, A. R. A newly emerging visible light-responsive BiFeO3 perovskite for photocatalytic applications: a mini review. Mater. Res. Bull. 90, 15–30 (2017).
Selvarajan, S., Suganthi, A., Rajarajan, M. & Arunprasath, K. Highly efficient BiVO4/WO3 nanocomposite towards superior photocatalytic performance. Powder Technol. 307, 203–212 (2017).
Dang, V. D. et al. S-scheme N-doped carbon dots anchored g-C3N4/Fe2O3 shell/core composite for photoelectrocatalytic trimethoprim degradation and water splitting. Appl. Catal. B 320, 121928 (2023).
Shanavas, S. et al. Development of high efficient Co3O4/Bi2O3/rGO nanocomposite for an effective photocatalytic degradation of pharmaceutical molecules with improved interfacial charge transfer. J. Environ. Chem. Eng. 10, 107243 (2022).
Shi, W. et al. Enhancement of synergistic effect photocatalytic/persulfate activation for degradation of antibiotics by the combination of photo-induced electrons and carbon dots. Chem. Eur. J. 433, 133741 (2022).
Shen, J., Qian, L., Huang, J., Guo, Y. & Zhang, Z. Enhanced degradation toward Levofloxacin under visible light with S-scheme heterojunction In2O3/Ag2CO3: internal electric field, DFT calculation and degradation mechanism. Sep. Purif. Technol. 275, 119239 (2021).
Xin, D., Xian, M. & Chiu, P. C. New methods for assessing electron storage capacity and redox reversibility of biochar. Chemosphere 215, 827–834 (2019).
An, X. et al. Sequential photocatalytic degradation of organophosphorus pesticides and recovery of orthophosphate by biochar/α-Fe2O3/MgO composite: A new enhanced strategy for reducing the impacts of organophosphorus from wastewater. Chem. Eur. J. 435, 135087 (2022).
Li, R. et al. Activation of peroxymonosulfate by Fe doped g-C3N4/graphene under visible light irradiation for Trimethoprim degradation. J. Hazard. Mater. 384, 121435 (2020).
Jiang, T. et al. Bi25FeO40/Bi2O2CO3 piezoelectric catalyst with built-in electric fields that was prepared via photochemical self-etching of Bi25FeO40 for 4-chlorophenol degradation. J. Clean. Prod. 341, 130908 (2022).
Zhang, D., Zhang, R., Liu, J., Pu, X. & Cai, P. 3D/2D ZnIn2S4/BiFeO3 as S-scheme heterojunction photocatalyst for boosted visible-light hydrogen evolution. J. Am. Ceram. Soc. 106, 4785–4793 (2023).
Zhang, Q., Bai, J., Li, G. & Li, C. Synthesis and enhanced photocatalytic activity of AgI-BiOI/CNFs for tetracycline hydrochloride degradation under visible light irradiation. J. Solid State Chem. 270, 129–134 (2019).
Prabhu, S., Cindrella, L., Kwon, O. J. & Mohanraju, K. Green synthesis of rGO-WO3 composite and its efficient photoelectrochemical water splitting. Int. J. Hydrog. Energ. 42, 29791–29796 (2017).
Ahn, M., Lee, J. & Lee, W. Nanofiber-based composite cathodes for intermediate temperature solid oxide fuel cells. J. Power Sources 353, 176–182 (2017).
Chakhtouna, H., Benzeid, H., Zari, N., Qaiss, A. E. K. & Bouhfid, R. Recent progress on Ag/TiO2 photocatalysts: photocatalytic and bactericidal behaviors. Environ. Sci. Pollut. Res. 28, 44638–44666 (2021).
Li, Y. et al. Enhanced magnetic and photocatalytic properties of BiFeO3 nanotubes with ultrathin wall thickness. Vacuum 184, 109867 (2021).
Wu, L., Sui, W., Dong, C., Zhang, C. & Jiang, C. One-dimensional BiFeO3 nanotubes: preparation, characterization, improved magnetic behaviors, and prospects. Appl. Surf. Sci. 384, 368–375 (2016).
Ammar, S. H., Ali, F. D., Hadi, H. J. & Jabbar, Z. H. Visible-light-induced photodegradation of ofloxacin antibiotic by facile assembled BiFeO3/Bi-modified g-C3N4 heterojunctions. Mater. Sci. Semicond. Process. 171, 108026 (2024).
Arora, M., Chauhan, S., Sati, P. C., Kumar, M. & Chhoker, S. Evidence of spin-two phonon coupling and improved multiferroic behavior of Bi1-xDyxFeO3 nanoparticles. Ceram. Int. 40, 13347–13356 (2014).
Wang, L. et al. Sample-size resonance, ferromagnetic resonance and magneto-permittivity resonance in multiferroic nano-BiFeO3/paraffin composites at room temperature. J. Magn. Magn. Mater. 421, 71–75 (2017).
Zhang, Y. et al. Water flow induced piezoelectric polarization and sulfur vacancy boosting photocatalytic hydrogen peroxide evolution of cadmium sulfide nanorods. Appl. Catal. B 331, 122714 (2023).
He, S., Chen, Y., Li, X., Zeng, L. & Zhu, M. Heterogeneous photocatalytic activation of persulfate for the removal of organic contaminants in water: a critical review. Acs. Est. Eng. 2, 527–546 (2022).
Ning, J. et al. Experiment and DFT study on the photocatalytic properties of La-doped Bi2WO6 nanoplate-like materials. Appl. Surf. Sci. 579, 152219 (2021).
Jia, M. et al. Magnetic heterojunction of oxygen-deficient Ti3+-TiO2 and Ar-Fe2O3 derived from metal-organic frameworks for efficient peroxydisulfate (PDS) photo-activation. Appl. Catal. B 298, 120513 (2021).
Chavan, C., Kamble, S., Murthy, A. V. R. & Kale, S. N. Ampicillin-mediated functionalized gold nanoparticles against ampicillin-resistant bacteria: strategy, preparation and interaction studies. Nanotechnology 31, 215604 (2020).
Shukla, A., Khan, E., Tandon, P. & Sinha, K. Study of vibrational spectra and hydrogen bonding network in dimeric and tetrameric model of ampicillin using DFT and AIM approach. J. Mol. Struct. 1131, 225–235 (2017).
Grau, E. N., Román, G., Juan, J., Compañy, A. D. & Simonetti, S. Advance on adsorption of amino-functionalized silica nanocarrier for the delivery of therapeutic ampicillin as drug model. Inorg. Chem. Commun. 123, 108346 (2021).
Dou, M., Wang, J., Ma, Z., Gao, B. & Huang, H. Origins of selective differential oxidation of β-lactam antibiotics with different structure in an efficient visible-light driving mesoporous g-C3N4 activated persulfate synergistic mechanism. J. Hazard. Mater. 426, 128111 (2022).
Zhou, C., Wang, Y., Chen, J. & Niu, J. Porous Ti/SnO2-Sb anode as reactive electrochemical membrane for removing trace antiretroviral drug stavudine from wastewater. Environ. Int. 133, 105157 (2019).
Tang, J., Wang, P., Xie, Z., Hu, B. & Wang, Z. Effect of iron plaque on antibiotic uptake and metabolism in water spinach (Ipomoea aquatic Forsk.) grown in hydroponic culture. J. Hazard. Mater. 417, 125981 (2021).
Hajiabadi, M. G., Zamanian, M. & Souri, D. Williamson-Hall analysis in evaluation of lattice strain and the density of lattice dislocation for nanometer scaled ZnSe and ZnSe: Cu particles. Ceram. Int. 45, 14084–14089 (2019).
Qu, Z. et al. Preparation of a coated Z-scheme and H-type SrTiO3/(BiFeO3@ZnS) composite photocatalyst and application in degradation of 2,4-dichlorophenol with simultaneous conversion of Cr(VI). Sep. Purif. Technol. 240, 116653 (2020).
Shao, L., Pan, B., Hou, R., Jin, Y. & Yao, Y. User-friendly microfluidic manufacturing of hydrogel microspheres with sharp needle. Biofabrication 14, 025017 (2022).
Liu, N. et al. Z-scheme heterojunction ZnS/WO3 composite: Photocatalytic reduction of uranium and band gap regulation mechanism. J. Colloid Interface Sci. 630, 727–737 (2023).
Frisch, M. J. et al. Gaussian 16 Rev. C.01[Computer Software]. 2016. Wallingford, CT.
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Ministry of Ecology and Environment of the People’s Republic of China. The guldelines for the hazard evaluation of new chemical substances. https://www.mee.gov.cn/ywgz/fgbz/bz/bzwb/gthw/qtxgbz/200406/t20040601_68392.shtml (2004).
Wang, Y. et al. Degradation of benzophenone-4 by peroxymonosulfate activated with microwave synthesized well-distributed CuBi2O4 microspheres: theoretical calculation of degradation mechanism. Appl. Catal. B 290, 120048 (2021).
Acknowledgements
We would like to thank Editage (https://www.editage.cn/) for English language editing. This work was supported by One Health Interdisciplinary Research Project, Ningbo University (HY202203), and Zhejiang Province Basic Public Welfare Research Program, Zhejiang China (LTGS24E080001).
Author information
Authors and Affiliations
Contributions
Bing Li, Zhiying Dong, and Li Jiang contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Junxin Yu, Tianyu Gu, Ruiying Wang, Zhexuan Li, Xiaohui Zhu, and Weiming Zhu. The first draft of the manuscript was written by Junxin Yu. Bing Li commented on previous versions of the manuscript. Bing Li undertook the funding of the project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
Li Bing reports financial support was provided by Ningbo University. Li Bing, Dong Zhiying and Li Jiang reports a relationship with Ningbo University that includes: employment. The other authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yu, J., Gu, T., Wang, R. et al. Low toxicity mechanistic insights into Z-scheme WO3/BiFeO3/DSB photocatalysts for efficient ampicillin degradation. npj Clean Water 8, 34 (2025). https://doi.org/10.1038/s41545-025-00442-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41545-025-00442-w
