
Abstract
Accurate identification of true biological signals from diverse undesirable variations in large-scale transcriptomes is essential for downstream discoveries. Here we develop a universal deep neural network, called DeepAdapter, to eliminate various undesirable variations including batch, platform, purity and other unknown sources from transcriptomic data. Our approach automatically learns the corresponding denoising strategies to adapt to different situations. The data-driven strategies are flexible and highly attuned to the transcriptomic data that requires denoising, yielding reduced undesirable variation originating from batches, sequencing platforms and biosamples with varied purity beyond manually designed schemes. Comprehensive evaluations across multiple batches, different RNA measurement technologies and heterogeneous biosamples demonstrate that DeepAdapter can robustly correct diverse undesirable variations and accurately preserve biological signals, the faithful gene expression patterns that facilitate reliable biomarker discovery, transcriptomic network analysis and comprehensive biological characterization. Our findings indicate that DeepAdapter can act as a versatile tool for the comprehensive denoising of the large and heterogeneous transcriptome across a wide variety of application scenarios.
Similar content being viewed by others


Main
A cell’s transcriptome reveals its cellular state and the regulatory mechanisms of its molecular underpinnings through quantification of the dynamically transcribed RNA1. An enormous amount of transcriptomic data has been accumulated over the past two decades2,3,4. These heterogeneous datasets provide invaluable resources and present computational challenges for delivering comprehensive biological insights from integrated analyses due to the presence of diversified unwanted variations.
These undesirable variations fall into four categories: variabilities from notorious batch effects5, sequencing platforms6,7, heterogeneous biosamples8,9 and other unknown technical differences. Batch variations arising from different runs at different time points represent the prevailing technical factors10. Platform variations include intra- and interplatform systematical experimental bias. Intraplatform variations are caused by varied sequencing depth over orders of magnitude6,11, while interplatform variations originate from different sequencing technologies, for example, RNA-seq and microarray12. Purity variations refer to the bias among profiles sequenced from heterogeneous biosamples with diversified cellular composition, such as cancer cell lines and tumour tissues8,9. In addition to these three well-defined types of variation, many unknown variations exist that are obscured and difficult to correct. More challenging is that these undesirable variations often coexist, making the quest for true biological discoveries more complicated.
Existing methods remove one of these variations with nearly manually designed strategies to constrained situations for integrated transcriptomic analysis. Batch removal methods hypothesize that the true biological signals and non-biological noises exhibit either a linear13,14 or orthogonal5,15 relationship and mitigate the batch variations by subtracting the estimated batch noises. Still, the rigid assumption falls short in correcting the large and complex transcriptomic datasets featuring co-existing non-orthogonal variations. Normalization methods are designed to eliminate intraplatform variations from profiles sequenced by the same platform with the implicit assumption that transcriptomic readouts are proportional to some scaling factors16,17. Correspondingly, variation removal is implemented by simply dividing all gene readouts in one sample using a defined scale factor18,19. When datasets come from different platforms, the interplatform gene readouts cannot be corrected with one simple scale factor. For purity variations, bulk deconvolution methods successfully estimate the relative cellular abundances from heterogeneous biosamples20. Yet, deconvolution methods fail to reconstruct the denoised transcriptomic spectra, showing limitations in correcting composition differences across biosamples. As for unknown variations, manual schemes fail to tackle this kind of variation due to the high dependence on pre-defined assumptions.
Here we develop a deep neural network, called DeepAdapter, to robustly remove undesirable variations from large and heterogeneous transcriptomes. Tailored to adapt to various application scenarios automatically, DeepAdapter learns the latent space where distances of biosamples that are collected from diverse data sources yet should carry similar biological signals are minimized. DeepAdapter requires no previous information, endowing it with an advantage for eliminating unknown variations. These flexibilities facilitate the automatic learning of diverse denoising strategies, making it suitable for various application scenarios.
We evaluated DeepAdapter thoroughly on multiple tasks, and it considerably outperforms state-of-the-art methods in variation correction and biological signal conservation. For batch variations, by exploiting transcriptomic datasets derived from multiple batches, we reveal that DeepAdapter can eliminate conventional batch variations and conserve distinct donor-wise signals. For platform variations, we use large RNA-seq and microarray profiles of the same cancer cells and demonstrate that DeepAdapter can remove these variations and advance cancer subtype identification across different platforms. For purity variations, by employing RNA-seq profiles of cancer cell lines and tumour tissues, we illustrate that DeepAdapter can correct variations, including but not limited to those caused by varied tumour purity and immune infiltration, enhancing lineage identification and reproducing the associations between prognostic marker gene expression and clinical survival outcomes across heterogeneous biosamples. For unknown/mixed variations, we conceptualize the blind information of other technical factors, such as laboratory sites and library preparation protocols, as the unknown variations and demonstrate that DeepAdapter can eliminate these undesirable variations and recover true gene co-expression signals.
Results
Datasets with undesirable variations
Transcriptomic data with batch variations
We include 2 sets of transcriptomic datasets to study the batch variations: (1) Batch-LINCS: RNA-seq profiles of cardiomyocyte-like cell lines across 4 batches are sourced from the LINCS DToxS project21. The transcriptomic profiles are sequenced from 709 samples within 4 donors. The gene set is the intersection of 10,112 transcripts across 4 batches. (2) Batch-Quartet: RNA-seq profiles of B-lymphoblastoid cell lines spanning 21 batches are sourced from the Quartet project22. The profiles are collected from 252 samples within 4 donors. The gene set is the intersection of 58,395 transcripts across 21 batches.
Transcriptomic data with platform variations
We include cell lines sequenced by RNA-seq and microarray technologies to study the variations caused by technology revolutions. Microarray of cell lines is sourced from the Genomics of Drug Sensitivity in Cancer (GDSC) project3, which covers 948 samples spanning 30 cancer types. RNA-seq of cell lines is sourced from CCLE (22Q2 project)4, which covers 1,406 samples within 32 cancer types. The gene set is the intersection of 16,308 transcripts between microarray and RNA-seq. The curated dataset is referred to as Platform–GDSC–CCLE.
Transcriptomic data with purity variations
We include RNA-seq data of cell lines and tumour tissues to study the variations caused by heterogeneity in different biosamples. RNA-seq of cell lines comes from CCLE (19Q4 project)4, which covers 1,249 samples within 32 cancer types, and RNA-seq of tumour tissues comes from The Cancer Genome Atlas (TCGA) project2, which covers 12,236 samples spanning 32 cancer types. The transcriptomic data cover 36,631 protein-coding genes derived from the intersection between cell lines and tumour tissues. The curated dataset is referred to as Purity–TCGA–CCLE.
Transcriptomic data with unknown/mixed variations
We utilize Batch-Quartet, TCGA and Batch-LINCS datasets to illustrate unknown/mixed variations. For Batch-Quartet, to simulate unknown variations, we blind DeepAdapter to the information on library preparation protocols, RNA-seq devices and laboratory sites. For TCGA, we use information on different processing dates and batches as the unknown/mixed variations in the same manner. Similarly, for Batch-LINCS, we utilize information on replicate order as the unknown/mixed variations. Batch-LINCS features 56 drug treatments and 190 unique [donor, drug] samples, with each sample sequenced in 1–11 technical replicates within the same batch.
Overview of DeepAdapter
DeepAdapter is specifically designed to remove a wide range of different variations in transcriptomics data, thereby enabling meaningful biological discoveries with aligned large-scale datasets. These variations can arise from different sources such as batch effects10, sequencing technology revolutions (for example, from microarray to RNA-seq)23, inherent heterogeneity of biosamples (for example, cell lines and tumour tissues)9 and so on.
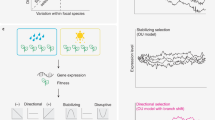
Despite being corrupted by diverse unwanted variations, we assume that paired samples (such as the transcriptome data obtained from microarray and RNA-seq for the same cell lines) inherently carry identical biological signals. Any differences between them lie in the presence of unwanted variations. Complete removal of all unwanted variations will make them indistinguishable. On the basis of this hypothesis, DeepAdapter utilizes autoencoder architecture to learn a latent space from large-scale transcriptomes. In this space, all the paired samples cannot be distinguished. This enables the simultaneous elimination of multiple variations without relying on specific assumptions regarding the source of variations. Moreover, each individual sample in the latent space must not only be closest to its corresponding pair, but must also be capable of being reconstructed back to its original form, guaranteeing the preservation of underlying biological signal (Fig. 1a).

a, Diverse sources of unwanted variations in transcriptome and the architecture of DeepAdapter. DeepAdapter is developed in an autoencoder, where variations removal is performed in the latent space between the encoder and the decoder. Specifically, a discriminator and a triplet network are trained to remove the unwanted variations. b, Alignment performances of different methods across batches (batch variations). c, Alignment performances of different methods across microarray and RNA-seq sequencing technologies (platform variations). d, Alignment performances of different methods across tumour cells and tumour tissues (purity variations).
Structure of DeepAdapter
DeepAdapter is inspired by the success of deep adversarial autoencoder (AAE)24 to extract low-dimensional intrinsic features. We designed DeepAdapter to consist of an encoder (E), a decoder (D), a discriminatory network (F) and a triplet neural network (T).
Following the concept of AAE, a min–max adversarial game between encoder and discriminator is established. The encoder learns a function to map the original transcriptomic profile from two sources (such as microarray data and RNA-seq data) to a latent space (𝑙). The discriminator then computes the probability that a sample in the latent space is from one of the sources. The encoder is trained to maximally confuse the discriminator into believing that samples in the latent space originate from a single data distribution rather than from multiple distinct sources. In direct competition with the encoder, the discriminator is trained to accurately distinguish the source of the data (Methods). Meanwhile, the triplet neural network trained with metric learning minimizes the distances between paired samples, which have mutually similar biological information but are collected from different data sources (Methods). Last, the decoder reconstructs the learned latent vector back to the original transcriptomic profile as accurately as possible. The resulting reconstructed data serve as the output of data correction by DeepAdapter (Methods).
The ablation studies on adversarial learning and deep metric learning further provide additional validations of their corresponding roles in variation removal and signal conservation (Supplementary Table 1).
Baseline comparison of alignment performance
We compare the performances on variation removal with quantile normalization (QuanNorm)25, Combat13, MNN5, Harmony26, PRPS6, Scanorama27, BBKNN28, AutoClss29 and scVI30. We utilize UMAP31 to visualize the results and alignment score32 to assess the quantitative performances (Fig. 1b–d). The raw data are greatly affected by the undesirable variations described above, with an alignment score of 0~0.030 (Fig. 1b–d). Strikingly, DeepAdapter dramatically eliminates these variations and exhibits a substantial improvement in alignment performance compared with baseline methods: (1) the top-3 alignment scores of 0.856/0.816/0.680 are for DeepAdapter/scVI-optim/scVI-origin across batches, (2) the top-3 alignment scores of 0.836/0.833/0.806 are for MNN/DeepAdapter/PRPS across sequencing platforms and (3) the top-3 alignment scores of 0.961/0.908/0.844 are for DeepAdapter/scVI-optim/Harmony across biosamples with different purities.
Next, we dive deeper into the correction results from our model in the three different scenarios described above. With these examples, we demonstrate the effectiveness of DeepAdapter in removing diverse variations and preserving biological signals for large and heterogeneous transcriptomic datasets.
Correcting batch variations
Batch effects are the common challenge in transcriptomic investigations, stemming from technical factors such as instrumental calibration and reagent variability. Remarkably, these variations often obscure true biological signals, introducing complexity and confounding subsequent analyses.
DeepAdapter eliminates batch variations
To assess performance in correcting batch variations, we employ DeepAdapter to correct the batch effects in Batch-LINCS and Batch-Quartet datasets. Obviously, raw transcriptomic data show clear separations among batches (Fig. 2b,e) with poor alignment scores (0.307/0.011) and average silhouette width (ASW)15 scores (0.462/0.326) for Batch-Quartet and Batch-LINCS, respectively (Fig. 2a,d). With DeepAdapter, the evaluation metrics have been largely improved, with alignment scores of 0.731/0.856 and ASW scores of 0.572/0.543 across Batch-Quartet (4 batches, Fig. 2a,b) and Batch-LINCS (21 batches, Fig. 2d,e), respectively.

a, Quantitative metrics of batch correction and biological signals conservation for Batch-Quartet data. b, UMAP analysis of raw and aligned data across 21 batches for Batch-Quartet data. c, UMAP analysis of aligned data across 4 donors for Batch-Quartet data. d, Quantitative metrics of batch correction and biological signals conservation for Batch-LINCS data. e, UMAP analysis of raw and aligned data across 4 batches for Batch-LINCS data. f, UMAP analysis of aligned data across 4 donors for Batch-LINCS data.
DeepAdapter conserves the distinct donor-wise signals
Donor sources are representative biological signals. We observe that DeepAdapter clearly separates samples from distinct donors into clusters, showing a satisfactory performance of biological signals conservation. The results are further supported by the normalized mutual information (NMI)33 and adjusted rand index (ARI)34 analyses. Specifically, DeepAdapter makes a noteworthy improvement in NMI score, progressing from 0.142/0.207 to 0.695/0.344, and ARI score, advancing from 0.042/0.126 to 0.634/0.346, for Batch-Quartet (Fig. 2c and Supplementary Fig. 2) and Batch-LINCS (Fig. 2f and Supplementary Fig. 2), respectively. These findings illustrate the robust efficacy of DeepAdapter in accurately preserving biological signals across batches.
Correcting platform variations
Sequencing technologies for transcriptomics have evolved rapidly from microarray to RNA-seq. Integrated analyses of datasets from these two technologies remain challenging due to their inherent variability difference. Microarray platforms detect the signals with sophisticated probe sets, while RNA-seq technologies record the counts of copy numbers for transcripts. Consequently, microarray measures the continuous values of transcripts following the normal Gaussian distribution, and RNA-seq records the integer counts, thereby making the expression profiling incomparable and hampering meaningful biological discoveries.
DeepAdapter eliminates platform variations
To evaluate the performance of DeepAdapter in eliminating platform variations, we use DeepAdapter to remove the non-biological effects between microarray and RNA-seq datasets and generate an integrated gene expression dataset across sequencing platforms. As expected, raw data show an obvious separation between microarray and RNA-seq technologies with a poor alignment score of 0 and ASW score of 0.265 (Fig. 3a,b), consistent with the differential distributions in gene expression (Fig. 3d). Correspondingly, DeepAdapter substantially improves the alignment/ASW score to 0.833/0.496 (Fig. 3a,c) and reduces the divergence score from 226.14 to 2.41 across microarray and RNA-seq technologies (Fig. 3d).

a, Quantitative metrics of platform correction and biological signals conservation. b,c, UMAP analysis of raw (b) and aligned (c) data across microarray and RNA-seq platforms. Open and filled circles indicate the training and testing sets, respectively. d, Divergence score of raw and aligned data. Shaded areas denote minimal and maximal density across expression level. e, UMAP analysis of aligned data across cancer types. f, UMAP analysis of aligned data for molecular subtypes. SCLC, small-cell lung cancer. g, Correlation distribution of the identical tumour cells profiled by microarray and RNA-seq across cancer types. h, Cross-sequencing-platform classification performances of raw (dashed lines) and aligned (solid lines) data using DeepAdapter.
DeepAdapter preserves cancer types across platforms
To assess the effectiveness of DeepAdapter in preserving biological variations, we first evaluate whether DeepAdapter erased information about known cancer types and subtypes across microarray and RNA-seq datasets during the correction process. As demonstrated in Fig. 3e, DeepAdapter corrects much of the systematic variations while reproducing clear cancer-type clusters across different platforms, including lymphoma, myeloma, leukaemia, skin cancer, brain cancer, bone cancer, neuroblastoma, colon/colorectal cancer, breast cancer, head and neck cancer, and kidney cancer (Supplementary Fig. 4). Quantitative analyses confirm that our method improves the clustering of tumour types (Fig. 3a and Supplementary Fig. 3, NMI/ARI score from 0.383/0.177 to 0.503/0.326). To extend this analysis, we annotate the cancer subtype labels of aligned transcriptomic data for microarray and RNA-seq. The results reveal that molecular subtype information is aligned with DeepAdapter. For example, lung cancer and breast cancer subtypes are aligned in the same tumorous space (Fig. 3f). Correspondingly, leukaemia cell lines formed two distinct clusters: a myeloid cluster and a lymphocytic cluster (Fig. 3f) with B and T cells in distinguishable clusters (Fig. 3f).
In addition, we explore the similarity between corresponding transcriptomes that were measured by both microarray and RNA-seq techniques (n = 684). Interestingly, compared with the correlation of 0.798–0.831 in the original data, the correlation has been notably improved to 0.978–0.993 after removing the platform variation using DeepAdapter (Fig. 3g).
A huge number of transcriptomic data are available today through various platforms. However, they are difficult to integrate and, therefore, not fully utilized. For example, modern artificial intelligence and machine learning capabilities rely heavily on the availability of large amounts of high-quality data. We thus assessed the potential to integrate datasets from different sources. To simulate this situation, we design the cross-platform classification experiment, where machine learning models for cancer-type classification are trained on only microarray datasets, and then tested with RNA-seq data strictly. Strikingly, after the correction of undesirable variation by DeepAdapter, machine learning models extensively improve the classification performances. In the experiment of training on microarray datasets and testing using RNA-seq datasets (Fig. 3h and Supplementary Fig. 5), the accuracies (0.464/0.590/0.686) for classifying 25/12/5 cancer types notably increase, compared with limited accuracies (0.106/0.103/0.252) in the unaligned data. The improved accuracies of multiclassification are also observed in the counterpart setting of training on RNA-seq datasets and testing on microarray datasets (Fig. 3g). In addition, we compare gene expression profiles between pan-cancer cell lines using gene set enrichment analysis (GSEA)35. Multiple cancer-related gene modules36 are significantly enriched after correction (Supplementary Fig. 6).
Taken together, our results indicate that DeepAdapter has successfully preserved transcriptomic features representative of these biological variations.
Correcting purity variations
Tumour masses are highly heterogeneous structures composed of both malignant and non-malignant cells37. Due to the heterogenicity of the tumour samples, numerous bulk transcriptomic data accumulated over the past two decades are compromised by the presence of normal cells to varying degrees. Deconvolution methods have successfully demonstrated the feasibility of inferring cell type compositions from bulk RNA-seq integrated with single-cell RNA-seq as previous knowledge38. However, extracting the tumour-specific profiles by eliminating purity variations beyond estimating cellular composition stands as a challenging but pioneering endeavour.
DeepAdapter removes purity variations
To challenge the performance of DeepAdapter in removing purity variations, we apply DeepAdapter to align the gene expression profiles measured from cancer cell lines and human tumour tissues (Fig. 4). The underlying hypothesis is that if transcriptomic differences between tumour cell lines and tumour masses are corrected by DeepAdapter, then the majority of the eliminated signals will be purity variations. To test this hypothesis, we compare the transcriptomic data before and after alignment by DeepAdapter, and find that cell lines and tumour tissues are well aligned, with alignment/ASW scores (Fig. 4a) impressively improved from 0.030/0.285 to 0.961/0.499 (Fig. 4b,c). Interestingly, additional analyses validate that this result is largely due to the removal of purity variations. To ensure rigour, we apply two metrics (tumour purity score and immune score)39 to assess tumour purity and, more specifically, the proportion of infiltrating stromal and immune cells from transcriptomic data. DeepAdapter remarkably improves the estimated tumour purity (Fig. 4g and Supplementary Fig. 7) and decreases the presence of immune cells for 96.9% (31/32) of aligned cancer types (Fig. 4h,i and Supplementary Fig. 7a–d). In addition, the decrease in immune cells is further confirmed using CIBERSORTx40,41. Of the cancer types, 81.3% (26/32) show a reduced presence of immune cells after alignment by DeepAdapter (Supplementary Fig. 7e,f).

a, Quantitative metrics of purity correction and biological signals conservation. b,c, UMAP analysis of raw (b) and aligned (c) data across tumour cells and tumour tissues. Open and filled circles denote the training and testing sets, respectively. d, UMAP analysis of aligned data across lineages. e, Correlation distribution of profiles sequenced from cancer cells and tumour tissues across cancer types. f, Cross-biosample classification performances of raw (dashed lines) and aligned (solid lines) data using DeepAdapter. g, Analysis of tumour purity for 32 cancer types with raw and aligned data. h, Analysis of immune score for 32 cancer types with raw and aligned data. Each dot denotes one sample. Significance levels were calculated between raw and aligned groups using two-sided t-test. *p < 0.05, **p < 0.01. i, Tumour purity and immune score of individual cancer types. Each dot corresponds to the metrics (immune score (IS), tumour purity (TP)) for each descriptor. Diameter is proportional to the significance level (−log10(p)) of the changed TP. The orange region indicates cancers with low TP and high IS. The green region indicates cancers with high TP and low IS.
DeepAdapter improves lineage fidelity across biosamples
To confirm that meaningful biological features are retained, we inspect whether the information about the tumour tissue origin is preserved. As demonstrated in Fig. 4d, the corrected gene expression profiling of tumour tissues is clustered with the cell lines of the corresponding tissue origin. Notably, the intralineage similarity between cell lines and tissues is improved from 0.872 to 0.964 across lineage types (Fig. 4e and Supplementary Fig. 8).
To confirm this observation, we design the cross-biosample classification experiment. We train the cross-biosample classifier with only transcriptomic data of cell lines and test with tumour tissues strictly. DeepAdapter largely improves the accuracy by 37.352% on average. More specifically, DeepAdapter substantially improves accuracies to 0.516/0.696/0.831/0.891 for classifying 18/8/5/2 lineages, compared with 0.158/0.174/0.325/0.360 for the unaligned data (Fig. 4f) with a limited number of training samples (169/255/304/274).
DeepAdapter reproduces the prognostic markers
Undesirable variations can compromise the downstream identification of prognostic markers from transcriptomic data of patient tumour tissues6. For example, SUCLG2P2, GPC1, CISH and ANKZF1 are identified as survival-associated genes in colon adenocarcinoma (COAD)6,42. However, no such association was detected in unaligned TCGA COAD data (n = 262, Fig. 5a). Other examples are FBXL14 and PKP2 in rectum adenocarcinoma (READ) and STAB1 in breast cancer (BRCA), whose associations with survival have been previously reported6,42, but these associations were also obscured in uncorrected datasets (Fig. 5b,c). Surprisingly, after DeepAdapter correction, we found that these associations were all reproduced (Fig. 5).

Overall survival analysis of gene expression for (a) COAD, (b) READ and (c) BRCA. First row: survival analysis of raw transcriptomic data. Second row: survival analysis of aligned transcriptomic data.
Correcting unknown/mixed variations
In addition to the three well-known variations mentioned above, independent transcriptome studies inevitably introduce a large number of other unwanted variations from a variety of sources. These variations are difficult to define and eliminate, leading to misleading discoveries.
DeepAdapter eliminates unknown variations
Batch-Quartet study involves 252 samples within 4 donors collected in 8 laboratory sites using 2 library preparation protocols (Poly(A) selection and ribosomal rRNA depletion) and 2 RNA-seq devices (Illumina NovaSeq and MGI DNBSEQ-T7) in 21 batches22. Both alignment score and UMAP analysis have identified that all of these technical variations introduced unwanted variation into the transcriptomic data, with the variation caused by library preparation having the greatest impact (Fig. 6a). Library preparation protocol variations are clearly visible in the UMAP analysis of the raw data (Fig. 6b). Notably, these unknown variations, as well as batch variations, are greatly reduced in DeepAdapter-aligned data (Fig. 6a–c and Supplementary Fig. 9). We systematically examine the transcriptomic profiles collected from different laboratories and devices but by the same protocol. The results reveal that the laboratory/device variations have persisted even after ruling out the major deviations brought about by the protocol, further confirming the complicated mixed bias introduced by multiple technical variations (Fig. 6a and Supplementary Fig. 10). Interestingly, DeepAdapter adaptively learns from the dataset and efficiently eliminates the laboratory/device variations in this new situation (Supplementary Fig. 10). These results robustly illustrate DeepAdapter’s efficacy in simultaneously removing multiple mixed/unknown variations whose sources are challenging to identify.

a, Quantitative metrics of known (batch) and unknown (library protocols, laboratory sites and devices) variations for Batch-Quartet data. b,c, UMAP analysis of raw (b) and aligned (c) data across library preparation protocols. d, p-value distribution of gene ontology (GO) enrichment analysis in sig2sig, sig2sig+ns2sig and sig2sig+sig2ns genes between cancer and control cell lines. The central lines indicate the median. The lower and upper box bounds indicate the first and third quartiles, respectively. The whiskers show data within 1.5 times the interquartile (IQR). The error bars denote standard errors. The number of p values from GO enrichment analysis ranges from 719 to 2,054 across cancer types. The p values were calculated using Fisher’s exact test. e, Correlation analysis for the expression level of CNOT1 and E2F4 in raw transcriptomic data. f,g, Correlation analysis and variation analysis of 5 processing dates (f) and 44 experimental batches (g) for CNOT1 and E2F4 gene expression in raw transcriptomic data. i, Correlation analysis for CNOT1 and E2F4 gene expression in aligned transcriptomic data. j,k, Correlation analysis and variation analysis of 5 processing dates (j) and 44 experimental batches (k) for the expression level of CNOT1 and E2F4 in aligned transcriptomic data. h,l, Coefficient of variation analysis for dates (h) and batches (l).
DeepAdapter reconstructs differentially expressed genes
We investigate the differential genes between cancer and control cell lines measured by RNA-seq. Across 22 cancer types, the majority of genes (53.2%, sig2sig group) remain significant before and after correction (Supplementary Fig. 11). Interestingly, a small number of genes changes: 11.3% of differentially expressed genes (DEGs) in the raw data no longer showed significant differences (sig2ns group). Conversely, new DEGs (24.9%) are identified after alignment (ns2sig group). This suggests the presence of confounding effects from undesirable variations in the identification of DEGs. To examine the effects of varied gene significances, we consider that genes (sig2sig) identified as DEGs in both raw and aligned datasets represent true biological signals. We then examine which groups of genes (sig2ns or ns2sig) carry similar biological signals to the sig2sig group. To do so, we compare the results from gene ontology (GO) enrichment analysis43 of three gene sets: sig2sig only, sig2sig+sig2ns and sig2sig+ns2sig. If the genes carry the true biological signals, their inclusion should increase the enrichment of biological function; conversely, if they have confounding effects, they will decrease this enrichment. Notably, we found that when newly identified DEGs (ns2sig, account for 24.9% of the whole DEGs) are included, the significance of the true biological functions (sig2sig) was further increased across all cancer types (Fig. 6d). In addition, the addition of DEGs removed after correction (sig2ns) leads to a decreased enrichment (Fig. 6d). Together, these results illustrate the confounding effects of undesirable variation on DEG analysis and show the effectiveness of DeepAdapter in eliminating these variations and extracting real biological differences.
DeepAdapter reproduces gene co-expression relationships
Undesirable variations can compromise gene co-expression analyses. For example, CNOT1 (CCR4-NOT transcription complex subunit 1) and E2F4 (E2F transcription factor 4) are demonstrated as the correlated gene pair for BRCA in both RNA-seq and microarray transcriptomic data6. Yet, the co-expression analysis between CNOT1 and E2F4 shows a limited correlation coefficient of 0.537 in unaligned TCGA data (Fig. 6e). After DeepAdapater alignment, a strong correlation between the expression levels of both genes is detected (𝜌 = 0.849, Fig. 6i). Further analysis reveals that this is greatly affected by unwanted variation from complex sources: batches, processing dates and potentially other sources. By regrouping the raw data by processing dates and batches separately, the average correlation of these two genes within each group is largely improved (𝜌 = 0.690/0.688 for 5 date groups/44 batch groups, Fig. 6f–h) compared with the overall correlation (𝜌 = 0.537). After alignment, the regrouped data also show a high average correlation (𝜌 = 0.863/0.865 for dates/batches, Fig. 6j–l). These results show that DeepAdapter corrects the unknown variations caused by complex mixed factors without previous knowledge.
DeepAdapter reduces unknown deviation within replicates
Finally, we examine whether there are unknown variations present among replicates. For Batch-LINCS, at the sample-wise level, the raw transcriptomic data exhibit good repeatability. Interestingly, after correction by DeepAdapter, the correlations among all the replicates further increase (Supplementary Fig. 12), indicating that variations are present and effectively reduced. At the gene-wise level, we investigate the deviation of single-gene expression within technical replicates of the same sample. Notably, the deviation of single-gene expression decreases within replicates significantly (p < 10−4 for all testing samples, Supplementary Fig. 13). The results illustrate DeepAdapter’s capability to reduce deviation among replicates, underscoring the importance of DeepAdapter in cleaning up the transcriptomic data before downstream analysis.
Discussion
In summary, we propose a universal framework to eliminate variations caused by a single factor or a combination of factors, such as batches, platforms and tumour purities. Using transcriptomic datasets obtained from LINCS, Quartet, TCGA, GDSC and CCLE, we reveal that DeepAdapter is efficient and robust in removing these variations and preserving meaningful biological variability for downstream analysis.
By carefully analysing these large-scale transcriptomic data above, we illustrate the presence of numerous undesirable variations from different sources in the data, influencing downstream analysis and distorting biological insights. These variations tend not to exist in isolation, but widely coexist in the data, including from a single independent study and multiple different studies. In such situations, prevailing models that specialize in handling a specific type of undesirable variation5 are limited. A suitable model must be versatile in efficiently identifying and addressing different types of unwanted variation. We take advantage of deep generative networks coupled with adversarial learning and contrastive learning to automatically correct multiple types of undesirable variation. Our results clearly show that DeepAdaptor can remove different sources of unwanted variation simultaneously. Therefore, we believe that it will be a valuable tool for biological scientists to better integrate and analyse large-scale transcriptomes.
The versatility of our model substantially advances integrative investigations of large and heterogeneous transcriptomes. Continuous innovation in RNA profiling technologies has resulted in the accumulation of extensive datasets. However, due to undesirable variations introduced by different technologies, they are not fully used. Our approach offers a practical solution to systematically eliminate these variations, enabling deep mining by integrating the historical datasets generated by early-developed technologies.
A remarkable feature of DeepAdapter is its capability to reconstruct transcriptomic profiles of pure tumour cells from bulk RNA data of highly heterogeneous tumour tissue by removing the purity variations. Although single-cell RNA-seq technology enables the detection of transcriptomic signatures of tumour cells, the cost has largely limited its clinical application. As an alternative, our work has demonstrated the feasibility of inferring the transcriptomic profiles of tumour cells from bulk RNA by eliminating the inevitable interferences from normal cells. We suggest that DeepAdapter could be used as a powerful tool for advancing clinical molecular diagnostics and precision medicine.
Currently, one limitation of our design is that the DeepAdapter framework, based on deep neural networks, relies on large amounts of high-quality data. Given the inherent constraints of transcriptomic data in accurately identifying and quantifying cell types, we highly recommend using standard reference transcriptomes as controls. Moreover, in many real-world scenarios, the sample sizes are inherently limited. While we highlight the effectiveness of our DeepAdapter with transfer learning strategy in overcoming the challenges of learning with limited sample size (Supplementary Fig. 14), it is worth noting that the revolution in deep learning techniques and the accumulation of large high-quality datasets offer superior solutions for the future.
Methods
Datasets
Transcriptomic data
The transcriptomic data included gene expression datasets across batches, sequencing platforms and biosamples. For batch variations, RNA-seq profiles of cell lines were sourced from the LINCS (http://lincsportal.ccs.miami.edu/dcic-portal/) and Quartet (https://docs.chinese-quartet.org) projects. For platform variations, microarray of cell lines was obtained from the Genomics of Drug Sensitivity in Cancer (GDSC) project (https://www.cancerrxgene.org). RNA-seq of cell lines came from DepMap (22Q2 project, https://depmap.org/portal/). For purity variations, RNA-seq of cell lines came from DepMap (19Q4 project, https://depmap.org/portal/), and RNA-seq of tumour tissues came from The Cancer Genome Atlas (TCGA) project2 (https://www.cancer.gov/ccg/research/genome-sequencing/tcga).
Survival data
The survival data of the TCGA project were downloaded from the GDC Data Portal (https://portal.gdc.cancer.gov/).
Batch and processing date annotations
The annotations of batches and processing dates for the TCGA project were downloaded from the MBatch Omic Browser (https://bioinformatics.mdanderson.org/MQA/).
Known potential prognostic genes
The genes with significance in overall survival analysis were obtained from two sources: (1) the presented results of the PRPS project6, which aims to remove variations caused by tumour purity and batch effects for the TCGA project; (2) targeted research on glycolysis-related gene sets in prognosis analysis for colon adenocarcinoma (COAD) and rectal adenocarcinoma (READ)42. Specifically, there exists 8, 6 and 3 known potential prognostic genes for COAD, READ and BRCA, respectively: (1) SUCLG2P2, GPC1, CISH, ANKZF1, BRAF, P4HA1, STC2 and PCK1 for COAD; (2) TSTA3, RAB18, PTPN14, PKP2, FBXL14 and CSGALNACT2 for READ; and (3) ZEB2, STAB1 and ESRRA for BRCA.
Data split
For batch variations, the biosamples across partial batches were split as the training set (N = 624), while the samples across all batches were extracted as the testing set (N = 85). For platform variations, the biosamples with both the RNA-seq and microarray profiles were split as the testing set (N = 1,368), while the remaining biosamples with one-platform sequencing were extracted as the training set (N = 986). For purity variations, biosamples were randomly split into 80%/20% training/testing sets (N = 10,788/2,697 for training/testing sets).
Preprocessing of transcriptomic profiles
Sample normalization and log normalization were utilized in data preprocessing. Let \({S}_{i}=\sum _{l}{x}_{{il}}\) denote the sum over all genes for sample \({x}_{i}\). In sample normalization, every gene expression profile was divided by \({S}_{i}\) and multiplied by a constant 10,000 as follows:
log transformation was calculated as \({x}_{{il}}=\log \left({x}_{{il}}+1\right)\).
The architecture of DeepAligner
DeepAligner was designed in an autoencoder structure and unwanted variations were removed in the latent space between encoder and decoder. The encoder E takes the gene expression vector as the input x and extracts the latent vector with fully connected layers. With the extracted latent vector l projected into a low-dimensional space, DeepAligner corrects the artefacts with adversarial learning and deep metric learning. After removing the unwanted variations, the decoder D reconstructs the transcriptomic data \({x}^{{\prime} }\) by projecting the latent vector back into the original high-dimensional space.
Adversarial learning removes the artefacts by introducing a discriminator F, which is designed to detect unwanted variations. During adversarial learning, the discriminator F aims to minimize the classification loss by refining its ability to identify data sources. At the same time, the autoencoder strives to maximize the classification loss by enhancing its capability to generate latent vectors where the paired samples are indistinguishable to confuse the discriminator.
Deep metric learning removes the artefacts by minimizing the distance between the anchor and the positive and maximizing the distance between the anchor and the negative. DeepAligner automatically defines the positive samples with mutual nearest neighbours5 without any additional biological annotations as follows:
where m and n refer to the extracted MNN pair, \({\rm{NN}}\left({x}_{n}^{b},{X}^{a}\right)\) refers to the nearest neighbours in source a of sample n from source b, and \({\rm{NN}}\left({x}_{m}^{a},{X}^{b}\right)\) refers to the nearest neighbours in source b of sample m from source a. The negative is defined as the randomly selected samples from the same source, excluding the sample itself.
Loss function
The loss function includes 3 parts: the reconstruction loss of the autoencoder, the adversarial loss of the discriminator and the triplet loss of the deep metric learning. Let l denote the latent vector extracted from raw profile 𝑥 by encoder E:
The deep metric learning minimizes the distances between paired samples, which have mutually similar biological information but are collected from different data sources as follows:
where \({\bf{l}}_{a},{\bf{l}}_{p}\) and \({\bf{l}}_{n}\) denote the latent vectors of the anchor, the positive and the negative samples, d refers to the distance metric measured by Euclidean distance, and m indicates the margin value of 1. Paired samples denote the anchor and positive samples that are mutual nearest neighbours.
The discriminator aims to minimize the classification loss as follows:
where CE refers to the cross-entropy loss between the true label of the data source (\({y}^{{\rm{s}}}\)) and the predicted source from the latent vector (l) by the discriminator (F).
The decoder is trained to reconstruct the learnt latent vector back to the original transcriptomic profile as accurately as possible. The reconstruction loss of the autoencoder is as follows:
where E refers to the encoder, D denotes the decoder, and MAE indicates the mean absolute error between the raw and corrected profiles.
Based on competitive learning optimization, the min–max game works as follows:
where \({\rm{\lambda }}=0.01\) refer to the weight of adversarial learning in the total loss function. The autoencoder aims to minimize the total loss (\(L\)), thereby maximizing the discriminator loss (\({{\rm{loss}}}_{{\rm{disc}}}\)). Conversely, the discriminator seeks to maximize \(L\), effectively minimizing \({{\rm{loss}}}_{{\rm{disc}}}\).
In this manner, DeepAdapter achieves two main goals: (1) eliminating unwanted variations in the latent representations and (2) avoiding signal distortion while eliminating variations.
Training of DeepAdapter
DeepAdapter was trained with epochs = 150,000 using Adam optimizer with \({\beta }_{1}=0.9\) and \({\beta }_{2}=0.98\). The learning rate was first increased linearly to the maximum learning rate of \(5\times {10}^{-4}\) and then decreased linearly to the minimum learning rate of \({10}^{-5}\). The batch size was set as 256. The whole model was trained with single Nvidia GeForce RTX 3090 Ti.
Evaluation metrics
Two sets of evaluation metrics were included in this work: (1) variation removal and (2) biological signal conservation.
Variation removal metrics included alignment score32 and modified average silhouette width (ASW) score15. The basic intuition of variation removal is that if the samples are well aligned, the neighbours of any sample are evenly distributed across data sources. Alignment score measures the distribution of samples from different sources around one sample. The alignment score was calculated as follows:
where k refers to the number of nearest neighbours, N refers to the total number of samples, and \(\bar{x}\) refers to the average number of nearest neighbours belonging to the same data source. The original ASW score is the average value of all silhouette widths of all samples and was defined as follows:
where i refers to the sample i and the silhouette width measures the similarity consistency within clusters:
where a and b refer to the intracluster and nearest-cluster distance for sample i. Thus, the modified ASW for assessing variation correction measures the opposite aspect of within-cluster consistency and was defined as follows:
Biological signal conservation metrics included normalized mutual information (NMI)33 and adjusted rand index (ARI)34. Both NMI and ARI compare the overlap between biological annotations and clustered annotations calculated from the integrated dataset. NMI was calculated with entropy as follows:
where Y and C denote the biological and clustered annotations, H(Y) and H(C) refer to the entropy of biological and clustered annotations, and I(Y;C) indicates the mutual information between biological and clustered annotations. ARI was calculated as follows:
where \({n}_{{Y}_{i}{C}_{j}}\) refers to the number of samples of the biological annotation \({Y}_{i}\) assigned to cluster \({C}_{j}\), \({n}_{{Y}_{i}}\) refers to the number of samples in biological annotation \({Y}_{i}\), and \({n}_{{C}_{j}}\) refers to the number of samples in the cluster annotation \({C}_{j}\).
Jensen–Shannon divergence (JSD) was used to measure the similarity of gene expression distributions for matched RNA-seq and microarray of cell lines. JSD was calculated as follows:
where \(M=1/2(P+Q)\), D refers to the Kullback-Leibler divergence and P and Q refer to the gene expression distributions for matched RNA-seq and microarray of cancer cell lines, respectively.
Ablation studies
The ablation studies on adversarial learning and deep metric learning were performed to investigate their individual contributions to integrated performances. The ablated experiments were conducted by disabling the corresponding adversarial learning and deep metric learning modules.
Baseline approaches
Quantile normalization was implemented with qnorm (v.0.8.1).
Combat13 corrects the unwanted variations with an empirical Bayes design and was implemented with pycombat (v.0.3.3, https://epigenelabs.github.io/pyComBat/).
MNN5 corrects bias with the correction vectors between MNN pairs and was implemented with mnnpy (v.0.1.9.5, https://pypi.org/project/mnnpy/).
Harmony26 employs an iterative algorithm on the PCA-embedded data until convergence and was implemented with scanpy (v.1.9.3, https://scanpy.readthedocs.io/en/stable/).
PRPS6 creates the pseudo-replicates of pseudo-samples derived from real samples that are estimated to be homogenous concerning library size, tumour purity and batch effects. We ran PRPS with the package (v.0.1.0, https://rdrr.io/github/AbhishekSinha28/tgcapkg/).
Scanorama27 extends mutual nearest neighbours5 with a panorama stitching algorithm on a low-dimensional embedding space. We ran Scanorama with the package scanorama (v.1.7.4, https://cb.csail.mit.edu/cb/scanorama/).
BBKNN28 builds a balanced neighbourhood graph across all batches, ensuring that every sample has an independent pool of neighbours. We ran BBKNN with the package bbknn (v.1.6.0, https://bbknn.readthedocs.io/en/latest/).
AutoClass29 extends the naïve autoencoder with a classifier: the autoencoder is applied to reconstruct profiles and the classifier is utilized to predict the virtual biological classes (the pseudo biological labels generated by K-means clustering). We ran AutoClass with the released codes (https://github.com/datapplab/AutoClass).
ScVI30 assumes that gene expression follows the negative binomial distribution and utilizes a variational inference model by deep neural networks. We ran scVI with the package scVI (v.0.6.8, https://pypi.org/project/scvi/). The original parameters for scVI include latent space dimensionality of 30, hidden units of 128, hidden layers of 2 and epochs of \(400\times (20,000/N)\), where \(N\) refers to the sample number of the dataset. A model trained with the original parameters was named scVI-origin. We further optimized the parameters of scVI for higher performances: latent space dimensionality of 128, hidden units of 256, hidden layers of 5 and epochs of 15,000. Correspondingly, a model trained with the optimized parameters was named scVI-optim.
DeepAdapter with transfer learning
We utilized the DeepAdapter well trained with 201 samples in Batch-Quartet as the pre-trained model for batch correction. To simulate the scenarios with a limited sample size, we randomly sampled 4, 8, 16, 32, 64 and 96 samples from another dataset (Batch-LINCS). Given that the fine-tuned dataset encompassed a different number of batch categories (4 batches) compared with the pre-trained dataset (21 batches), we modified the last layer of discriminatory network to classify 4 batch categories instead of 21. We unfroze all layers and trained the fine-tuned models for 5,000 epochs. This procedure was repeated 20 times, with performances assessed using an independent testing set of 24 samples.
GO enrichment analysis
We performed gene ontology (GO) enrichment analysis43 between cancer and control cell lines measured by RNA-seq. We ran GO analysis using the online enrichment analysis tool (https://geneontology.org/).
GSEA validation
We conducted gene set enrichment analysis35 (GSEA) of integrated cancer cell lines profiled by RNA-seq and microarray. The cancer-related gene sets were obtained from validated references36,44. We ran GSEA using the package gseapy45 (v.1.0.3, https://gseapy.readthedocs.io/en/latest/).
Tumour purity calculation
The inferred tumour purity of bulk gene expression profiles was calculated with ESTIMATE by inferring the proportion of stromal and immune cells39.
In addition, CIBERSORTx40 was utilized to calculate the presences of immune cells. To ensure the rigour and consistency of the platform, the ‘LM6’ matrix provided by CIBERSORTx (v.1.04) measured by RNA-seq41 was employed as the immune signature matrix.
Statistical analysis
Calculation of Pearson correlation coefficients was implemented with scipy (v.1.11.1, www.scipy.org). UMAP decomposition was implemented with umap-learn (v.0.5.3, https://umap-learn.readthedocs.io). AWS, NMI and ARI were implemented with scikit-learn (v.1.2.2, www.scikit-learn.org). Kaplan–Meier analysis was implemented with lifelines (v.0.27.7, https://lifelines.readthedocs.io). log rank test for survival analysis was implemented with scikit-survival (v.0.21.0, https://scikit-survival.readthedocs.io/en/stable/).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All transcriptomic datasets in this study are publicly available. RNA-seq profiles of Batch-LINCS and Batch-Quartet can be downloaded from the LINCS (http://lincsportal.ccs.miami.edu/dcic-portal/) and Quartet (http://chinese-quartet.org) projects. The microarray of cancer cell lines can be downloaded from the GDSC project (https://www.cancerrxgene.org/). RNA-seq of cancer cell lines can be downloaded from the CCLE project (https://depmap.org/portal/). RNA-seq of human tumour tissues can be downloaded from the TCGA project (https://www.cancer.gov/ccg/research/genome-sequencing/tcga). The pre-trained models can be downloaded from Zenodo at https://zenodo.org/records/14664454.
Code availability
The codes of DeepAdapter, Benchmarking methods, fine-tuning steps and detailed tutorials are available in GitHub at https://github.com/mjDelta/DeepAdapter (ref. 46). The Python version is 3.9.21.
References
Cieślik, M. & Chinnaiyan, A. M. Cancer transcriptome profiling at the juncture of clinical translation. Nat. Rev. Genet. 19, 93–109 (2018).
Weinstein, J. N. et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 45, 1113–1120 (2013).
Iorio, F. et al. A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754 (2016).
Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019).
Haghverdi, L., Lun, A. T., Morgan, M. D. & Marioni, J. C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018).
Molania, R. et al. Removing unwanted variation from large-scale RNA sequencing data with PRPS. Nat. Biotechnol. 41, 82–95 (2023).
Gagnon-Bartsch, J. A. & Speed, T. P. Using control genes to correct for unwanted variation in microarray data. Biostatistics 13, 539–552 (2012).
Domcke, S., Sinha, R., Levine, D. A., Sander, C. & Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 4, 2126 (2013).
Warren, A. et al. Global computational alignment of tumor and cell line transcriptional profiles. Nat. Commun. 12, 22 (2021).
Leek, J. T. et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 11, 733–739 (2010).
Sims, D., Sudbery, I., Ilott, N. E., Heger, A. & Ponting, C. P. Sequencing depth and coverage: key considerations in genomic analyses. Nat. Rev. Genet. 15, 121–132 (2014).
Wang, C. et al. The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nat. Biotechnol. 32, 926–932 (2014).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Zhang, Y., Parmigiani, G. & Johnson, W. E. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom. Bioinform. 2, lqaa078 (2020).
Büttner, M., Miao, Z., Wolf, F. A., Teichmann, S. A. & Theis, F. J. A test metric for assessing single-cell RNA-seq batch correction. Nat. Methods 16, 43–49 (2019).
Ledford, H. The death of microarrays? Nature 455, 847 (2008).
Fu, X. et al. Estimating accuracy of RNA-seq and microarrays with proteomics. BMC Genomics 10, 161 (2009).
Dillies, M.-A. et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief. Bioinform. 14, 671–683 (2013).
Bullard, J. H., Purdom, E., Hansen, K. D. & Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-seq experiments. BMC Bioinformatics 11, 94 (2010).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
van Hasselt, J. G. C. et al. Transcriptomic profiling of human cardiac cells predicts protein kinase inhibitor-associated cardiotoxicity. Nat. Commun. 11, 4809 (2020).
Yu, Y. et al. Quartet RNA reference materials improve the quality of transcriptomic data through ratio-based profiling. Nat. Biotechnol. 42, 1118–1132 (2024).
Stark, R., Grzelak, M. & Hadfield, J. RNA sequencing: the teenage years. Nat. Rev. Genet. 20, 631–656 (2019).
Makhzani, A., Shlens, J., Jaitly, N., Goodfellow, I. & Frey, B. Adversarial autoencoders. In International Conference on Learning Representations (ICLR) 2016 Workshop (2015).
Zhao, Y., Wong, L. & Goh, W. W. B. How to do quantile normalization correctly for gene expression data analyses. Sci. Rep. 10, 15534 (2020).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Hie, B., Bryson, B. & Berger, B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat. Biotechnol. 37, 685–691 (2019).
Polański, K. et al. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics 36, 964–965 (2019).
Li, H., Brouwer, C. R. & Luo, W. A universal deep neural network for in-depth cleaning of single-cell RNA-seq data. Nat. Commun. 13, 1901 (2022).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
McInnes, L., Healy, J. & Melville, J. UMAP: Uniform manifold approximation and projection for dimension reduction. Preprint at https://doi.org/10.48550/arXiv.1802.03426 (2018).
Butler, A., Hoffman, P., Smibert, P., Papalexi, E. & Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Hubert, L. & Arabie, P. Comparing partitions. J. Classif. 2, 193–218 (1985).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Segal, E., Friedman, N., Koller, D. & Regev, A. A module map showing conditional activity of expression modules in cancer. Nat. Genet. 36, 1090–1098 (2004).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Chu, T., Wang, Z., Pe’er, D. & Danko, C. G. Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat. Cancer 3, 505–517 (2022).
Yoshihara, K. et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 4, 2612 (2013).
Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782 (2019).
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M. & Alizadeh, A. A. in Cancer Systems Biology: Methods and Protocols (ed. von Stechow, L.) 243–259 (Springer, 2018).
Liu, Z. et al. A glycolysis-related two-gene risk model that can effectively predict the prognosis of patients with rectal cancer. Hum. Genomics 16, 5 (2022).
Mi, H., Muruganujan, A., Casagrande, J. T. & Thomas, P. D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 8, 1551–1566 (2013).
Su, A. I. et al. Large-scale analysis of the human and mouse transcriptomes. Proc. Natl Acad. Sci. USA 99, 4465–4470 (2002).
Fang, Z., Liu, X. & Peltz, G. GSEApy: a comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 39, btac757 (2023).
Zhang, M. A self-adaptive and versatile tool for eliminating multiple undesirable variations from large-scale transcriptomes. Source code. Zenodo https://zenodo.org/records/15338597 (2025).
Acknowledgements
This study was performed at Yale University and Westlake University, where the partial experimental design, data interpretation and manuscript revision were carried out, and at FosunLead, Inc., where the partial experimental design, model design and manuscript drafting were carried out. The work was supported by the Fellowship of Outstanding Doctoral Graduate, financed by Shanghai Jiao Tong University to M.Z. This work was supported by a grant from the National Natural Science foundation of China (U21A20201); a grant from the National Key R&D Program of China (2022YFE0204900); a grant for the construction of Key Laboratory of Growth Regulation and Translational Research of Zhejiang Province; grants from the Science Technology Department of Zhejiang Province (2020E10027, 2021ZY1019, 2022ZY1005); Research Program No. 202208011 of Westlake Laboratory of Life Sciences and Biomedicine; Research Program No. WU2023C020 of Research Center for Industries of the Future, Westlake University; and the Westlake Education Foundation.
Author information
Authors and Affiliations
Contributions
M.Z., T.X., B.C. and D.Y. conceived and designed the study. M.Z. developed DeepAdapter and produced figures and documentation. M.Z. and D.Y. contributed to the experimental design. M.Z. performed experiments and statistical analysis. D.Y. provided insights into the application of the model. M.Z., L.Y., Xinbo Wang, Y.Y., S.Z., S.Y., Xinyu Wang, B.C., Q.L., Z.Z., Y.S., Y.Z., W.W., H.Z., W.S. and D.Y. contributed to data interpretation. M.Z. and S.Y. contributed to data acquisition. M.Z. and D.Y. wrote the manuscript. M.Z. and D.Y. reviewed and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Biomedical Engineering thanks Xiaohui Fan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–14 and Table 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, M., Yan, L., Wang, X. et al. A self-adaptive and versatile tool for eliminating multiple undesirable variations from large-scale transcriptomes. Nat. Biomed. Eng (2025). https://doi.org/10.1038/s41551-025-01466-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41551-025-01466-w
This article is cited by
-
A self-adaptive and versatile tool for eliminating multiple undesirable variations from large-scale transcriptomes
Nature Biomedical Engineering (2025)
