
Abstract
Chaperone-mediated autophagy (CMA) declines in ageing and neurodegenerative diseases. Loss of CMA in neurons leads to neurodegeneration and behavioural changes in mice but the role of CMA in neuronal physiology is largely unknown. Here we show that CMA deficiency causes neuronal hyperactivity, increased seizure susceptibility and disrupted calcium homeostasis. Pre-synaptic neurotransmitter release and NMDA receptor-mediated transmission were enhanced in CMA-deficient females, whereas males exhibited elevated post-synaptic AMPA-receptor activity. Comparative quantitative proteomics revealed sexual dimorphism in the synaptic proteins degraded by CMA, with preferential remodelling of the pre-synaptic proteome in females and the post-synaptic proteome in males. We demonstrate that genetic or pharmacological CMA activation in old mice and an Alzheimer’s disease mouse model restores synaptic protein levels, reduces neuronal hyperexcitability and seizure susceptibility, and normalizes neurotransmission. Our findings unveil a role for CMA in regulating neuronal excitability and highlight this pathway as a potential target for mitigating age-related neuronal decline.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout








Similar content being viewed by others

Data availability
There are no restrictions on data availability in this Article. All information is included in the Article. The proteomic data were deposited at ProteomeXchange via the PRIDE partner repository with the dataset identifiers PXD057545 (lysosomal degradation in CMA-deficient mice) and PXD057677 (sarkosyl-insoluble fraction following CMA upregulation). Animal models and cells lines generated in this study will be provided on request under both-parties signed institutional materials transfer agreements. Source data are provided with this paper.
References
Nixon, R. A. The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997 (2013).
Bourdenx, M. et al. Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 184, 2696–2714 (2021).
Menzies, F. M. et al. Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93, 1015–1034 (2017).
Hara, T. et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 (2006).
Komatsu, M. et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884 (2006).
Fleming, A. et al. The different autophagy degradation pathways and neurodegeneration. Neuron 110, 935–966 (2022).
Yamamoto, H., Zhang, S. & Mizushima, N. Autophagy genes in biology and disease. Nat. Rev. Genet. 24, 382–400 (2023).
Kaushik, S. & Cuervo, A. M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 19, 365–381 (2018).
Khawaja, R.R. et al. Sex- and cell type-specific loss of chaperone-mediated autophagy across tissues with aging. Nat. Aging 5, 691–708 (2025).
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T. & Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295 (2004).
Orenstein, S. J. et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 16, 394–406 (2013).
Dong, S. et al. Chaperone-mediated autophagy sustains haematopoietic stem-cell function. Nature 591, 117–123 (2021).
Kaushik, S. & Cuervo, A. M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 17, 759–770 (2015).
Schneider, J. L., Suh, Y. & Cuervo, A. M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 20, 417–432 (2014).
Yang, Q. et al. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 323, 124–127 (2009).
Park, C., Suh, Y. & Cuervo, A. M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat. Commun. 6, 6823 (2015).
Kaushik, S. et al. Chaperone-mediated autophagy regulates adipocyte differentiation. Sci. Adv. 8, eabq2733 (2022).
Juste, Y. R. et al. Reciprocal regulation of chaperone-mediated autophagy and the circadian clock. Nat. Cell Biol. 23, 1255–1270 (2021).
Valdor, R. et al. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat. Immunol. 15, 1046–1054 (2014).
Reynolds, C. A. et al. Restoration of LAMP2A expression in old mice leads to changes in the T cell compartment that support improved immune function. Proc. Natl Acad. Sci. USA 121, e2322929121 (2024).
Jahn, R. & Fasshauer, D. Molecular machines governing exocytosis of synaptic vesicles. Nature 490, 201–207 (2012).
Sudhof, T. C. Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell 171, 745–769 (2017).
Cesca, F., Baldelli, P., Valtorta, F. & Benfenati, F. The synapsins: key actors of synapse function and plasticity. Prog. Neurobiol. 91, 313–348 (2010).
Luscher, C., Nicoll, R. A., Malenka, R. C. & Muller, D. Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat. Neurosci. 3, 545–550 (2000).
Sudhof, T. C. The cell biology of synapse formation. J. Cell Biol. 220, e202103052 (2021).
Nikoletopoulou, V. & Tavernarakis, N. Regulation and roles of autophagy at synapses. Trends Cell Biol. 28, 646–661 (2018).
Hill, S. E. & Colon-Ramos, D. A. The journey of the synaptic autophagosome: a cell biological perspective. Neuron 105, 961–973 (2020).
Griffey, C. J. & Yamamoto, A. Macroautophagy in CNS health and disease. Nat. Rev. Neurosci. 23, 411–427 (2022).
Hoyer, M. J. et al. Combinatorial selective ER-phagy remodels the ER during neurogenesis. Nat. Cell Biol. 26, 378–392 (2024).
Tang, G. et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143 (2014).
Lieberman, O. J. et al. Cell-type-specific regulation of neuronal intrinsic excitability by macroautophagy. eLife 9, e50843 (2020).
Overhoff, M. et al. Autophagy regulates neuronal excitability by controlling cAMP/protein kinase A signaling at the synapse. EMBO J. 41, e110963 (2022).
Kuijpers, M. et al. Neuronal autophagy regulates presynaptic neurotransmission by controlling the axonal endoplasmic reticulum. Neuron 109, 299–313 (2021).
Hernandez, D. et al. Regulation of presynaptic neurotransmission by macroautophagy. Neuron 74, 277–284 (2012).
Nikoletopoulou, V., Sidiropoulou, K., Kallergi, E., Dalezios, Y. & Tavernarakis, N. Modulation of autophagy by BDNF underlies synaptic plasticity. Cell Metab. 26, 230–242 (2017).
Kallergi, E. et al. Dendritic autophagy degrades postsynaptic proteins and is required for long-term synaptic depression in mice. Nat. Commun. 13, 680 (2022).
Compans, B. et al. NMDAR-dependent long-term depression is associated with increased short term plasticity through autophagy mediated loss of PSD-95. Nat. Commun. 12, 2849 (2021).
Chen, J.-H. et al. Reduced lysosomal density in neuronal dendrites mediates deficits in synaptic plasticity in Huntington’s disease. Cell Rep. 42, 113573 (2023).
Caballero, B. et al. Acetylated tau inhibits chaperone-mediated autophagy and promotes tau pathology propagation in mice. Nat. Commun. 12, 2238 (2021).
McCabe, M. et al. Small molecule disruption of RARα/NCoR1 interaction inhibits chaperone-mediated autophagy in cancer. EMBO Mol. Med. 17, 1716–1755 (2025).
Kallergi, E. et al. Profiling of purified autophagic vesicle degradome in the maturing and aging brain. Neuron 111, 2329–2347 (2023).
Costenla, A. R., de Mendonca, A. & Ribeiro, J. A. Adenosine modulates synaptic plasticity in hippocampal slices from aged rats. Brain Res. 851, 228–234 (1999).
Lin, J. W. et al. Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat. Neurosci. 3, 1282–1290 (2000).
Azarnia Tehran, D. et al. Selective endocytosis of Ca2+-permeable AMPARs by the Alzheimer’s disease risk factor CALM bidirectionally controls synaptic plasticity. Sci. Adv. 8, eabl5032 (2022).
Bissen, D., Foss, F. & Acker-Palmer, A. AMPA receptors and their minions: auxiliary proteins in AMPA receptor trafficking. Cell Mol. Life Sci. 76, 2133–2169 (2019).
Diogenes, M. J. et al. Enhancement of LTP in aged rats is dependent on endogenous BDNF. Neuropsychopharmacology 36, 1823–1836 (2011).
Shankar, S., Teyler, T. J. & Robbins, N. Aging differentially alters forms of long-term potentiation in rat hippocampal area CA1. J. Neurophysiol. 79, 334–341 (1998).
Gomez-Sintes, R. et al. Targeting retinoic acid receptor alpha-corepressor interaction activates chaperone-mediated autophagy and protects against retinal degeneration. Nat. Commun. 13, 4220 (2022).
Anastacio, H. T. D., Matosin, N. & Ooi, L. Neuronal hyperexcitability in Alzheimer’s disease: what are the drivers behind this aberrant phenotype?. Transl. Psychiatry 12, 257 (2022).
Dong, S. et al. Monitoring spatiotemporal changes in chaperone-mediated autophagy in vivo. Nat. Commun. 11, 645 (2020).
Grueninger, F. et al. Phosphorylation of Tau at S422 is enhanced by Aβ in TauPS2APP triple transgenic mice. Neurobiol. Dis. 37, 294–306 (2010).
Zhang, Y. et al. LAMP2A-mediated neuronal hyperexcitability by enhancing NKAβ1 degradation underlies depression-induced allodynia. Cell Rep. 44, 115489 (2025).
Chen, W. et al. Pharmacological modulation of autophagy for epilepsy therapy: opportunities and obstacles. Drug Discov. Today 28, 103600 (2023).
Fassio, A. et al. Emerging role of the autophagy/lysosomal degradative pathway in neurodevelopmental disorders with epilepsy. Front. Cell Neurosci. 14, 39 (2020).
Perucca, E., Perucca, P., White, H. S. & Wirrell, E. C. Drug resistance in epilepsy. Lancet Neurol. 22, 723–734 (2023).
Brodie, M. J., Barry, S. J., Bamagous, G. A., Norrie, J. D. & Kwan, P. Patterns of treatment response in newly diagnosed epilepsy. Neurology 78, 1548–1554 (2012).
Hui, K. K. et al. GABARAPs dysfunction by autophagy deficiency in adolescent brain impairs GABAA receptor trafficking and social behavior. Sci. Adv. 5, eaau8237 (2019).
Yang, L. et al. Stress dynamically modulates neuronal autophagy to gate depression onset. Nature 641, 427–437 (2025).
Celone, K. A. et al. Alterations in memory networks in mild cognitive impairment and Alzheimer’s disease: an independent component analysis. J. Neurosci. 26, 10222–10231 (2006).
Dickerson, B. C. et al. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology 65, 404–411 (2005).
Bassett, S. S. et al. Familial risk for Alzheimer’s disease alters fMRI activation patterns. Brain 129, 1229–1239 (2006).
Diering, G. H., Gustina, A. S. & Huganir, R. L. PKA–GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron 84, 790–805 (2014).
Mirza, F. J. & Zahid, S. The role of synapsins in neurological disorders. Neurosci. Bull. 34, 349–358 (2018).
Rosahl, T. W. et al. Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375, 488–493 (1995).
Fassio, A. et al. SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum. Mol. Genet. 20, 2297–2307 (2011).
Schmid, E. M. & McMahon, H. T. Integrating molecular and network biology to decode endocytosis. Nature 448, 883–888 (2007).
Gu, M. et al. AP2 hemicomplexes contribute independently to synaptic vesicle endocytosis. eLife 2, e00190 (2013).
Wang, X., Zhang, C., Szabo, G. & Sun, Q. Q. Distribution of CaMKIIα expression in the brain in vivo, studied by CaMKIIα–GFP mice. Brain Res. 1518, 9–25 (2013).
Feng, R. et al. Deficient neurogenesis in forebrain-specific presenilin-1 knockout mice is associated with reduced clearance of hippocampal memory traces. Neuron 32, 911–926 (2001).
Madrigal-Matute, J., Cuervo, A. M. & Sluimer, J. C. Chaperone-mediated autophagy protects against atherosclerosis. Autophagy 18, 2505–2507 (2022).
Van Erum, J., Van Dam, D. & De Deyn, P. P. PTZ-induced seizures in mice require a revised Racine scale. Epilepsy Behav. 95, 51–55 (2019).
Gambardella, J. et al. Ketone bodies rescue mitochondrial dysfunction via epigenetic remodeling. JACC Basic Transl. Sci. 8, 1123–1137 (2023).
Aniento, F., Roche, E., Cuervo, A. M. & Knecht, E. Uptake and degradation of glyceraldehyde-3-phosphate dehydrogenase by rat liver lysosomes. J. Biol. Chem. 268, 10463–10470 (1993).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Aguilan, J. T., Kulej, K. & Sidoli, S. Guide for protein fold change and P-value calculation for non-experts in proteomics. Mol. Omics 16, 573–582 (2020).
Szklarczyk, D. et al. The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–d646 (2023).
Milacic, M. et al. The Reactome Pathway Knowledgebase 2024. Nucleic Acids Res. 52, D672–D678 (2023).
Krämer, A., Green, J., Pollard, J. Jr & Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530 (2014).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Gilles, J. F., Dos Santos, M., Boudier, T., Bolte, S. & Heck, N. DiAna, an ImageJ tool for object-based 3D co-localization and distance analysis. Methods 115, 55–64 (2017).
Khawaja, R. R. et al. Sex-specific and cell-type-specific changes in chaperone-mediated autophagy across tissues during aging. Nat. Aging 5, 691–708 (2025).
Kirchner, P. et al. Proteome-wide analysis of chaperone-mediated autophagy targeting motifs. PLoS Biol. 17, e3000301 (2019).
Acknowledgements
We thank all of the members of our laboratory for their guidance, assistance and feedback. Special thanks to D. Sulzer and J. D. Surmeier for their feedback on the studies pertinent to calcium dynamics and electrophysiology recordings. We thank A. P. Aghasadeghi for his assistance in automating batch analyses and quantifications. We thank the Einstein Analytical Imaging and Neuroscience core facilities for access to the confocal microscope and cryostat. This work was supported by grants from the National Institutes of Health (grant numbers AG021904 and AG054108 to A.M.C.; AG031782 to A.M.C., L.S. and P.E.C.; MH125772 to P.E.C.; and HL146691 and DK123259 to G.S.) and the generous support of the JPB Foundation (A.M.C. and L.G.), Rainwaters Foundation (A.M.C. and L.G.), Hevolution Foundation (A.M.C., L.G. and S.S.), Backus Foundation (A.M.C.), Robert and Renee Belfer (A.M.C.), the Grace Science Foundation (S.K.), the American Heart Association (grant number 24IPA1268813) as well as the Monique Weill–Caulier and Irma T. Hirschl Trusts (G.S.). R.R.K. was supported by an IRACDA-BETTR grant and T32 Kirschstein-NRSA.
Author information
Authors and Affiliations
Contributions
R.R.K. designed, coordinated and performed all behavioural, biochemical and image-based experiments; analysed and graphed the data; prepared the first draft of the paper and edited it to include feedback. K.L. assisted with the animal procedures, primary neuron culture, RT–qPCR and western blots. E.G., A. Salek and H.R.M. performed, with assistance from M.B., the electrophysiological recordings and analysis. J.G. assisted with the calcium recordings. A. Scrivo and J.M.-P. performed the MEA experiments. Z.N.K., S.G., R.C. and L.S. performed the proteomic analyses. B.C. assisted with the animal behaviour tests. I.T. assisted with the initial generation and maintenance of the CKhL2AOE mouse line. S.S. supervised the proteomic studies. G.S. supervised the calcium recordings. S.K. assisted with subcellular fractionation and lysosomal isolation, measurement of proteasome activity and provided feedback on the paper. L.G. supervised the MEA experiments, contributed expertise and provided feedback. P.E.C. designed and supervised the electrophysiology studies, contributed expertise and provided feedback. A.M.C. conceived and directed the study, secured funding, assisted with data analysis and interpretation. All of the authors critically read the final draft of the paper.
Corresponding authors
Ethics declarations
Competing interests
A.M.C. is a co-founder and scientific advisor for the autophagy programme at Life Biosciences. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Cell Biology thanks Ling-Qiang Zhu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Neuronal hyperactivity and disruption of calcium homeostasis in CMA-deficient neurons.
a, Extracellular recordings of burst percentage (left) and network burst percentage (right) of L2AKO and control (Ctrl) primary neurons (obtained from female and male pooled brains) in MEA plates at DIV10. Open and closed circles indicate wells from the two independent experiments. b–g, Quantification of number of spikes, number of bursts, average burst frequency (b), average number of spikes per burst (c), burst percentage (d), network burst frequency, number of spikes per network burst, network burst percentage (e), mean firing rate (f) and synchrony index (g) of L2AKO and Ctrl primary neurons in MEA plates at DIV15. Open and closed circles indicate wells from the two independent experiments. h, Ratiometric measurement of cytosolic Ca2+ dyes, Fluo-4-AM (F1) and Fura-Red (F0), to quantify intracellular Ca2+-release following addition of ionomycin (1.5 μM) at indicated times marked by the vertical dotted lines. Imaging performed in Ca2+-free solution in DIV12 L2AKO and Ctrl primary neurons (female and male pooled). F1/ F0 ratiometric quantification (left) and area under the curve (AUC) (right) are shown Boxes: median and 25th and 75th percentiles; whisker ends: 25th and 75th percentiles ±1.5 times the IQR. i,j, Cytosolic Ca2+ quantified in female (i) and male (j) mouse primary neurons using Fluo-4-AM following KCl-induced depolarization F1quantification (left) and AUC (right) are shown. Boxes: median and 25th and 75th percentiles; whisker ends: 25th and 75th percentiles ±1.5 times the IQR. k, Baseline Ca2+ quantified in female and male primary neurons using Fluo-4-AM. From left to right in each graph: n = [12] (a–g) wells from 2 independent experiments per group; n = [3] (h), [6] (i), [5 Ctrl, 7 L2AKO] (j) independent experiments per group; n = [10, 8] (k left), [8, 11] (k right) wells from 3-4 independent experiments per group. Two-tailed unpaired Student’s t-test (a–g,h right, i right, j right, k), or two-way ANOVA test (h left, i left, j left) are used. Data are presented as mean values ±SEM.
Extended Data Fig. 2 Loss of CMA does not affect synaptic number, dendritic spines or passive neuronal properties.
a,b, Quantification of different subtypes of biocytin-labelled dendritic spines per μm (a) and as a percentage of total spines (b) in CA1 pyramidal neuronal processes in hippocampal slices in 10 months old female and male control (Ctrl) and excitatory neuron-specific LAMP2A knockout (CKL2AKO) mice. c–e, Representative images (c) and quantification of MAP2+ neuronal processes (d) and Bassoon+Homer1+ neuronal synapses (e) in DIV12 primary neurons (obtained from female and male pooled brains). Scale bars: 50 μm (left) and 10 μm (right). f–h, Representative images (f) and quantification of LAMP1+ endolysosomes in processes (g) and in the synapses (LAMP1+Homer1+; h). Scale bars: 50 μm (left) and 10 μm (right). i–k, Input resistance (i) resting membrane potential (j) and input and output function plotted as number of spikes in response to injected current (k) obtained from whole-cell recordings of CA1 neurons from hippocampal slices of Ctrl and CKL2AKO male and female mice at 10 months of age. l, Extracellular field recordings in CA1 neurons of theta-burst stimulation (TBS)-induced LTP in hippocampal slices from 10 months old male mice. Right: quantification of average fEPSP amplitude between 61–80 mins of recordings. From left to right in each graph: n = [6 each] (a left, b left), [8 Ctrl, 6 CKL2AKO] (a right, b right) neurons from 3–4 mice per group; n = [3 each] (d,e), [4, 5] (g,h) independent experiments per group; n = [11 Ctrl females, 13 CKL2AKO females, 14 Ctrl males, 11 CKL2AKO males] (i–k) neurons from 3-4 mice per group; n = [8, 9] (l) slices from 4-5 mice per group. No statistical significance detected for any of the indicated measurements using two-way ANOVA (a,b) and two-tailed unpaired Student’s t-test (d,e,g–j). Data are presented as mean values ±SEM.
Extended Data Fig. 3 Consequences of CMA blockage on the synaptic proteome.
a, Comparative proteomics of whole-brain ex vivo slices from 9 months old male and female control (Ctrl) and L2AKO mice incubated with or without lysosomal protease inhibitors (20 mM ammonium chloride and 400 μM leupeptin, NL) for 20 h. Timeline for ex vivo slice culture and NL incubation of the brain slices is shown. b,c, Representative immunoblot (b) and quantification (c) of lysosomal degradation of LC3-II (changes in protein levels upon NL addition relative to (left) or minus untreated (right)). n = [6] mice per group (c). Ponceau staining is shown as loading control. The dotted line in c indicates no changes relative to untreated samples (NL/untreated = 1). d, Proteins detected by mass spectrometry as increased (>1.5-fold, orange) or reduced (<0.8- fold, blue) in L2AKO females or male brains relative to controls. e,f, GO terms (e) and STRING plots (f) of the pathways enriched in proteins with increased steady-state levels in L2AKO brains compared to Ctrl female and male brains. All GO terms are statistically enriched with P < 0.05. Data are presented as mean values ±SEM. False discovery rate (FDR) test was used to calculate values in e.
Extended Data Fig. 4 CMA contributes to lysosomal degradation of proteins important for synaptic neurotransmission.
Comparative proteomics of whole-brain ex vivo slices from 9 months old male and female control (Ctrl) and L2AKO mice incubated with or without lysosomal protease inhibitors (20 mM ammonium chloride and 400 μM leupeptin, NL) for 20 h. a, STRING plots of pathways enriched in proteins no longer degraded in lysosomes in the L2AKO female or male mice brain compared to controls. b, IPA analysis pathways with significant enrichment in proteins undergoing LAMP2A-dependent lysosomal degradation (CMA substrates) in both females and males. c,d, IPA analysis for cellular pathways with enrichment of proteins displaying CMA degradation in females (c) or males (d). e, Heat maps showing fold change of pre-synaptic protein levels in aggregates (pellet) compared to soluble cytosol in CKL2AKO male mice brain relative to Ctrl. n = 3 mice per group. f, Synapsin-I (Syn1), Synapsin-II (Syn2) and Synaptotagmin-1 (Syt1) mRNA levels by qRT-PCR of female and male Ctrl and L2AKO mice hippocampus. g, Representative immunoblots (left) for p62 in whole-brain ex vivo slices from 9 months old female and male Ctrl and L2AKO mice incubated with lysosomal protease inhibitors (20 mM ammonium chloride and 400 μM leupeptin, NL) for 20 h. Lysosomal degradation, calculated as the increase in protein levels upon NL addition, is shown on right. Ponceau staining is shown as loading control for the immunoblots. h, Chymotrypsin-like activity of the 20S and 26S proteasome in female and male Ctrl and L2AKO mice whole-brain expressed as area under the curve. n = [5] (f); [6] (g), [4] (h) mice per group. No statistical significance detected for any of the indicated measurements using wo-tailed unpaired Student’s t-test. All GO terms are statistically enriched with P < 0.05. Data are presented as mean values ±SEM. P values in b–d were calculated using the hypergeometric test.
Extended Data Fig. 5 Impact of acute pharmacological inhibition of CMA on the synaptic proteome.
a, Timeline for treatment of whole-brain slices with the CMA inhibitor CIM7 (5 µM, 24 h) or vehicle (Veh.) and incubated with or without lysosomal protease inhibitors (20 mM ammonium chloride and 400 μM leupeptin, NL) for 20 h. b, Transcriptional CMA score (left) and heat map (right) of normalized expression (z-scoring) of CMA network genes (organized by the indicated functional groups) measured by qRT-PCR in female (triangles) and male (open circles) mice brains. c,d, Comparative proteomics analysis of proteins elevated by >1.5 folds in L2AKO and CIM7 treated brain slices relative to their respective controls (Ctrl) in females (c) and males (d). e, Representative immunoblot (top) and quantification (bottom) of lysosomal degradation of Synapsin-I in ex vivo whole-brain slices from WT mice upon NL incubation with CIM7 or vehicle as in a. The dotted lines in e indicate no changes relative to untreated samples (NL/untreated = 1). n = [4] (b),[6] (e) mice per group. Two-tailed paired Student’s t-test is used. Data are presented as mean values ±SEM.
Extended Data Fig. 6 Comparison of lysosomal degradation of the synaptic proteome by CMA and macroautophagy.
a, Comparison of synaptic and synapses-related proteins degraded by CMA in female and male mice whole brains with those reported degraded by macroautophagy in males in a previously published study36. b,c, IPA pathway analysis enrichment (GO terms; b) and graphical representation generated by Reactome of number of proteins (c) undergoing lysosomal degradation by both CMA and macroautophagy or the ones unique to each of two pathways. d,e, Heat maps showing lysosomal degradation (NL/None) for membrane proteins (d) and major subunits of the indicated membrane receptors and channels (e) in L2AKO and control (Ctr) brains. Grey: no lysosomal degradation. mGluRs: metabotropic glutamate receptors, VGKCs, VGSCs, VGCCs: voltage gated potassium, sodium or calcium channels. P values in b were calculated using the hypergeometric test.
Extended Data Fig. 7 Regulation of AMPARs and GABAAR synaptic levels by CMA.
a–f, Representative images of MAP2+GluA1+ neuronal processes (a) and GluA1+ neuronal soma (c) in L2AKO and control (Ctrl) primary neurons (obtained from female and male pooled brains) following incubation with CTZ (25 μM, 1 h prior to collection) or AMPA (100 μM, 30 min prior to collection) and Leupeptin (200 μM, 24 h prior to collection). Inset: higher magnification of boxed areas. Scale bars: 50 μm (lower magnification panels) or 20 μm (insets). Quantification of GluA1 lysosomal degradation in processes (b) or soma (d,e) upon addition or not of CTZ and changes in GluA1 levels in soma following CTZ treatment (f). n = 28–48 fields from 4–5 independent experiments per group. g–i, Representative images of MAP2+Bassoon1+GABAAR β1+ (g) and MAP2+vGluT1+GABAAR β2,3+ (h) neuronal processes and quantification (i,j) of total levels (left and middle panels) and lysosomal degradation (right panels) in L2AKO and Ctrl primary neurons (from female and male pooled brains) following incubation with Leupeptin. Insets: higher magnification. Scale bars: 50 μm (lower magnification panels) or 5 μm (insets). The dotted lines in b, e, f, i(right) and j(right) indicate no changes relative to untreated samples (NL or CTZ/untreated = 1). From left to right in each graph: n = [30, 40] (b left), [39, 40] (b right), [31, 32, 31, 37, 38, 37] (d), [30, 34] (e), [32, 38] (f), [16 each] (i), [16, 20] (j left, j middle), [15, 14] (j right) fields from 4–8 independent experiments per group. Two-way ANOVA with Tukey’s multiple comparison test (d) or two-tailed unpaired Student’s t-test (b,e,f,i,j) are used. Data are presented as mean values ±SEM.
Extended Data Fig. 8 Sexual dimorphism in regulation of AMPARs and NMDARs synaptic levels by CMA.
a–d, Representative images of GluA1+ processes (a) and soma (c) in female and male L2AKO and control (Ctrl) primary neurons. Scale bars: 50 μm (lower magnification panels) or 5 μm (insets). Quantification of soma GluA1 levels (b), and lysosomal degradation in processes (d left) or in soma (d right). The dotted lines in d indicate no changes relative to untreated samples (NL/untreated = 1). e,f, Quantification of cytosolic Ca2+ with Fluo-4AM (F1) in L2AKO and Ctrl female (e) and male (f) primary neurons stimulated with AMPA (100 μM) after addition of Cyclothiazide (CTZ, 150 μM) at times indicated by dotted lines. Average area under the curve values are shown on the right. Boxes: median and 25th and 75th percentiles; whisker ends: 25th and 75th percentiles ±1.5 times the IQR. g, GluA1 (Gria1) mRNA levels by qRT-PCR of female and male Ctrl and L2AKO mice hippocampus. h, Immunoblot (top) and quantification (bottom) of surface biotinylation of GluA1 in whole-brain ex vivo slices from Ctrl and L2AKO mice. i–k, Representative immunoblot (i) and quantification of total levels (j) and lysosomal degradation (k) of GluN1 (also known as NR1) in ex vivo whole-brain slices from 7–9 months old female and male Ctrl and L2AKO upon NL incubation in presence or not of inhibitor of endocytosis (800 μM Dynasore, Dyn) for 20 h. Ponceau staining is shown as loading control for the immunoblots. l, Representative immunoblot (left) and quantification (right) of GluN1 levels in hippocampus of CKL2AKO and Ctrl female and male mice. m, GluN1 (Grin1) mRNA levels by qRT-PCR in female and male Ctrl and L2AKO mice hippocampus. n–p, Representative immunoblot (n), total levels (o) and lysosomal degradation (p) of GluA2 in whole-brain slices from 7–9 months old female and male L2AKO and Ctrl mice following incubation with lysosomal protease inhibitors (NL) and/or endocytosis inhibitor (Dyn). q, GluA2 (Gria2) mRNA levels by qRT-PCR in female and male Ctrl and L2AKO mice hippocampus. From left to right in each graph: n = [20, 24] (b left), [20, 32] (b right), [19, 20] (d left), [20, 20] (d right) fields from 5–8 independent experiments per group; n = [6, 5] (e,f) independent experiments; n = [5 each] (g), [6 each] (h), [11 each] (j left), [12 each] (j right), [11, 11, 4, 4] (k left), [12, 12, 4, 4] (k right), [8, 4] (l left), [6, 5] (l right), [5 each] (m), [7 each] (o left), [10, 9] (o right), [7, 7, 4, 4] (p), [5 each] (q) mice per group. Two-tailed unpaired Student’s t-test (b,d,e right,f right, g,h,j,l,m,o,q), or two-way ANOVA (e left,f left,k,p) with Tukey’s multiple comparison test are used. Data are presented as mean values ±SEM.
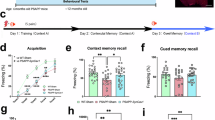
Extended Data Fig. 9 Effect of CMA activation on synaptic density, long-term memory and seizure susceptibility.
a–h, Representative images (a,e) and quantification of PSD95+vGluT1+ puncta (b–d) and Homer1+SV2+ puncta (f–h) in stratum radiatum in hippocampus of young (Y, 4–6 months old) h2AOE and control (Ctrl) female and male mice. Scale bars: 50 μm (lower magnification panels) or 10 μm (insets). i–k, Quantification of seizure severity (i), frequency (j) and duration (k) following PTZ (40mg kg−1) injection in 4–6 months old female and male hL2AOE and Ctrl mice. l,m, Long-term memory quantified in the novel-arm Y-maze test as percentage of entries made (right panels) or percentage of time spent (left panels) in novel arm in young (6 months) or old (23 months) female (l) and male (m) mice that received CA77.1 or vehicle 4 months prior to the memory assessment. n, Representative full field images of immunostaining for Dendra, LAMP1 (L1), and human Tau (hTau) in hippocampal CA1 pyramidal neurons from 4 months old male KFERQDendra; ×3TG (expressing APPSwe, PS2N141I, and hTauP301L), and quantification of KFERQDendra+LAMP1+ and LAMP1+ puncta separately in female and mice as fold of sex-matched controls in the same images. Scale bar: 50 μm. o, Quantification of seizure duration (left) or severity (right) following PTZ (40 mg kg−1) injection in 6 months old females (open triangles) and males (closed circles) ×3TG mice, 2 months after daily administration of CA77.1 or vehicle (Veh.). The doted lines in n indicate no changes relative to control (x3TG/Ctrl = 1). From left to right in each group: n = [4 Ctrl females, 6 hL2AOE females, 4 Ctrl males, 5 hL2AOE males] (b–d,f–h), [7, 13] (i–k left), [16, 14] (i–k right), [9, 8, 7] (l), [5, 8, 9] (m), [4, 5] (n), [11, 5, 9] (o left), [16, 11, 12] (o right) mice. Two-tailed unpaired Student’s t-test (b–d,f–k,n), two-way ANOVA with Tukey’s multiple comparison test (l,m) or one-way ANOVA with Tukey’s multiple comparison test (o) are used. Data are presented as mean values ±SEM.
Extended Data Fig. 10 Effect of pharmacological activation of CMA in the hippocampal proteome of an AD mouse model.
Comparative quantitative proteomic analysis of the hippocampus regions of 6 months old Ctrl and ×3TG mice receiving vehicle or CA77.1 (oral, 5 days / week, 30 mg kg−1 daily dose for 2 months). a,b, Heat map of z-score (a) and log2FC plot (b) of changes in the total proteome of ×3TG mice brains completely or partially corrected by CA77.1 treatment. Only proteins with significant changes in any of the three groups relative to the others were included in this analysis. c,d, STRING plot (c) and IPA (d) of the pathways enriched in proteins increased (>1.5 folds) in ×3TG aggregates compared to Ctrl, but no longer detected in the aggregate fraction of ×3TG mice hippocampus upon CA77.1 treatment. e, IPA-predicted activation in glutamate-receptor signalling pathway based on the proteins increased by at least 1.5 folds in the total proteome from ×3TG vehicle mice. n = 6 Ctrl Veh., 5 ×3TG Veh., 5 ×3TG CA77.1 mice. P values in d were calculated using the hypergeometric test.
Supplementary information
Supplementary Tables 1–4
Supplementary Table 1. KFERQ-like motifs in proteins identified as CMA substrates in both male and females, only males or only females. Supplementary Table 2. Source of antibodies used in this work. Supplementary Table 3. Light microscopy reporting table. Supplementary Table 4. Primers used in this work.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 3
Unprocessed western blots.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 4
Unprocessed western blots.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 5
Unprocessed western blots.
Source Data Fig. 6
Statistical source data.
Source Data Fig. 7
Statistical source data.
Source Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 1
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 3
Unprocessed western blots.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 4
Unprocessed western blots.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 5
Unprocessed western blots.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 8
Unprocessed western blots.
Source Data Extended Data Fig. 9
Statistical source data.
Source Data Extended Data Fig. 10
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Khawaja, R.R., Griego, E., Lindenau, K. et al. Chaperone-mediated autophagy regulates neuronal activity by sex-specific remodelling of the synaptic proteome. Nat Cell Biol 27, 1688–1707 (2025). https://doi.org/10.1038/s41556-025-01771-1
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41556-025-01771-1
This article is cited by
-
Chaperone mediated autophagy is deficient in spinal motoneurons of ALS patients with TDP-43 proteinopathy
Acta Neuropathologica Communications (2026)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}