
Abstract
Photochemistry involves elementary steps in which single electrons are transferred, but artificial photosynthesis requires multi-electron reactions. This discrepancy necessitates light-driven charge accumulation, which has so far proved very difficult to achieve without sacrificial redox reagents. Here we report a molecular donor–photosensitizer–acceptor compound in which light absorption leads to the reversible accumulation of two positive and two negative charges. The resulting photoproduct forms with an overall quantum yield of 37%, has a lifetime of more than 100 ns and stores 3.0 eV of energy. The use of a structurally well-defined molecular compound provides fundamental insights into how light-driven multi-electron transfer can generally be performed efficiently and sustainably, at irradiance levels orders of magnitude below those required in comparable systems. This represents a step towards more application-oriented research on solar fuels from fundamental studies of photoinduced (single) electron transfer.

Similar content being viewed by others

Main
The first elementary redox reaction step of natural photosynthesis is the light-induced transfer of an electron from a chlorophyll pigment to a pheophytin molecule and onwards to a quinone. This results in a so-called charge-separated state (CSS), which is composed of the oxidized chlorophyll and the reduced quinone1. Hundreds of previous studies have mimicked this photoinduced charge separation in artificial donor–photosensitizer–acceptor (D–PS–A) compounds and considered it an important step towards artificial photosynthesis2,3,4. However, separating a single oxidation equivalent from a single reduction equivalent is obviously still a long way from turning water and carbon dioxide into biomass or any useful solar fuel.
Fuel-forming reactions require multiple electrons, whereas photoinduced elementary reaction steps are usually single electron transfer events5. Research on artificial photosynthesis that aims to convert solar energy into energy-rich chemical fuels therefore requires overcoming several fundamental challenges. These include the efficient harvesting of light at low irradiances, the transfer and accumulation of multiple redox equivalents2,6,7,8,9 and selective catalytic systems for substrate reduction and, ideally, water oxidation10. For the reduction of CO2 in particular, multi-electron transfer appears to be essential5,6.
Against this background, fundamental research on photoinduced electron accumulation often studied molecular systems that use large amounts of sacrificial redox reagents. These undergo irreversible reactions after donating an electron to drive the reaction in only one direction11,12,13,14,15,16,17,18,19,20. To avoid the use of these high-energy sacrificial reagents, recent studies have relied on bimolecular reactions with reversible electron donors such as ascorbate21,22. This is a useful strategy, even though some of the natural photosynthetic elementary reaction steps are also irreversible. Nonetheless, fully integrated covalent systems that do not require any additives remain of central interest, for example for the separation of redox equivalents across a membrane23.
Charge-separated states in D–PS–A compounds (Fig. 1a) represent the simplest form of photochemically stored energy24, but these states are inherently unstable due to spontaneous electron transfer reactions occurring in the reverse direction25,26. Charge recombination typically occurs within a few nanoseconds but can be decelerated to the microsecond regime by elongating the distance between the electron donor and the acceptor27,28,29,30,31. This timescale is then in principle suitable for the slower onward reactions involved in artificial photosynthesis. However, the accumulation and storage of multiple electrons or holes in such fully integrated D–PS–A systems is substantially more difficult than the formation of single electron–hole pairs.

a, Energy-wasting photoinduced charge recombination is favoured over energy-storing charge accumulation. b, Simultaneous excitation of both photosensitizer units in D–PS–A–PS–D molecules circumvents the destructive processes from a. c, The sequential excitation pattern (achievable at low irradiances) in such D–PS–A–PS–D molecules cannot lead to substantial charge accumulation. d, A general strategy for charge accumulation via sequential photon absorption at much lower irradiances. Blue arrows denote excitation with light; black horizontal arrows are simple reaction arrows. The initial electron–hole pair is stored peripherally, away from the photosensitizer. Secondary excitation of PS allows faster reaction with nearby electron donors and acceptors, despite greater driving force for unwanted light-induced charge recombination compared to light-induced charge accumulation. Legend: in general, for simplicity and comparability, reductive quenching of the excited state is shown. Depending on the actual donor and acceptor, oxidative quenching may also occur. The general principles apply to both cases.
Without the use of external redox reagents, only a handful of previous studies have achieved either the accumulation of two reduction or two oxidation equivalents in purely molecular systems, but never both32,33,34,35. Furthermore, all of these D–PS–A–PS–D-type systems reported to date lack the ability for efficient electron accumulation through sequential excitation (see above), which is considered a key requirement for a functional system under low solar irradiance7,8. One study separated two electrons from two holes (electron vacancies), but used TiO2 nanoparticles for electron accumulation36. The separation of two electrons and two holes has not been achieved in fully molecular systems or been performed in a sequential manner.
A major challenge for the accumulation of electrons is overcoming the second absorbed photon to induce charge recombination (Fig. 1a)7,32,37,38,39,40. When the chromophore of a CSS is excited, the electron acceptor is in its reduced form (A•−), which is much more reducing than the originally intended electron donor. As a result, the excited state (*PS) in a CSS is quenched mainly by the reduced electron acceptor (A•−) and not by the intended electron donor. As a consequence, the electron–hole pair, previously stabilized by the spatial separation of D•+ and A•−, is brought back into mutual proximity and the electron on PS•− recombines very quickly with the hole on D•+. The absorption of a second photon is therefore usually not productive, but rather triggers a fast, energy-wasting charge recombination, finally resulting in A and D in their redox neutral forms and PS in its ground state (Fig. 1a).
All known fully molecular systems capable of accumulating redox equivalents without external redox reagents are of the D–PS–A–PS–D type, in which two electrons are eventually stored on a central acceptor flanked by two photosensitizers (Fig. 1b)32,33,34,35,41. These previous studies addressed the challenge of photoinduced charge recombination by the simultaneous excitation of both photosensitizers. Since the formation of a typical CSS in such systems usually occurs on the order of 10 ps (ref. 42), the second PS must absorb a photon within these 10 ps, which requires a very high irradiance. The solar irradiance is orders of magnitude too low for this purpose, and sunlight therefore allows only sequential excitation strategies7,8. However, under sequential excitation of such D–PS–A–PS–D-type molecules, the same energy-wasting charge recombination as discussed above is prevalent and other unproductive processes may occur (Fig. 1c).
To overcome these severe conceptual obstacles to achieving efficient light-driven charge accumulation at more moderate irradiances, a different molecular design is needed. Our aim was to allow for the sequential absorption of two photons, where the second excitation leads to charge separation despite the greater driving force for the unwanted photoinduced charge recombination. Since electron transfer becomes slower with increasing distance, we identified a faster electron transfer from a closer donor (kinetics) as a possible mechanism to outweigh the larger driving force (thermodynamics) of the reduced acceptor (Fig. 1d). Thus, if the redox equivalents are stored further away from the photosensitizer, the immediate environment of the photosensitizer can be restored to its original state, including an electron donor in close proximity. In addition, this far separation of charges in the CSS-2 will also increase the lifetime of this CSS-2 and thus provide more time for the absorption of the second photon. This concept would, in principle, also allow for the integration of catalysts at the periphery, as outlined earlier in a forward-looking paper by Odobel and co-workers7.
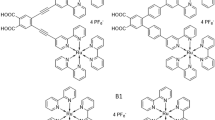
Against this background, we designed a molecular pentad comprised of two electron donors, one photosensitizer and two electron acceptors (Fig. 2b). We hypothesized that primary excitation would establish a long-lived CSS (CSS-2 in Fig. 2d), with a long distance (44 Å) between the oxidation (D2•+) and the reduction equivalent (A2•−). When exciting the photosensitizer in this constellation again, its nearest-neighbour redox partners D1 and A1 are as yet uncharged. This is expected to allow the accumulation of two oxidation equivalents on the donor branch in the form of D1•+ and D2•+, as well as the accumulation of two reduction equivalents on the acceptor branch in the form of A1•− and A2•− (CSS-3 in Fig. 2d). Oxidative and reductive equivalents are therefore ultimately accumulated on adjacent redox units. This is conceptually different from the mutually independent charges targeted in dendrimer-based or polymer-based approaches43,44. Furthermore, charge accumulation in our pentad involves sequential excitation of the same photosensitizer, and the two consecutive charge separation steps involve the same primary donor and acceptor units in an overall system that provides a clear redox gradient.

a,b, Molecular structures highlighting the individual electron donor (D1, D2) and acceptor (A1, A2) units in different colours. c,d, Energy-level diagrams of the key photoproducts and their corresponding energies in the triad (c) and the pentad (d). The final charge-separated state (CSS) in the triad corresponds to the first charge-separated state in the pentad (CSS-1), which spontaneously evolves onwards to CSS-2. The central hypothesis of this study was that a secondary excitation event then leads to CSS-3 in the pentad. The two ϕ values indicate quantum yields. The quantum yields and their determination via relative actinometry are discussed in the main text. PS, photosensitizer.
Results
Establishing the redox chain in the molecular pentad
Our covalently linked D2–D1–PS–A1–A2 pentad (Fig. 2b) consists of a triarylamine (TAA) as D2, a phenothiazine (PTZ) as D1, a [Ru(bpy)3]2+ complex as PS (bpy = 2,2′-bipyridine), an anthraquinone (AQ) as A1 and a naphthalene diimide (NDI) as A2. The synthesis procedures and complete characterizations of the pentad and the triad from Fig. 2a,b as well as the reference compounds from Fig. 3a are in the Supplementary Information.

a, Reference molecules of the individual redox-active units of the triad and the pentad. b, Cyclic voltammograms of the first reduction of the reference electron acceptors ref-AQ (orange) and ref-NDI (red) along with the first oxidation of the reference electron donors ref-TAA (blue) and ref-PTZ (green) (top) and cyclic voltammogram of the main ligand of the pentad (with all organic redox-active units present but no Ru(II) coordinated) showing the same redox events and additionally the reduction of NDI•− to NDI2− (bottom). The dotted vertical lines indicate the midpoint between the peak potentials in the oxidative and reductive sweeps, respectively. c, Difference UV–vis absorption spectra obtained after electrochemical oxidation of ref-PTZ to ref-PTZ•+ (green) and after reducing ref-AQ to ref-AQ•− (orange). d, Difference spectra obtained after electrochemical oxidation of ref-TAA to ref-TAA•+ (blue) and after reducing ref-NDI to ref-NDI•− (red). The dashed black trace is the linear combination of the two spectra using a scaling factor to take the different extinction coefficients of the ref-TAA•+ and ref-NDI•− radicals into account. The resulting sum spectrum is the expected UV–vis absorption signature of the CSS-2 in Fig. 2d (D2•+–D1–PS–A1–A2•−). e, UV–vis transient absorption spectra of the pentad in dry Ar-saturated CH3CN at 20 °C after excitation with 460 nm laser pulses (approximately 17 mJ) of approximately 10 ns duration, time-integrated for 200 ns after the indicated delay times (purple traces). The expected sum spectrum from d (black dashed trace) is included for direct comparison. The asterisk marks the signature of TAA•+ (see text for details).
Cyclic voltammetry of the individual donor and acceptor units (Fig. 3a) verifies that D2 is a stronger donor than D1 and A2 is a stronger acceptor than A1 (Fig. 3b). When integrated into the backbone of the pentad, the redox properties remain essentially unaltered (lower half of Fig. 3b and Supplementary Table 1). The ref-PTZ•+ (D1•+) and ref-TAA•+ (D2•+) radical cations are easily distinguishable from one another by ultraviolet–visible (UV–vis) absorption spectroscopy (green trace in Fig. 3c versus blue trace in Fig. 3d). Analogously, the ref-AQ•− (A1•−) and ref-NDI•− (A2•−) radical anions are spectrally very different from one another (orange trace in Fig. 3c versus red trace in Fig. 3d), as well as from the radical cation spectra.
Based on the redox potentials from Fig. 3b and the known properties of [Ru(bpy)3]2+ (ref. 45), the energy-level diagram in Fig. 2d can be established (see Supplementary Fig. 11 for details). Following excitation of [Ru(bpy)3]2+, we expect the formation of a primary CSS (CSS-1) comprised of PTZ•+ (D1•+) and AQ•− (A1•−). In the reference triad, this corresponds to the final observable photoproduct (CSS in Fig. 2c), but the pentad is designed such that there are spontaneous onward electron transfer reactions. TAA (D2) is able to reduce PTZ•+ (D1•+) (ΔGET0 = −0.2 eV) and AQ•− (A1•−) is able to reduce NDI (A2) (ΔGET0 = −0.3 eV) to form CSS-2 in Fig. 2d.
Achieving long-lived single electron–hole separation
Using laser pulses of ~10 ns duration, a 20 µM solution of the pentad in Ar-saturated CH3CN was excited at 460 nm into the [Ru(bpy)3]2+ unit. The transient UV–vis absorption spectrum recorded without time delay provides direct evidence for the formation of the anticipated CSS-2 photoproduct (Fig. 3e) and thus validates the electron transfer steps anticipated in Fig. 2d. The formation of NDI•− is observable by transient absorption bands that match the spectroelectrochemical signature of NDI•− from Fig. 3d (red trace). The peak at 390 nm in the spectra from Fig. 3e (marked by an asterisk) signals the formation of TAA•+ where this radical cation’s absorption is not masked by an overlapping negative signal caused by the disappearance of charge-neutral NDI. The main absorption band of TAA•+ at 960 nm (Fig. 3d) is outside the detectable spectral window of the nanosecond transient absorption instrument used here, but is observable when using a femtosecond transient absorption spectrometer (see below).
All transient absorption features of TAA•+ and NDI•− decay uniformly within approximately 1 ms (Fig. 3e). Intermolecular electron transfer processes become non-negligible at such long timescales, and therefore the transient absorption decays become multi-exponential (Supplementary Fig. 24). From concentration-dependent experiments (Supplementary Table 2), we extract a lifetime of 120 µs for CSS-2 in the pentad. Thus, CSS-2 forms within 10 ns and stores 1.3 eV (Fig. 2d) in the form of an electron–hole pair separated over 44 Å for 120 µs. There are only a few molecular systems with charge-separated states with such long lifetimes27,28,29,30.
Double excitation experiment for charge accumulation
The very long-lived CSS-2 forms a good basis for exploring accumulative electron transfer upon secondary excitation of the pentad. For this purpose, we designed a cw-pump–pump–probe experiment, in which a continuous-wave (cw) pump laser is used to generate a steady population of CSS-2, followed by a secondary excitation with a pulsed pump laser and a probe pulse of adjustable delay46. This was achieved by directing the beam of the cw laser into a commercial pump–probe spectrometer (Fig. 4a,b). The use of a cw laser instead of a pulsed laser for primary excitation is advantageous, because it allows a greater concentration of CSS-2 to form. This increases the sensitivity for the detection of the CSS-3 (Fig. 2d).

a, Schematic representation of the commercial transient absorption spectroscopy setup, into which an external continuous-wave (cw) laser has been integrated as an additional pump. The result is a cw-pump–pump–probe setup, where one pump (447 nm) is continuously irradiating the solution and the other pump (460 nm) is pulsed. b, Photograph of the actual setup from a with the directions of the individual beams shown by coloured lines. c, Arrangement of the three different beams at the cuvette, to highlight the fact that the cw pump beam is approximately seven times larger than the pulsed pump beam. d, Measurement and processing of transient absorption spectra in classical pump–probe experiments. The unpumped spectrum (i) is subtracted from the spectrum recorded after a pump pulse (ii) providing the transient absorption (difference) spectrum (iii). The grey background in d and e highlights that the two respective spectra ((ii) in d; (i) in e) are identical. The blue background in e highlights the cw-pump–pump–probe experiment as a whole. e, In the cw-pump–pump–probe experiment, the cw pump laser continuously forms some photoproducts (red and blue in i), which become part of the background signal. Therefore, the actual observable of the cw-pump–pump–probe experiment is not the sum of all photoproducts, but only of those photoproducts formed by the pulsed pump laser (orange and green in iii).
In conventional pump–probe experiments, the spectrum recorded prior to the pump pulse serves as a background (Fig. 4d(i)) and is subtracted from the spectrum recorded with a given time delay after the pump pulse (Fig. 4d(ii)). The resulting transient difference spectrum (Fig. 4d(iii)) shows the signatures of the photoproducts formed by the pump pulse. In the cw-pump–pump–probe experiment from Fig. 4a,b, the final observable (Fig. 4e(iii)) corresponds to the photoproducts formed by the pulsed pump laser, while the photoproducts formed by the cw-pump laser become part of the background (Fig. 4e(i)). In other words, the spectral signature of the CSS-2 is removed with the background and, consequently, when aiming at the detection of CSS-3, one can merely expect to see the signatures of D1•+ and A1•− whilst the signatures of the simultaneously present D2•+ and A2•− will be in the background.
The cw laser pumps a sample volume of approximately 4 mm3 with an average photon flux of approximately 1018 s−1 (Supplementary Section 13). The pulsed pump laser beam is roughly seven times smaller than the cw pump laser (overlap volume is approximately 0.6 mm3) and is aligned to the centre of the cw laser (Fig. 4c). For a pentad concentration of 300 µM, we estimate that the cw-pump laser provides one photon per pentad molecule roughly every 300 µs (Supplementary Section 13). Given a lifetime of CSS-2 in the same time regime (120 µs, see above), this means that one can anticipate nearly quantitative formation of CSS-2 in the pentad molecules present in the 0.6 mm3 overlap volume irradiated by the cw-pump beam. In other words, the cw-pump–pump–probe experiment in Fig. 4a,b offers the advantage that essentially all pentad molecules present in the beam of the cw laser can be promoted to CSS-2.
Achieving double charge separation
Before turning to the cw-pump–pump–probe experiments described in Fig. 4, we investigated the processes after single excitation in more detail with conventional pump–probe experiments. First, a transient absorption spectrum of the CSS in the reference triad from Fig. 2a was recorded (Fig. 5a, light blue). This CSS (Fig. 2c) is comprised of PTZ•+ (650 nm) and AQ•− (565 nm) as is evident when comparing to the spectroelectrochemical data obtained for ref-PTZ•+ and ref-AQ•− (Fig. 3c). These same spectroscopic features are expected for the initial intermediate photoproduct in the pentad (CSS-1 in Fig. 2d). Furthermore, these spectral signatures are expected to appear in CSS-3 of the pentad. The spectrum from the reference triad in Fig. 5a therefore serves as a reference for CSS-1 and CSS-3 in the pentad.

a, Transient absorption spectra of the CSS-2 in the pentad and the CSS in the triad. The CSS of the triad is composed of PTZ•+ and AQ•− and serves as a spectroscopic reference for the CSS-1 and CSS-3 in the pentad. The spectra were time-integrated over the first 200 ns after the excitation of a 20 µM solution of the pentad or a 16 µM solution of the triad in dry, Ar-saturated CH3CN. Laser pulses of approximately 10 ns duration at 460 nm were used. b, Transient absorption spectra of a 300 µM solution of the pentad in dry dimethylformamide after excitation with 460 nm pulses of approximately 250 fs duration. The CSS-1 can be observed from 5 to 100 ps, after which the spectroscopic signatures spontaneously evolve into those of CSS-2. c, Transient absorption spectra recorded as described in b while the solution is continuously irradiated with an external cw laser of 447 nm. The spectroscopic signature of this CSS-3 is similar to the one of CSS-1 but cannot evolve any further, because TAA•+ cannot be further oxidized by PTZ•+ and NDI•− cannot be further reduced by AQ•−. d, Schematic representation of the photoproducts on the µs timescale after single excitation of the triad (CSS) and the pentad (CSS-2) as observed in a. e, Schematic representation of the photoproducts observed on the sub-nanosecond timescale in the pump–probe (b, purple) and the cw-pump–pump–probe (c, green) experiments.
Since the CSS-1 in the pentad is only a short-lived intermediate on the way to the 120 µs-lived CSS-2, we used pump–probe experiments with picosecond time resolution for its detection. Following excitation of the pentad at 460 nm, the transient absorption spectrum measured with a delay of 5 ps (Fig. 5b) contains the spectroscopic signatures of PTZ•+ (D1•+) and AQ•− (A1•−), as is evident from the comparison to the spectrum of the reference triad in Fig. 5a (light blue trace). After a delay of 100 ps, a new spectral signature begins to evolve and after a delay of 5 ns, the resulting spectrum in Fig. 5b is identical to that obtained from the nanosecond transient absorption experiment (Fig. 5a, dark blue), in which TAA•+ (D2•+) and NDI•− (A2•−) are observable as confirmed by the data in Fig. 3d,e. Overall, the data in Fig. 5b document the sequential formation of CSS-1 and CSS-2 in the pentad. More importantly, the data reveal the spectral signature for the PTZ•+ (D1•+) and AQ•− (A1•−) radical ion pair that is expected as part of CSS-3.
With this important piece of information, we then turned to the cw-pump–pump–probe experiments introduced above (Fig. 4). In addition to the conventional pump–probe method, there is now a blue cw laser (447 nm, 460 mW) that excites the pentad to generate a constant population of CSS-2. At the employed output power, the cw laser provides approximately 103 times more photons than the Sun and essentially all pentad molecules present within the probed sample volume are excited to CSS-2. When the sample is then further excited with 460 nm pulses of 250 fs duration, the transient absorption spectrum observed after 5 ps (Fig. 5c) is essentially identical to that obtained with the conventional pump–probe experiment in which PTZ•+ and AQ•− are observed. When the delay time is increased to 5 ns in the cw-pump–pump–probe experiment, the transient absorption spectrum remains largely unchanged (Fig. 5c). This is in contrast to the results in Fig. 5b obtained with conventional pump–probe spectroscopy (without cw laser), in which PTZ•+ and AQ•− spontaneously evolve to TAA•+ and NDI•− with time. This sole observation of PTZ•+ and AQ•− in the cw-pump–pump–probe experiment is exactly what is expected when the target state CSS-3 (Fig. 2d) is reached under these conditions, as discussed above (Fig. 4e). Since the cw-pump laser forms a steady and essentially quantitative population of CSS-2 comprised of the TAA•+/NDI•− couple in the probed pentad molecules (that becomes part of the baseline signal of the cw-pump–pump–probe experiment, Fig. 4e), the pulsed pump laser can only produce the additional PTZ•+/AQ•− signals, regardless of time delay. When temporarily blocking the cw-pump laser beam, the spectral features of TAA•+ and NDI•− immediately appear (Supplementary Fig. 27), confirming that the pentad stays intact and no substantial photodecomposition has occurred.
The spectra in Fig. 5b,c were recorded from the same sample using identical excitation pulse powers, and they show essentially identical signal intensities for CSS-1 and CSS-3 (approximately 2 mΔOD at 600 nm). This strongly suggests that the initial formation of CSS-1 and the formation of CSS-3 from CSS-2 are both similarly efficient steps. The quantum yield for the formation of CSS-2 (ΦCSS-2) is 61% (Supplementary Section 9), and therefore the quantum yield for the formation of CSS-1 (ΦCSS-1) must be at least 61% as well. When now using the experimental information that the formation of CSS-3 from CSS-2 is as efficient as the formation of CSS-1, one obtains an overall quantum yield of 37% for the formation CSS-3 (ΦCSS-3). Since the efficiency for both charge separation events is similar, we conclude that the actual charge accumulation event is not hindered by the charges already stored on D2 and A2. This result is corroborated by cyclic voltammetry measurements (Supplementary Table 1) in which charges on D2/A2 influenced the redox properties of D1/A1 only by less than 0.05 V.
The femtosecond spectrometer used for the cw-pump–pump–probe experiment permits monitoring of transient signals up to 7 ns. On this short timescale, the CSS-3 does not begin to decay, and consequently it seems reasonable to infer that CSS-3 has a lifetime of at least 100 ns. Intramolecular charge recombination by spontaneous electron transfer from AQ•− to PTZ•+ occurs with a time constant of 1,100 ns in the reference triad (Supplementary Fig. 14). On this basis, a CSS-3 lifetime between 100 and 1,100 ns seems plausible for the pentad. CSS-3 stores roughly 3.0 eV (Fig. 2d), which is about twice as much as that normally achieved in D–PS–A triads.
Conclusions
Donor–photosensitizer–acceptor compounds have been explored for over four decades47 to understand the initial events occurring in natural photosynthesis for their use in artificial photosynthesis3,7,8. Our pentad in Fig. 2b is a completely molecular example in which two positive charges can be separated from two negative charges. Many previous studies aimed to accomplish this goal7,8, but in most cases either only positive or only negative charges could be accumulated32,33,34,35,36, and often this furthermore necessitated the use of sacrificial redox reagents11,12,13,14,15,16,17,18,19,20 or at least the addition of excess reversible redox reagents21,22. In other cases, charge accumulation failed completely despite considerable synthesis efforts44.
The design of our pentad allows rapid (<10 ns) initial separation of a first electron–hole pair over a distance of 44 Å, leading to a long-lived (120 µs) CSS (CSS-2). The two redox units in immediate spatial proximity to the light-absorbing unit have returned to their initial charge-neutral redox states at that point. When exciting the photosensitizer in this constellation, productive onward electron transfer to form CSS-3 is kinetically favoured over energy-wasting photoinduced charge recombination. Thus, light-induced charge accumulation is facilitated, because energy-storing (overall thermodynamically upward) reaction steps are kinetically favoured over energy-wasting (thermodynamically downward) photoinduced reaction steps. The design principle of our pentad leads to an unprecedented scenario in which the charge accumulating reaction step proceeds with the same high quantum yield as the initial charge-separating step. We anticipate that this design principle will be broadly applicable to artificial photosynthetic systems based on light-induced charge accumulation. Future efforts in this direction will need to focus on the use of a pigment with high-energy excited states in combination with suitable catalysts (as D2 and A2) to ultimately provide sufficient redox power for multi-electron catalysis such as water splitting or CO2 reduction48.
The sequential excitation of the light-absorbing unit and the stepwise charging of the redox units in our pentad differs conceptually from previously researched artificial systems for charge accumulation, which are based on the simultaneous excitation of multiple light-absorbing units32,33,34,35,41. This is a big step forward towards applications under the low flux of photons provided by the Sun. When comparing the scenario of simultaneous excitation where the time that can pass between the absorption of two photons is between 10 ps and 10 ns as in previous systems32,33,34,35,42, we have elongated this time to 120 µs. Although this is still far away from an ideal lifetime on the seconds timescale (Supplementary Section 13)49, it represents a substantial improvement by a factor of 104 to 107. Thus, our strategy allows the use of irradiances that are five orders of magnitude lower than those used in comparable systems. Introducing additional fundamental concepts50, for example light-harvesting antennas2,7,8,9,51,52, seems to be necessary for efficient charge accumulation under solar irradiation. Nevertheless, this implementation of a sequential light-absorption strategy represents an important step towards harnessing relatively diffuse solar light for vectorial charge accumulation and subsequent sustainable formation of solar fuels.
Data availability
The data from the main paper are available from Figshare (https://doi.org/10.6084/m9.figshare.28839578) (ref. 53); all other data are in the Supplementary Information. Source data are provided with this paper.
References
Hohmann-Marriott, M. F. & Blankenship, R. E. Evolution of photosynthesis. Annu. Rev. Plant Biol. 62, 515–548 (2011).
Gust, D., Moore, T. A. & Moore, A. L. Solar fuels via artificial photosynthesis. Acc. Chem. Res. 42, 1890–1898 (2009).
Wasielewski, M. R. Photoinduced electron transfer in supramolecular systems for artificial photosynthesis. Chem. Rev. 92, 435–461 (1992).
Wang, D. et al. Molecular photoelectrode for water oxidation inspired by photosystem II. J. Am. Chem. Soc. 141, 7926–7933 (2019).
Appel, A. M. et al. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 113, 6621–6658 (2013).
Berardi, S. et al. Molecular artificial photosynthesis. Chem. Soc. Rev. 43, 7501–7519 (2014).
Pellegrin, Y. & Odobel, F. Molecular devices featuring sequential photoinduced charge separations for the storage of multiple redox equivalents. Coord. Chem. Rev. 255, 2578–2593 (2011).
Hammarström, L. Accumulative charge separation for solar fuels production: coupling light-induced single electron transfer to multielectron catalysis. Acc. Chem. Res. 48, 840–850 (2015).
Wasielewski, M. R. Self-assembly strategies for integrating light harvesting and charge separation in artificial photosynthetic systems. Acc. Chem. Res. 42, 1910–1921 (2009).
Proppe, A. H. et al. Bioinspiration in light harvesting and catalysis. Nat. Rev. Mater. 5, 828–846 (2020).
Schulz, M. et al. Photoinduced charge accumulation and prolonged multielectron storage for the separation of light and dark reaction. J. Am. Chem. Soc. 142, 15722–15728 (2020).
Konduri, R. et al. Ruthenium photocatalysts capable of reversibly storing up to four electrons in a single acceptor ligand: a step closer to artificial photosynthesis. Angew. Chem. Int. Ed. 41, 3185–3187 (2002).
Huang, P. et al. Photo-induced oxidation of a dinuclear Mn2II,II complex to the Mn2III,IV state by inter- and intramolecular electron transfer to RuIII tris-bipyridine. J. Inorg. Biochem. 91, 159–172 (2002).
Gotico, P. et al. Tracking charge accumulation in a functional triazole‐linked ruthenium‐rhenium dyad towards photocatalytic carbon dioxide reduction. ChemPhotoChem 5, 654–664 (2021).
Li, X.-B. et al. Self-assembled framework enhances electronic communication of ultrasmall-sized nanoparticles for exceptional solar hydrogen evolution. J. Am. Chem. Soc. 139, 4789–4796 (2017).
Matt, B. et al. Charge photo-accumulation and photocatalytic hydrogen evolution under visible light at an iridium(iii)-photosensitized polyoxotungstate. Energy Environ. Sci. 6, 1504–1508 (2013).
Kitamoto, K. et al. Molecular photo-charge-separators enabling single-pigment-driven multi-electron transfer and storage leading to H2 evolution from water. Inorg. Chem. Front. 3, 671–680 (2016).
Xie, Z.-L. et al. Photochemical charge accumulation in a heteroleptic copper(i)-anthraquinone molecular dyad via proton-coupled electron transfer. Chem. Sci. 14, 10219–10235 (2023).
Lefebvre, J.-F. et al. An artificial photosynthetic system for photoaccumulation of two electrons on a fused dipyridophenazine (dppz)–pyridoquinolinone ligand. Chem. Sci. 9, 4152–4159 (2018).
Wang, W. et al. Multi-electron visible light photoaccumulation on a dipyridylamine copper(ii)–polyoxometalate conjugate applied to photocatalytic generation of CF3 radicals. J. Am. Chem. Soc. 145, 12136–12147 (2023).
Mendes Marinho, S. et al. Time‐resolved interception of multiple‐charge accumulation in a sensitizer–acceptor dyad. Angew. Chem. Int. Ed. 56, 15936–15940 (2017).
Cruz Neto, D. H. et al. Time‐resolved mechanistic depiction of photoinduced CO2 reduction catalysis on a urea‐modified iron porphyrin. Angew. Chem. Int. Ed. 63, e202407723 (2024).
Bhosale, S. et al. Photoproduction of proton gradients with π-stacked fluorophore scaffolds in lipid bilayers. Science 313, 84–86 (2006).
Puntoriero, F. et al. In Comprehensive Inorganic Chemistry III (eds Reedijk, J. & Poeppelmeier, K. R.) 628–653 (Elsevier, 2023).
Chen, H.-Y. & Ardo, S. Direct observation of sequential oxidations of a titania-bound molecular proxy catalyst generated through illumination of molecular sensitizers. Nat. Chem. 10, 17–23 (2018).
Beiler, A. M. & Moore, G. F. Caught in the act. Nat. Chem. 10, 3–4 (2018).
Imahori, H. et al. Charge separation in a novel artificial photosynthetic reaction center lives 380 ms. J. Am. Chem. Soc. 123, 6617–6628 (2001).
Fukuzumi, S., Ohkubo, K. & Suenobu, T. Long-lived charge separation and applications in artificial photosynthesis. Acc. Chem. Res. 47, 1455–1464 (2014).
Borgström, M. et al. Light induced manganese oxidation and long-lived charge separation in a Mn2II,II−RuII(bpy)3−acceptor triad. J. Am. Chem. Soc. 127, 17504–17515 (2005).
Sampaio, R. N., Troian‐Gautier, L. & Meyer, G. J. A Charge‐separated state that lives for almost a second at a conductive metal oxide interface. Angew. Chem. Int. Ed. 57, 15390–15394 (2018).
Flamigni, L., Baranoff, E., Collin, J. & Sauvage, J. A triad based on an iridium(III) bisterpyridine complex leading to a charge‐separated state with a 120‐μs lifetime at room temperature. Chem. Eur. J. 12, 6592–6606 (2006).
O’Neil, M. P. et al. Picosecond optical switching based on biphotonic excitation of an electron donor-acceptor-donor molecule. Science 257, 63–65 (1992).
Orazietti, M., Kuss‐Petermann, M., Hamm, P. & Wenger, O. S. Light‐driven electron accumulation in a molecular pentad. Angew. Chem. Int. Ed. 55, 9407–9410 (2016).
Nomrowski, J. & Wenger, O. S. Exploiting potential inversion for photoinduced multielectron transfer and accumulation of redox equivalents in a molecular heptad. J. Am. Chem. Soc. 140, 5343–5346 (2018).
Imahori, H. et al. Synthesis and photophysical properties of porphyrin–tetracyanoanthraquinodimethane–porphyrin triad: photon-dependent molecular switching. Chem. Lett. 27, 721–722 (1998).
Karlsson, S. et al. Accumulative charge separation inspired by photosynthesis. J. Am. Chem. Soc. 132, 17977–17979 (2010).
Flamigni, L., Baranoff, E., Collin, J., Sauvage, J. & Ventura, B. Light intensity effects on photoinduced charge separation parameters in a molecular triad based on an iridium(iii) bis(terpyridine) unit. ChemPhysChem 8, 1943–1949 (2007).
Ha-Thi, M.-H. et al. Photoinduced electron transfer in a molecular dyad by nanosecond pump–pump–probe spectroscopy. Photochem. Photobiol. Sci. 17, 903–909 (2018).
Neumann, S., Kerzig, C. & Wenger, O. S. Quantitative insights into charge-separated states from one- and two-pulse laser experiments relevant for artificial photosynthesis. Chem. Sci. 10, 5624–5633 (2019).
Favereau, L. et al. A molecular tetrad that generates a high-energy charge-separated state by mimicking the photosynthetic Z-scheme. J. Am. Chem. Soc. 138, 3752–3760 (2016).
Bürgin, T. H. & Wenger, O. S. Recent advances and perspectives in photodriven charge accumulation in molecular compounds: a mini review. Energy Fuels 35, 18848–18856 (2021).
Hankache, J., Niemi, M., Lemmetyinen, H. & Wenger, O. S. Photoinduced electron transfer in linear triarylamine–photosensitizer–anthraquinone triads with ruthenium(ii), osmium(ii), and iridium(iii). Inorg. Chem. 51, 6333–6344 (2012).
Sykora, M., Maxwell, K. A., DeSimone, J. M. & Meyer, T. J. Mimicking the antenna-electron transfer properties of photosynthesis. Proc. Natl. Acad. Sci. U.S.A. 97, 7687–7691 (2000).
Kleine, A. et al. From molecular to polymeric donors: prolonged charge separation in modular photoredox-active Ru(ii) polypyridyl-type triads. Inorg. Chem. 63, 23233–23247 (2024).
Arias-Rotondo, D. M. & McCusker, J. K. The photophysics of photoredox catalysis: a roadmap for catalyst design. Chem. Soc. Rev. 45, 5803–5820 (2016).
Pfund, B., Gejsnæs-Schaad, D., Lazarevski, B. & Wenger, O. S. Picosecond reactions of excited radical ion super-reductants. Nat. Commun. 15, 4738 (2024).
Moore, T. A. et al. Photodriven charge separation in a carotenoporphyrin–quinone triad. Nature 307, 630–632 (1984).
Amthor, S. et al. A photosensitizer–polyoxometalate dyad that enables the decoupling of light and dark reactions for delayed on-demand solar hydrogen production. Nat. Chem. 14, 321–327 (2022).
Blankenship, R. E. Molecular Mechanisms of Photosynthesis (Wiley Blackwell, 2014).
Schlau-Cohen, G. S. Principles of light harvesting from single photosynthetic complexes. Interface Focus 5, 20140088 (2015).
Gobbato, T. et al. Enhancing oxygenic photosynthesis by cross-linked perylenebisimide “quantasomes”. J. Am. Chem. Soc. 144, 14021–14025 (2022).
Frischmann, P. D., Mahata, K. & Würthner, F. Powering the future of molecular artificial photosynthesis with light-harvesting metallosupramolecular dye assemblies. Chem. Soc. Rev. 42, 1847–1870 (2013).
Brändlin, M., Pfund, B. & Wenger, O. S. Data of figures contained in the paper titled ‘Photoinduced double charge accumulation in a molecular compound’. figshare https://doi.org/10.6084/m9.figshare.28839578 (2025).
Acknowledgements
O.S.W. acknowledges funding by the Swiss National Science Foundation (grant number 208446). We thank D. Gejsnæs-Schaad for preparing the graphical abstract figure.
Author information
Authors and Affiliations
Contributions
M.B. designed, synthesized and characterized all molecules and conducted all spectroscopic and electrochemical experiments. B.P. designed the cw-pump–pump–probe experiment and assisted with the execution of this key experiment. O.S.W. conceived the project, helped design the pentad and provided guidance. M.B. and O.S.W. wrote the manuscript with input from B.P.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Chemistry thanks Sven Rau, Ken Sakai and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–75 and Supplementary Tables 1 and 2.
Source data
Source Data Fig. 3
Cyclic voltammograms, spectroelectrochemical and transient absorption spectroscopy data.
Source Data Fig. 5
Transient absorption spectroscopy data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Brändlin, M., Pfund, B. & Wenger, O.S. Photoinduced double charge accumulation in a molecular compound. Nat. Chem. 17, 1777–1784 (2025). https://doi.org/10.1038/s41557-025-01912-x
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41557-025-01912-x
