
Abstract
The adoption of perovskite solar cells (PSCs) requires improved resistance to high temperatures and temperature variations. Hole-selective self-assembled monolayers (SAMs) have enabled progress in the performance of inverted PSCs, yet they may compromise temperature stability owing to desorption and weak interfacial contact. Here we developed a self-assembled bilayer by covalently interconnecting a phosphonic acid SAM with a triphenylamine upper layer. This polymerized network, formed through Friedel–Crafts alkylation, resisted thermal degradation up to 100 °C for 200 h. Meanwhile, the face-on-oriented upper layer exhibited adhesive contact with perovskites, leading to a 1.7-fold improvement in adhesion energy compared with the SAM–perovskite interface. We reported power conversion efficiencies exceeding 26% for inverted PSCs. The champion devices demonstrated less than 4% and 3% efficiency loss after 2,000 h damp heat exposure (85 °C and 85% relative humidity) and over 1,200 thermal cycles between −40 °C and 85 °C, respectively, meeting the temperature stability criteria outlined in the International Electrotechnical Commission 61215:2021 standards.
Similar content being viewed by others

Main
Perovskite solar cells (PSCs) have garnered interest among researchers owing to their outstanding power conversion efficiency (PCE) and low-cost energy-efficient production1,2,3,4,5. However, their market entry hinges on proving long-term stability under various environmental stressors. Temperature plays a crucial role in PSC stability, with potential thermal degradation from volatile organic components, ion migration and delicate interfaces at elevated temperatures6,7,8. Furthermore, temperature variations can cause inelastic stress accumulation and mechanical failure due to mismatched thermal expansion coefficients between the layers in devices8,9,10.
In the silicon photovoltaic industry, thermal and thermo-mechanical degradation are evaluated through damp heat (85 °C and 85% relative humidity (RH)) and thermal cycling (between −40 °C and 85 °C) tests, according to the International Electrotechnical Commission (IEC) 61215:2021 standards11, respectively. So far, there are limited reports of efficient PSCs enduring both accelerated ageing tests12,13,14.
Recent advances in the performance and stability of PSCs can partly be attributed to the use of self-assembled monolayers (SAMs) as hole-selective contacts (HSCs) in an inverted device architecture15,16,17,18,19. SAM formation occurs spontaneously through hetero-condensation reactions between phosphonic acids and hydroxylated metal oxide surfaces20,21. This modulates the electronic structures of metal oxides, reducing the interfacial energy-level mismatch. Compared with conventional hole-transport layers incorporating mobile and hygroscopic dopants, the SAM approach enhances the stability of PSCs12,19,22,23,24.
Nevertheless, temperature stability presents a concern for hole-selective SAMs; commonly used carbazole-containing phosphonic acids are susceptible to thermal disordering and desorption at temperatures over 65 °C (refs. 6,19,25). In addition, the contact between perovskites and SAMs affects the adhesion toughness at the interface, thereby influencing the mechanical stability of PSCs against temperature variations10,19,26. Several strategies exist to tackle issues related to temperature stress. For example, tuning the anchoring of head groups to substrate surfaces could improve the high-temperature stability of SAMs18. Using co-adsorbents, bifunctional ligands and superwetting overlayers could establish adhesive contact between perovskites and SAMs, thereby inhibiting crack growth during thermal cycling16,17,19,22,27.
Given these promising developments, a holistic approach is warranted to simultaneously enhance the thermal and mechanical robustness of the SAM/perovskite interface. However, it may restrict the applicability of many classic SAMs having limited thermal stability6,16,21,28. Meanwhile, developing new hole-selective SAMs should account for head-group interactions, terminal-group compatibility with perovskites, and conformation and dipole moments, increasing the complexity in their design and synthesis18,21,29,30.
Here, we explored self-assembled bilayers (SABs) owing to their potential for more diverse functional tuning. SABs represent a basic form of self-assembled multilayers, consisting of distinct monolayers with varying constituents—interconnected via covalent or ionic bonds31,32. This layer-by-layer assembly stabilizes the labile monolayer by introducing rigid components and enables control over film termination33. However, although bilayer structures have been reported for PSCs, they rely on secondary van der Waals interactions21,34. We reasoned that typical covalent-bonded SAB formation requires the chemical activation of bottom monolayer surfaces to regenerate reactive sites (for example, hydroxyl and phosphonic acid groups)15,17,35, yet this process is incompatible with hole-selective SAMs that feature a conjugated terminal group.
SAB formation via Friedel–Crafts alkylation
We used a benchmark hole-selective SAM, composed of 2-(9H-carbazol-9-yl)ethyl)phosphonic acid (2PACz), as the base layer for developing SABs. To enable the covalent attachment of a sequentially deposited upper layer, we investigated C–C bond-forming reactions for the carbazole terminal group. It is noted that common organic synthesis methods, such as C–H activation and Suzuki coupling of halogenated carbazoles, rely on transition metal catalysts36,37; however, these could introduce metal contamination into thin-film photovoltaic processing. Therefore, we focused on metal-free Friedel–Crafts alkylation.
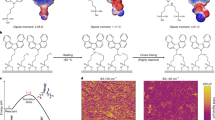
An alkylating agent, 4,4′,4′-tris(acetoxymethylene)triphenylamine (TATPA), was deposited on 2PACz-modified indium tin oxide (ITO) substrates using solution processing. TATPA was utilized in photo-cross-linking reactions, where photoacids catalyse the heterolytic cleavage of the C–O bond, yielding a reactive carbocation that acts as an electrophile for carbazole substitution, specifically at the nucleophilic C3 or C6 positions (Fig. 1a)38. This process culminates in the Friedel–Crafts alkylation of triphenylamine (TPA) to carbazoles, forming a polymer network (Supplementary Fig. 1). We postulated that these cross-linking reactions might spontaneously proceed on 2PACz SAMs with phosphonic acids serving as the proton donor38,39.

a, The proposed mechanism for the coupling reaction between TATPA and 2PACz. b, XPS spectra of TATPA, 2PACz and SAB films, where the solid lines represent the fits to the data and the dashed lines show the individual components, each fitted to a Gaussian distribution. c, 1H NMR spectra of TATPA, NECz and a mixture of NECz, TATPA and 2PACz. Inset: chemical structures of TATPA, NECz and 2PACz are shown with labels corresponding to NMR peaks38. d,e, Two-dimensional GIWAXS patterns of 2PACz (d) and SAB (e). IP, in-plane; OOP, out-of-plane. Diffraction signals with q > 2.0 Å−1 are attributed to ITO substrates. f, A schematic diagram illustrating the structure of SAB.
We investigated the chemical transformation of TATPA on both bare ITO and ITO–2PACz surfaces using X-ray photoelectron spectroscopy (XPS) and attenuated total-reflection Fourier transform infrared spectroscopy (ATR-FTIR). The C 1s XPS spectrum revealed a distinct peak at 289.2 eV on the TATPA-coated ITO surface, attributable to the carbonyl carbons of TATPA, alongside characteristic peaks for C–N and C–C/C–H bonds, signifying limited chemical reactivity (Fig. 1b and Supplementary Fig. 2). By contrast, the carbonyl peak was absent in the 2PACz–TATPA bilayer after thermal annealing at 100 °C, indicative of the decomposition of ester groups during on-substrate, solid-state reactions (Supplementary Fig. 3a). ATR-FTIR corroborated these findings, showing a diminished ester carbonyl stretching band of TATPA (at 1,740 cm−1) upon its interaction with the ITO–2PACz surface (Supplementary Fig. 3b). We suggested that the dissociation of TATPA was initiated by the residual P–OH groups within the 2PACz layer, as indicated by the presence of P–OH vibrations (at 947 cm−1 and 1,024 cm−1) in the corresponding ATR-FTIR spectra (Supplementary Fig. 4)17. The formation of carbocation intermediates was further indicated by the methoxylation of decomposed TATPA using deuterated methanol, probably facilitating Friedel–Crafts alkylation (Supplementary Note 1 and Supplementary Figs. 5 and 6).
To directly probe cross-linking reactions, we conducted time-of-flight secondary ion mass spectrometry (TOF-SIMS) measurements on substrates (Supplementary Fig. 7a). The mass spectrum for the ITO–2PACz substrate revealed no detectable molecular fragments with mass-to-charge (m/z) ratios exceeding 700 (Supplementary Fig. 7b). By contrast, the 2PACz–TATPA bilayer exhibited multiple peaks surpassing the mass ranges of the individual components. Notably, distinct peaks centred at m/z 862.4 and 804.3 were identified, which we attribute to Friedel–Crafts-type coupling products linked by methylene bridges (Supplementary Fig. 7c).
Nuclear magnetic resonance (NMR) was performed as a non-destructive method to validate the molecular structures. We initially prepared the NMR sample by scraping off a mixed film of TATPA and 2PACz (in a 1:1 weight ratio) to collect the powder. However, difficulties in dissolving these solids into dimethyl sulfoxide (DMSO) or chlorobenzene (CB) hindered the analysis of liquid-state NMR (Supplementary Fig. 8). To mitigate excess polymerization, N-ethylcarbazole (NECz), the non-acidic analogue of 2PACz, was utilized as the main reactant, with a small quantity of 2PACz (1:100 weight ratio to NECz) retained to catalyse TATPA decomposition. The proton (1H) NMR spectrum of this adjusted mixture revealed a singlet at δ = 4.00 ppm (that is, the Ar–CH2–Ar protons)38 and the absence of the methylene signal of TATPA (δ = 5.01 ppm; H-3), indicative of electrophilic aromatic substitution. This is supported by the up-field shift in aromatic proton signals, transitioning from H-1 (δ = 6.99 ppm) and H-2 (δ = 7.30 ppm) of TATPA to H-d (δ = 6.86 ppm) and H-c (δ = 7.13 ppm) in the cross-linked product, respectively (Fig. 1c). A similar trend was observed for the protons at the C4 and C5 positions (that is, H-aromatic) of carbazoles (Fig. 1c and Supplementary Figs. 9–12).
In addition, we explored a less reactive analogue of TATPA, 4-(di-p-tolylamino)benzyl acetate (DTBA), which contains a single ester group (Supplementary Fig. 13). Its surface reaction with 2PACz is expected to yield a discrete molecule, (2-(3,6-bis(4-(di-p-tolylamino)benzyl)-9H-carbazol-9-yl)ethyl)phosphonic acid (DTBA–2PACz), thereby simplifying product identification (Supplementary Fig. 13). We cast a mixed film of 2PACz and DTBA onto glass substrates and annealed it at 100 °C. High-resolution mass spectrometry confirmed the formation of DTBA–2PACz, detecting the characteristic [C56H52N3O3P + H]+ ion at m/z 846.3819 in positive ion mode and the [C56H52N3O3P–H]− ion at m/z 844.3678 in negative ion mode, both corresponding to their calculated molecular weights (Supplementary Fig. 14). 1H NMR analysis of the dissolved film in deuterated DMSO revealed the methylene bridge proton at δ = 4.00 ppm (Supplementary Fig. 15). Heteronuclear single quantum coherence spectroscopy validated this structural assignment, showing the 1H–13C correlation at δH = 4.00 ppm and δC = 40.97 ppm (Supplementary Fig. 16)38. 1H–1H homonuclear correlation spectroscopy further identified correlations between the methylene proton (δ = 4.00 ppm) and neighbouring protons from the carbazole and TPA units at δ = 7.15 ppm and δ = 8.03 ppm, respectively (Supplementary Fig. 17), supporting the Friedel–Crafts alkylation mechanism (Supplementary Note 2). In addition, another alkylating agent, (([1,1′-biphenyl]-4,4′-diylbis(azanetriyl))tetrakis(benzene-4,1-diyl))tetrakis(methylene) tetraacetate, bisTATPA), which features a linked TPA core, was shown to react with 2PACz (Supplementary Fig. 18). The SAB approach was also found to be generally applicable to various phosphonic acid SAMs that allow electrophilic attack by carbocations (Supplementary Note 3 and Supplementary Figs. 18–23)
The molecular arrangement and crystalline structure of the surface layers were characterized by grazing-incidence wide-angle X-ray scattering (GIWAXS). For the 2PACz monolayer, we observed a scattering arc pattern extending from the horizontal direction (at qxy = 1.56 Å−1) to an azimuthal angle of 52.5°, suggesting a vertical alignment of 2PACz molecules, albeit with a distribution in tilt angles (Fig. 1d, Supplementary Fig. 24 and Supplementary Note 4)40,41. This in-plane ordering is probably driven by π–π stacking among 2PACz molecules, as indicated by the diminishing molecular tilt in a densely packed reference film (Supplementary Fig. 25 and Supplementary Note 4). The deposition of TATPA disrupted the lateral crystallinity of the 2PACz layer, indicated by the disappearance of the in-plane diffraction peak (Fig. 1e). In addition, molecular tilt broadening was observed in the high-density reference film treated with TATPA (Supplementary Fig. 26).
Here, predominant interactions involve C–C bond formation between 2PACz and TATPA, resulting in a horizontal arrangement of TPA units atop 2PACz. The face-on orientation was confirmed by a π–π stacking peak along qz—corresponding to a ~0.4 nm periodicity of the repeating TPA units—observed at a higher TATPA solution concentration (Supplementary Fig. 27 and Supplementary Note 5). Surface reorganization was further indicated by Kelvin probe force microscopy (KPFM) measurements (Supplementary Fig. 28); the annealed 2PACz/TATPA bilayer exhibited a narrower distribution in contact potential difference (CPD) compared with their unannealed counterpart (Supplementary Fig. 29), attributed to more uniform molecular coverage6. Combining these results, we posited that the TATPA treatment led to a covalently interconnected SAB structure, with non-correlated 2PACz as the surface-bound molecule, linked to a planar TPA layer as the film termination (Fig. 1a,f).
Temperature stability of molecular contacts
We utilized KPFM to evaluate the thermal stability of molecular contacts on ITO surfaces. Surfaces modified with SAMs and SABs exhibited comparable CPDs, averaging 375 mV for both samples (Fig. 2a,b). We ascribed this to the minor impact of the TPA layer on work-function changes, possibly due to its in-plane orientation and, thus, negligible net dipole in the out-of-plane direction42,43. Following thermal annealing at 100 °C for 200 h, thermal degradation was observed in 2PACz SAMs, indicated by an increase in the CPD variations from 23 mV to 60 mV. By contrast, SAB-modified surfaces maintained a uniform potential distribution in the respective CPD map (with a CPD variation value of 30 mV), suggesting reduced structural changes in molecular contacts (Fig. 2c–f).

a–d, Surface potential images of the molecular contacts. a,b, Pristine ITO/SAM (a) and ITO/SAB (b) before thermal annealing at 100 °C for 200 h. c,d, ITO/SAM (c) and ITO/SAB (d) after thermal annealing at 100 °C for 200 h. Scale bars, 500 nm. e,f, Statistical potential distributions of ITO–2PACz (e) and ITO–SAB (f) surfaces. Gaussian distributions are fitted to the data curves. g, Illustrations of the sample preparation procedure for measuring molecular contact coverage, both before and after applying thermal cycling stress. h, The areal density of various molecular contacts, as determined using an electrochemical approach.
The thermal cycling stability of molecular contacts was then assessed through cyclic voltammetry (CV) measurements. The areal density of SAMs, measured before and after undergoing 1,000 temperature cycles from −40 °C to 85 °C, decreased from 3.15 × 1013 to 1.47 × 1013 molecules cm−2, indicating a 53% loss in coverage (Fig. 2g,h, Supplementary Fig. 30 and Supplementary Note 6). In addition, energy-dispersive X-ray spectroscopy revealed an increase in phosphorus content from 0.4 wt% to 1.6 wt% on the perovskite side of the buried interface after thermal cycling (Supplementary Fig. 31). Conversely, the polymerized network within SABs conferred better resistance to desorption under temperature variations, resulting in a lesser coverage loss of 32% and reduced attachment of phosphonic acids to perovskites (Supplementary Fig. 31)17,44. It is also noted that SABs exhibited a higher initial areal density of 2.45 × 1013 molecules cm−2 compared with SAMs, which we ascribed to the inclusion of electrochemically active TPA moieties45,46.
Characterization of the perovskite–SAM or perovskite–SAB interface
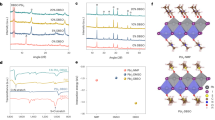
We fabricated FA0.84MA0.11Cs0.05Pb(I0.987Br0.013)3 perovskite films on SAM- or SAB-modified ITO substrates via an anti-solvent-assisted one-step solution process. Top-view scanning electron microscopy (SEM) images revealed similar average grain sizes (675 nm for SAM versus 720 nm for SAB) and pinhole-free surfaces for both films (Supplementary Fig. 32). GIWAXS analysis displayed nearly identical scattering patterns, with the (100) planes of black-phase perovskites preferentially oriented along 45° and 135° azimuthal angles, and a distinct PbI2 impurity peak at qz = 0.9 Å−1 (Fig. 3a,b and Supplementary Fig. 33). Despite these similarities in surface morphology and bulk structure, perovskites on SAB-modified substrates demonstrated a prolonged photoluminescence (PL) decay lifetime in time-resolved PL (TRPL) measurements, with averages increasing from 484 ns to 1,018 ns across four independent spots per sample (at an excitation flux of 718 nJ cm−2) (Fig. 3c and Supplementary Fig. 34), compared with those on SAM-modified substrates. This is indicative of suppressed non-radiative recombination, a finding further corroborated by PL quantum yield results (Fig. 3d, Supplementary Fig. 35 and Supplementary Note 7). Moreover, using the transmission line model47, the SAB–perovskite interfaces exhibited a lower contact resistance of 1.18 × 106 Ω cm compared with 2.43 × 106 Ω cm for the SAM-based counterparts (Fig. 3e and Supplementary Fig. 36). We concluded that the interface between perovskites and SABs was less defective and showed improved electrical properties relative to the perovskite–SAM interface.

a,b, GIWAXS patterns of perovskite films deposited on SAM (a) and SAB (b). c–f, TRPL spectra (c), PL spectra (d), TLM-measured resistance (e) and statistical distribution of adhesion energies from four measurements (f) for devices incorporating perovskite–SAM or perovskite–SAB interfaces. In e, the lines represent linear fits; In the boxplot in f, the medians (central lines), averages (hollow data points), quartiles and 1.5× interquartile ranges (whiskers) are shown; P in f represents load force. g,h, SEM images of buried perovskite surfaces delaminated from molecular contacts after thermal cycling tests (On SAM (g) and On SAB (h)). i,j, Electron density difference plots for perovskite/edge-on 2PACz SAM (i) and perovskite/face-on TPA moiety of SAB (j) interfaces, visualized using Visualization for Electronic and Structural Analysis (VESTA) software. Colour coding for atoms: Pb (grey), I (purple), C (brown), N (sliver grey) and H (white). A 3 × 4 periodicity with optimized lattice parameters of 19.012 Å × 25.630 Å was used in the xy plane.
Ultraviolet (UV) photoelectron spectroscopy was utilized to determine the valence band maximum (VBM) and work function of perovskites and HSCs (Supplementary Fig. 37). The conduction band minimum was estimated using the optical bandgap from UV–visible spectroscopy. For ITO–SAB substrates, the work function and VBM were found to be 60 meV and 100 meV lower, respectively, compared with ITO–SAM substrates. Consequently, the VBM offset between the SAB and perovskites was reduced by 40 meV relative to SAM, indicative of reduced energy losses at the buried interfaces29,30.
The mechanical integrity of the perovskite–molecular contact interface was evaluated using double cantilever beam (DCB) fracture mechanics tests. We prepared the DCB specimen by bonding a glass strip onto a poly(methyl methacrylate)-protected perovskite layer using brittle epoxy (Supplementary Fig. 38). Adhesion energy (GC) was obtained from force–displacement measurements following a reported methodology10. The average GC at the perovskite–SAB interface was 2.68 ± 0.33 J m−2 across four rounds of tests, 74% higher than that at the perovskite–SAM interface (1.54 ± 0.49 J m−2 for four rounds of tests), suggesting enhanced mechanical stability (Fig. 3f). Similar results were observed for fluorine-doped tin oxide (FTO) substrates, showing an average GC of 1.85 ± 0.40 J m−2 for perovskite–SAM to 2.94 ± 0.27 J m−2 for perovskite–SAB interfaces, respectively (Supplementary Fig. 39). We thereby exposed perovskites on various substrates to 1,000 thermal cycles, subsequently peeling off the films to examine the fracture surfaces. SEM analysis showed morphological degradation on surfaces in contact with SAM–ITO substrates, characterized by voids and non-conductive impurities (appearing as bright particles) scattered across the SEM image (Fig. 3g)21,48. Encouragingly, fracture surfaces from SAB–ITO substrates displayed no evident void formation and fewer impurity phases localized at grain boundaries, indicative of intimate contact between SAB and perovskites (Fig. 3h).
To gain insights into the interaction mechanisms at different interfaces, we performed density functional theory (DFT) calculations. Adsorption configurations were constructed with the molecular plane of 2PACz oriented perpendicularly to the perovskite surface and the TPA unit aligned parallel. Electron density difference plots unveiled localized interactions at the 2PACz–perovskite interface, with electron density transfer from H atoms in the carbazole terminal group to Pb atoms on the perovskite surface, accompanied by intramolecular polarization of 2PACz (Fig. 3i). By contrast, a maximized contact area at the TPA–perovskite interface led to extensive intermolecular charge transfer (Fig. 3j). This resulted in interaction energies of −4.41 eV, −1.71 eV and −0.72 eV for the π plane of TPA at dihedral angles of 0°, 30° and 60°, respectively, stronger than at the 2PACz–perovskite interface (−0.34 eV; Supplementary Fig. 40). The enhanced interlayer bonding contributes to improved mechanical stability and electronic properties at the SAB/perovskite interface (Fig. 3i,j).
Solar cell performance and stability
We fabricated PSCs using an inverted device configuration: ITO–SAM or SAB–perovskite–fullerene (C60)/bathocuproine (BCP)/Ag (Fig. 4a). Figure 4b shows the current–voltage (I–V) characteristics of the highest-performing SAM and SAB devices, measured in both forward and reverse scan directions. The champion SAB device exhibited a PCE of 26.3%, with an open-circuit voltage (VOC) of 1.174 V, a short-circuit current (JSC) of 26.2 mA cm−2 and a fill factor (FF) of 85.5% from the forward I–V scan, showing no performance hysteresis. This represents improvements over the PCEs of 24.4% (reverse scan) and 24.0% (forward scan) for the champion SAM device (Fig. 4b). We noted that the JSC values from the I–V sweeps match the integrated JSC derived from the incident photon-to-current efficiency (IPCE) measurements: 25.5 mA cm−2 for the SAM device and 26.1 mA cm−2 for the SAB device (Fig. 4c); furthermore, the PCE values agree with those from the steady-state power output (SPO) measurements (Fig. 4d). An average PCE of 26.1% was recorded among a batch of 20 SAB devices, surpassing the average PCE of 24.1% obtained in 20 SAM devices, with enhancements across all photovoltaic parameters (Supplementary Fig. 41 and Supplementary Tables 1 and 2).

a, A schematic illustration of the inverted PSCs. b, I–V curves for the champion inverted PSCs based on SAM and SAB interface. c,d, IPCE spectra (c) and SPO (d) for PSCs based on SAM and SAB interfaces. e, Forward and reverse I–V scan results tested at the Shanghai Institute of Microsystem and Information Technology (SIMIT). f, I–V curves at 1 cm2 device active area for PSCs based on SAM and SAB interfaces. g–i, PCE evolution for the PSCs based on SAM and SAB interface under 1-sun MPPT following the ISOS-L-1 protocol (g), under dark conditions and 85 °C, 85% RH following the ISOS-D-3 protocol (h), and under dark, thermal cycling between −40 °C and 85 °C following the ISOS-T-3 protocol (i). Data in h and i are presented as mean values ± s.d. PCEs in h and i represent the average value from seven devices for each condition. The initial PCEs are noted in the inset of g, and initial average PCEs are stated in the insets of h and i. The error bars in h and i reflect the s.d. among seven devices.
We sent one SAB device to China National Accreditation Service, an accredited third-party institute for independent certification, which delivered a PCE of 26.08%, with a VOC of 1.185 V, a JSC of 26.27 mA cm−2 and a FF of 83.84%, positioning it among the most efficient PSCs so far (Fig. 4e and Supplementary Fig. 42). The SAB strategy is effective for larger-scale devices; we achieved a PCE of 24.7% for PSCs with an active area of 1 cm2, compared with 23.1% based on the SAM approach (Fig. 4f). When applied to phosphonic acid SAMs beyond 2PACz, it also improved PSC performance (Supplementary Figs. 43, 44). Furthermore, we fabricated triple-halide PSCs with a wide-bandgap of 1.68 eV on ITO–SAB substrates, achieving a champion PCE of up to 22.5%—suitable for tandem applications with silicon (Supplementary Fig. 45)49,50.
The stability of PSCs was evaluated according to the International Summit on Organic Photovoltaic Stability (ISOS) and industry-relevant IEC 61215:2021 protocols. No performance degradation was observed for SAM and SAB devices (with initial PCEs of 24.1% and 25.8%, respectively) when stored under nitrogen in dark conditions for 140 days (Supplementary Fig. 46). To prevent corrosion of the metal electrode, BCP was replaced with atomic layer-deposited tin oxides (SnO2) as the barrier layer for ISOS-L and IEC 61215:2021 test conditions. The SAB device exhibited better operating stability under 1-sun-equivalent light-emitting diode (LED) illumination at a device temperature of ~55 °C (ISOS-L-1I): a minimal relative PCE loss of <2% was recorded, in contrast to a 16% loss for the SAM device, following 1,000 h maximum power-point tracking (MPPT; Fig. 4g).
Accelerated ageing of PSCs was further conducted at elevated temperatures of 70 °C and 85 °C, following ISOS-L-2I protocols (Supplementary Fig. 47a,b)12,51. At 70 °C, the SAM device exhibited a relative efficiency decay rate of 0.032% h−1, which is over double the rate of 0.0127% h−1 for the SAB device. At 85 °C, the time to retain 80% of initial performance during MPPT (that is, t80) was 192 h for SAM devices, whereas for SAB devices, it extended by approximately 1.7-fold to 520 h. The activation energy (Ea) of operational degradation was estimated by assuming an Arrhenius temperature dependence of the degradation rates (Supplementary Fig. 47c,d)12. The apparent Ea for SAM- and SAB-based PSCs was determined to be 0.46 eV and 0.69 eV, respectively. Consequently, we extrapolated the t80 at 35 °C to be approximately 3,000 h for SAM and 22,000 h for SAB devices.
We then conducted damp heat and thermal cycling tests based on the IEC 61215:2021 protocols. To minimize moisture ingress, PSCs were encapsulated using glass-to-glass packaging, with butyl rubber doped with zeolite desiccant as the edge sealant, reinforced with a UV-cured epoxy seal (Supplementary Fig. 48). Device performance was periodically monitored during the accelerated ageing process. For the SAB approach, thermal stability was indicated by seven devices retaining an average 94% of their initial PCEs (averaging 24.3%) after 2,000 h of damp heat testing at 85 °C and 85% RH (Fig. 4h). Notably, the best SAB device demonstrated a relative PCE loss of less than 4% over 2,000 h, which is in line with the industry benchmark of a 5% loss over 1,000 h for silicon photovoltaics. By contrast, SAM PSCs displayed a reduction in average performance (among seven devices) to 76% during the damp heat testing period.
Thermal cycling tests followed a similar trend: the average PCE of seven SAB devices decreased to 94% of its initial value (24.5%) after 1,288 cycles between −40 °C and 85 °C. Champion device under thermal cycling tests maintained 97% of its initial efficiency, exceeding the IEC 61215 requirement of a 5% drop after 200 cycles (Fig. 4i). By contrast, seven SAM devices maintained an average of 79% of their initial performance over the same times of cycles. Electroluminescence characterization showed that SAB devices had a higher resistance to non-radiative recombination after 200 thermal cycles, with an external quantum efficiency of 3.77% at an applied current of 28 mA cm−2, compared with 1.52% for SAM devices over the same period (Supplementary Fig. 49). These results confirm the excellent thermal and thermo-mechanical stability of SAB and SAB–perovskite interfaces.
For comparison, we investigated a model bilayer-forming molecule, 2-(4-(bis(4-methoxyphenyl)amino)phenyl)-1-cyanovinyl)phosphonic acid (MPA-CPA), which relies on non-covalent interlayer interactions21. MPA-CPA-based PSCs were tested under MPPT at 85 °C and 1-sun-equivalent LED illumination, which exhibited a t80 of 210 h (2.5 times shorter than that for SAB devices) (Supplementary Fig. 50a). When subjected to thermal cycling between −40 °C and 85 °C, the MPA-CPA devices showed more than 10% degradation in performance after just 150 cycles, significantly underperforming compared with the SAB devices (Supplementary Fig. 50b). We summarized the reported PSCs undergoing damp heat and thermal cycling tests (Supplementary Fig. 51)7,8,9,12,13,21,24,28,52,53,54,55,56,57,58, showcasing excellent stability for our encapsulated SAB devices among both categories, achieved with PCEs >24% (Supplementary Tables 3 and 4)7,8,9,12,13,21,24,28,52,53,54,55,56,57,58.
Conclusions
This work demonstrated covalently bonded SAB as a new type of hole-selective molecular contact for PSCs. Bilayer structures are formed through on-substrate Friedel–Crafts alkylation, connecting TPA units with a phosphonic acid SAM via a methylene bridge. The surface chemistry approach is versatile, compatible with a range of SAM-forming molecules and alkylating agents. The resulting cross-linked network shields SABs from thermal desorption, resisting degradation at 100 °C for 200 h. Moreover, the TPA units exhibit adhesive contacts with perovskite surfaces, minimizing thermo-mechanical degradation at the buried interface. We achieved a certified PCE of over 26% for inverted PSCs, with less than 4% efficiency loss after 2,000 h of damp heat exposure (85 °C and 85% RH) and 3% efficiency loss after 1,200 thermal cycles between −40 °C and 85 °C for the encapsulated champion-stability devices. Multilayer molecular contacts that harness covalent interlayer connections enhance the resistance of PSCs to temperature-related stressors.
Methods
Materials
Anhydrous solvents including CB, N,N-dimethylformamide, DMSO and isopropanol were purchased from Sigma-Aldrich. Anhydrous ethanol was purchased from Aladdin. Lead iodide (PbI2, 99.99%), BCP (>99%) guanidinium bromide and 2PACz (>98%) were purchased from TCI. Formamidinium iodide (99.99%) was purchased from Great Cell Solar. Methylammonium bromide (99.5%), methylammonium iodide (99.5%), methylammonium chloride (99.5%), caesium iodide (CsI, 99.999%) and C60 (99%) were purchased from Xi’an Polymer Light Technology. 1,2-Dichlorobenzene, tetrabutylammonium hexafluorophosphate and other solvents and chemicals if not mentioned above were purchased from Adamas China. Conductive glasses, FTO (15 Ω per square), ITO (20 Ω per square) and the anti-reflective coating foil were purchased from Suzhou ShangYang Solar Technology.
Synthesis of TATPA
To a solution of 4,4′,4′-nitrilotribenzaldehyde (200 mg) in acetic anhydride (1.8 ml), N,N-diisopropylethylamine (0.8 ml) was added slowly in an ice-water bath under N2 protection. The ice-water bath was then removed, and the reaction mixture was stirred at 40 °C for an additional 2 h. Upon completion of the reaction, the mixture was cooled to room temperature before being slowly poured into a mixture of ice water and ethyl acetate. The mixture was stirred for 15 min before hydrochloric acid (5%) and sodium hydroxide (5%) were subsequently added. The organic layer was extracted multiple times and dried with anhydrous sodium sulfate. The crude product was purified by column chromatography on silica gel (ethyl acetate:ether = 1:2), and the solvent was then removed by rotary evaporation to yield the final product as a whitish solid. 1H NMR (400 MHz, DMSO-d6, TMS): δ = 7.30 (d, J = 8.5 Hz, 6H), 6.99 (d, J = 8.5 Hz, 6H), 5.01 (s, 6H), 2.05 (s, 9H). 13C NMR (400 MHz, DMSO-d6, TMS) (ppm): δ = 170.75, 147.28, 131.17, 130.21, 124.07, 65.64 and 21.22.
Device fabrication
Perovskite precursor solution was prepared using the following procedure: 9.0 mg of methylammonium bromide, 23.9 mg of CsI, 22.2 mg of methylammonium iodide, 292.3 mg of formamidinium iodide, 1,005 mg of PbI2 and 10.8 mg of methylammonium chloride were dissolved in 1.3 ml of a N,N-dimethylformamide and DMSO mixed solution (volume ratio: 4:1) and stirred for 1 h. Inverted PSCs use a device architecture of ITO–HSC–perovskite–C60–BCP–Ag. ShangYang technology ITO or FTO glass was sequentially cleaned using a 2% detergent aqueous solution, deionized water, acetone and ethanol, with sonication for 20 min for each solvent. After drying with nitrogen flow, UV–ozone was applied for further cleaning. The following procedures were conducted in a N2-filled glovebox with an oxygen content of less than 1 ppm and water content less than 0.1 ppm. The pristine 2PACz SAM was dissolved in ethanol at 0.5 mg ml−1 and spin-coated directly onto the precleaned transparent conductive oxide (TCO) surface, then annealed at 100 °C for 10 min. The SAB was prepared by sequentially depositing 2PACz and TATPA. 2PACz was first deposited on the TCO, followed by spin-coating TATPA on top of 2PACz at 0.5 mg ml−1 in ethanol. The bilayer was annealed at 100 °C for 10 min. The HSCs were cooled to room temperature before deposition of the perovskite layer. The perovskite precursor solution was then deposited on the freshly prepared ITO–HSC substrate using a two-step spin-coating method: the first step was conducted at 1,000 rpm with an acceleration rate of 200 rpm s−1 for 10 s and the second step at 5,000 rpm with an acceleration rate of 2,000 rpm s−1 for 30 s. At the 15th second, 200 µl of CB was applied. After spin-coating, the substrate was annealed at 100 °C for 50 min. The chlorine content was found to be reduced during thermal annealing, lowering the residual chlorine element to levels below the detection limits of energy-dispersive X-ray spectroscopy and XPS analyses (Supplementary Figs. 52 and 53). A thin passivation layer using guanidinium bromide in ethanol (0.5 mg ml−1) was applied on top of the perovskite layer. C60 (25 nm) and BCP (8 nm) were deposited sequentially at rates of 0.2 Å s−1 and 0.5 Å s−1, respectively. Finally, 100 nm of Ag was thermally evaporated using a shadow mask. The 1 cm2 inverted solar cells are fabricated using similar approach, although designs in the electrode layout and shadow masks are different. We note that the SAB strategy is effective for both FTO and ITO substrates.
Solar cell characterization
The I–V characteristics of solar cells were measured using a Keithley 2400 source meter under the illumination of a solar simulator (Newport, Class AAA) at a light intensity of 100 mW cm−2, which is calibrated with a reference solar cell (Newport). The active area is determined by a shadow mask (0.0553, 0.1001 or 1.000 cm2). External quantum efficiency spectra were recorded with a commercial system (Arkeo-Ariadne, Cicci Research) based on a 300 W xenon light source and a holographic grating monochromator (Cornerstone, Newport). Stabilized power output measurement was done by Keithley 2400 source meters under the same solar simulator.
Stability measurements of solar cells
For long-term operational stability measurements, the unencapsulated cells were placed in a homemade stability tracking station protected under N2 atmosphere. The illumination source was a white-light LED (Tianjin Mato, grade A) with intensity calibrated to match the 1-sun condition. The devices were kept at 50 °C, 70 °C and 85 °C. The maximum power point was recorded every hour, and the solar cells were kept at the maximum power point voltage during this interval. For long-term storage stability, the unencapsulated cells are stored in a N2-filled glovebox with oxygen and water contents less than 1 ppm and 0.1 ppm, respectively. We note that it is important not to store the devices into the same glovebox for device fabrication, since the solvent vapour will cause severe device performance decay. Damp heat stability and thermal cycle stability were measured with a programmable temperature and humidity control box from Jing Yu machine. The devices are sandwiched with two glasses, in which quartz was used at the illumination side. The device–quartz interface was filled and cured with a transparent epoxy glue to avoid additional reflection at the air gap. The device and back contact glass were contacted with a slice of indium metal that was chopped to a size similar to that of the pad. The device was carefully encapsulated twice preliminarily with polyisobutyl glue and then UV-cured glue59. A photograph of a final encapsulated device is shown in Supplementary Fig. 48. We note that the IEC 61215:2021 protocol is designed for silicon solar modules and involves a series of tests beyond damp heat and thermal cycling, including static mechanical load and hail tests. It is anticipated that specific test protocols will be developed for PSCs; however, the IEC 61215:2021 protocol is still widely used by the perovskite community to evaluate and compare solar cell stability.
XPS measurements
XPS spectra were recorded with Thermo Fisher Scientific ESCALAB Xi+ system with 180° double-focusing, hemispherical analyser. The system is equipped with a 128-channel detector and monochromatic small-spot XPS. An AlKα source was used for excitation and a pass energy of 50 eV was used for XPS acquisition. All data were analysed with Thermo Avantage software. For XPS sample preparation, the deposition of the HSC and perovskite photoactive layer follows the same protocol as the fabrication of an inverted PSC, as outlined in the device fabrication section. The precursor concentration was increased to 1 mg ml−1 for both 2PACz and TATPA to improve the signal-to-noise ratio.
1H NMR measurements
1H NMR was performed using a Bruker 400 MHz ASCEND ADVANCE III HD spectrometer. For sample preparation, initially, we prepared a mixed solution containing 5 mg ml−1 2PACz and 5 mg ml−1 TATPA and cast a thin film from this mixture. The concentration was increased tenfold to obtain sufficient sample for the NMR test. The film was heated at 100 °C for 10 min and subsequently scraped from the substrate, yielding a greenish solid. The greenish colour originated primarily from the unreacted carbocation intermediate phase (Supplementary Fig. 8)60. This solid is insoluble in deuterated CB and DMSO. Consequently, we utilized a model reaction containing mixed reactants of NECz, TATPA and 2PACz, as described in the main text and illustrated in Fig. 1c.
DFT calculations
Spin-polarized electronic structure calculations were performed using the plane-wave basis set approach as implemented in the Vienna ab initio simulation package (VASP)61. The projector augmented wave method was used to represent the ion–core electron interactions62. The valence electrons were represented with a plane-wave basis set with an energy cut-off of 450 eV. Electronic exchange and correlation were described with the Perdew–Burke–Ernzerhof functional63. The DFT-D3 method was used to correct for the van der Waals interaction64. A 25 Å vacuum space was used to avoid interactions between surface slabs. K-point meshes of 3 × 3 × 1 and 1 × 1 × 1 were used for the Brillouin zone sampling of FAPbI3 (001) surface and other isolated molecules65. The convergence criteria for the self-consistent electronic structure and geometry were set to 10−5 eV and 0.03 eV Å−1, respectively. Binding energies (Eb) were calculated by
where Ebinding_system, EFAPbI3_001 and Eadsorbate represent the total energies of the binding system, FAPbI3 (001) surface and adsorbate, respectively.
Mechanical testing
Adhesion energy testing follows the same procedure as previously reported10. In brief, the DCB specimens adopted structures of glass–TCO–HSC–perovskite–poly(methyl methacrylate) (PMMA)–epoxy–TCO–glass. For the control samples, HSC consists of 2PACz, and for the target samples, SAB is used. PMMA (350 kDa) was dissolved in CB, spin-coated on top of the perovskite layer and annealed at 100 °C for 5 min. A brittle epoxy was then applied on top of the PMMA layer before being laminated by another TCO glass. The DCB was conducted using the reported procedure. The displacement rate was set at 1 μm s−1 while simultaneously recording the load force (P) and displacement (Δ). The crack length, denoted as a, was estimated using
where B = 15 mm and E = 70 GPa represent the width and Young’s modulus of the glass substrate, respectively, and h = 1.1 mm is the half-thickness of the DCB specimen. The adhesion energy, GC, is given by
where PC is the load at the onset of nonlinearity in the P–Δ curve. We note that the loading was repeated five times, and the average GC values were calculated from the last four repetitions.
TLM measurements
In the TLM technique, the total resistance (Rtot) between two contacts with length (L) and width (W) is measured and plotted as a function of the contact spacing d. As d increases, the influence of the sheet resistance on the total resistance measurement also increases. Fitting the resulting curve to a linear function yields a straight line with slope k. The sheet resistance can be calculated from this slope using the following equation66:
At d = 0, the contact resistance RC can be calculated using
CV measurements
CV measurements were conducted using a three-electrode configuration with a potentiostat (DH7000C, DONGHUA). The working electrodes, either SAM or SAB covered TCO glasses, were prepared following the same protocol used for device fabrication. A platinum wire served as the counter electrode, and an Ag wire was used as the reference electrode. To understand the desorption of SAM or SAB at the TCO–perovskite interfaces during thermal cycling tests, the perovskite layer was exfoliated using an established procedure67. The inverted PSCs were first removed from their encapsulation. Subsequently, the metal contact was gently exfoliated using Scotch tape. A thin layer of PMMA was then spin-coated on top of the perovskite–C60–BCP layer. A drop of UV-curable glue (ThreeBond 3035B) was applied to the PMMA surface, and a flexible polyethylene terephthalate sheet was laminated onto the glue. The stack was cured under UV light (256 nm), facilitating the exfoliation of the polyethylene terephthalate–perovskite stack and leaving behind the substrate exposing the HSC, either 2PACz SAM or SAB. The areal density of 2PACz and 2PACz–TATPA was measured in 1,2-dichlorobenzene with a 0.1 M TBA+PF6− electrolyte. All potentials were referenced against the ferrocene redox couple, serving as an internal standard. The effective coverage of the SAM or SAB on the ITO surface was determined by the slope of the linear dependency of the oxidative peak intensity against the scan rate44
where ip,o is the oxidative peak current (A), ν is the voltage scan rate (V s−1), n is the number of electrons transferred, the Faraday constant F is 96 485.33 C mol−1, the universal gas constant R is 8.3144 J K mol, T is the temperature (K), the Avogadro constant NA is 6.022 × 1023 mol−1, A (cm2) is electrode area and Γ* (molecules cm−2) is the surface density that can be obtained by calculating the slope of ip,o versus ν.
Other characterizations
ATR-FTIR measurement was performed with Bruker VERTEX70, and samples were scanned in the spectral range of 400–4,000 cm−1. High-resolution mass spectrometry was measured on a Thermo Scientific Q Exactive with atmospheric pressure chemical ionization ion sources. TOF-SIMS surface chemical analyses were performed using a PHI nano TOF 3+ instrument (ULVAC-PHI), equipped with a bismuth liquid metal ion gun as a primary beam and gas cluster ion beam (GCIB)–Ar–Cs ion gun as a sputtering source. The data of TOF-SIMS were acquired by 30 keV Bi3++ pulsing ion source operated in the high mass resolution mode (Bunched mode), and the d.c. current of bismuth source was measured as 9.3 nA. Positive ion spectra were collected in 100 frames (10 min). Data processing was done with TOF-SIMS Data Reduction software (TOF-DR) software. KPFM was measured with Asylum Cypher S atomic force microscope (Oxford Instruments) with a Ti–Ir-coated ASYELEC and Nanosurf AG, Flex-Mount atomic force microscope. The excitation amplitude was 4 V, and the scan rate was set to 0.25 Hz. GIWAXS was performed at BL02U2 and BL17B1 beamline of Shanghai Synchrotron Radiation Facility (SSRF) using the X-ray energy of 10 KeV. Two-dimensional images were acquired by a PLATUS 2M detector mounted vertically at a distance ~290 mm from the sample with a grazing-incidence angle of 0.2° and an exposure time of 20 s. High-resolution SEM images were obtained using the GeminiSEM 500 with an accelerating voltage of 1.5 kV. TRPL spectra were measured using M-X355L (Orient KOJI) in 10 kHz and 350 ps duration light source for the measurement. The applied power density was 13.3 W cm−2, and excitation fluences were 12.8, 32.1, 64.1, 128 and 718 nJ cm−2.
Data availability
All data generated or analysed during this study are included in the Article and its Supplementary Information and Source data. Source data are provided with this paper.
References
Kojima, A., Teshima, K., Shirai, Y. & Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 131, 6050–6051 (2009).
Tan, S. et al. Stability-limiting heterointerfaces of perovskite photovoltaics. Nature 605, 268–273 (2022).
McMeekin, D. et al. Intermediate-phase engineering via dimethylammonium cation additive for stable perovskite solar cells. Nat. Mater. 22, 73–83 (2023).
You, S. et al. Radical polymeric p-doping and grain modulation for stable, efficient perovskite solar modules. Science 379, 288–294 (2023).
Domanski, K., Alharbi, E. A., Hagfeldt, A., Gratzel, M. & Tress, W. Systematic investigation of the impact of operation conditions on the degradation behaviour of perovskite solar cells. Nat. Energy 3, 61–67 (2018).
Li, Z. et al. Stabilized hole-selective layer for high-performance inverted p-i-n perovskite solar cells. Science 382, 284–289 (2023).
Azmi, R. et al. Damp heat-stable perovskite solar cells with tailored-dimensionality 2D/3D heterojunctions. Science 376, 73–77 (2022).
Jiang, Q. et al. Surface reaction for efficient and stable inverted perovskite solar cells. Nature 611, 278–283 (2022).
Li, G. et al. Highly efficient p-i-n perovskite solar cells that endure temperature variations. Science 379, 399–403 (2023).
Dai, Z. et al. Interfacial toughening with self-assembled monolayers enhances perovskite solar cell reliability. Science 372, 618–622 (2021).
IEC 61215-1:2021 Terrestrial Photovoltaic (PV) Modules—Design Qualification and Type Approval—Part 1: Test Requirements 2nd edn (International Electrotechnical Commission, 2021).
Jiang, Q. et al. Towards linking lab and field lifetimes of perovskite solar cells. Nature 623, 313–318 (2023).
Mei, A. et al. Stabilizing perovskite solar cells to IEC61215:2016 standards with over 9,000-h operational tracking. Joule 4, 2646–2660 (2020).
Khenkin, M. V. et al. Consensus statement for stability assessment and reporting for perovskite photovoltaics based on ISOS procedures. Nat. Energy 5, 35–49 (2020).
Al-Ashouri, A. et al. Conformal monolayer contacts with lossless interfaces for perovskite single junction and monolithic tandem solar cells. Energy Environ. Sci. 12, 3356–3369 (2019).
Peng, W. et al. Reducing nonradiative recombination in perovskite solar cells with a porous insulator contact. Science 379, 683–690 (2023).
Park, S. M. et al. Low-loss contacts on textured substrates for inverted perovskite solar cells. Nature 624, 289–294 (2023).
Tang, H. et al. Reinforcing self-assembly of hole transport molecules for stable inverted perovskite solar cells. Science 383, 1236–1240 (2024).
Yu, S. et al. Homogenized NiOx nanoparticles for improved hole transport in inverted perovskite solar cells. Science 382, 1399–1404 (2023).
Hotchkiss, P. J. et al. The modification ofindium tin oxide with phosphonic acids: mechanism of binding, tuning of surface properties, and potential for use in organic electronic applications. Acc. Chem. Res. 45, 337–346 (2012).
Zhang, S. et al. Minimizing buried interfacial defects for efficient inverted perovskite solar cells. Science 380, 404–409 (2023).
Zheng, X. et al. Co-deposition of hole-selective contact and absorber for improving the processability of perovskite solar cells. Nat. Energy 8, 462–472 (2023).
Turren Cruz, S., Hagfeldt, A. & Saliba, M. Methylammonium-free, high-performance, and stable perovskite solar cells on a planar architecture. Science 362, 449–453 (2018).
Chen, R. et al. Reduction of bulk and surface defects in inverted methylammonium- and bromide-free formamidinium perovskite solar cells. Nat. Energy 8, 839–849 (2023).
Zhao, Y. et al. Discovery of temperature-induced stability reversal in perovskites using high-throughput robotic learning. Nat. Commun. 12, 2191 (2021).
Li, X. et al. Constructing heterojunctions by surface sulfidation for efficient inverted perovskite solar cells. Science 375, 434–437 (2022).
He, R. et al. Improving interface quality for 1-cm2 all-perovskite tandem solar cells. Nature 618, 80–86 (2023).
Li, Z. et al. Organometallic-functionalized interfaces for highly efficient inverted perovskite solar cells. Science 376, 416–420 (2022).
Li, T. et al. Inorganic wide-bandgap perovskite subcells with dipole bridge for all-perovskite tandems. Nat. Energy 8, 610–620 (2023).
Tan, Q. et al. Inverted perovskite solar cells using dimethylacridine-based dopants. Nature 620, 545–551 (2023).
Woodruff, M. A. & Hutmacher, D. W. The return of a forgotten polymer—polycaprolactone in the 21st century. Prog. Polym. Sci. 35, 1217–1256 (2010).
Guzmán, E., Rubio, R. G. & Ortega, F. A closer physico-chemical look to the layer-by-layer electrostatic self-assembly of polyelectrolyte multilayers. Adv. Colloid Interface Sci. 282, 102197 (2020).
Zhang, X., Chen, H. & Zhang, H. Layer-by-layer assembly: from conventional to unconventional methods. Chem. Commun. 14, 1395–1405 (2007).
Pitaro, M. et al. Tuning the surface energy of hole transport layers based on carbazole self-assembled monolayers for highly efficient Sn/Pb perovskite solar cells. Adv. Funct. Mater. 34, 2306571 (2023).
Yao, Y. et al. Organic hole-transport layers for efficient, stable, and scalable inverted perovskite solar cells. Adv. Mater. 34, 2203794 (2022).
Aukland, M. H., Šiaučiulis, M., West, A., Perry, G. & Procter, D. Metal-free photoredox-catalysed formal C–H/C–H coupling of arenes enabled by interrupted Pummerer activation. Nat. Catal. 3, 163–169 (2020).
Saber, A. F., Sharma, S., Lee, J., El-Mahdy, A. F. M. & Kuo, S. Carbazole-conjugated microporous polymers from Suzuki–Miyaura coupling for supercapacitors. Polymer 254, 125070 (2022).
Wang, Y.-X. & Leung, M.-K. 4,4′,4′-Tris(acetoxymethylene)triphenylamine: an efficient photoacid promoted chemical cross-linker for polyvinylcarbozole and its applications for photolithographic hole-transport materials. Macromolecules 44, 8771–8779 (2011).
Topa, M. et al. One-component cationic photoinitiators based on coumarin scaffold iodonium salts as highly sensitive photoacid generators for 3D printing IPN photopolymers under visible LED sources. Polym. Chem. 11, 5261–5278 (2020).
Memon, W. A. et al. Precise control of crystal orientation of conjugated molecule enables anisotropic charge transport properties. Adv. Funct. Mater. 32, 2110080 (2022).
Ogle, J., Powell, D., Amerling, E., Smilgies, D.-M. & Whittaker-Brooks, L. Quantifying multiple crystallite orientations and crystal heterogeneities in complex thin film materials. CrystEngComm 21, 5707–5720 (2019).
Fuchs, F., Caffy, F., Demadrille, R., Mélin, T. & Grévin, B. High-resolution Kelvin probe force microscopy imaging of interface dipoles and photogenerated charges in organic donor–acceptor photovoltaic blends. ACS Nano 10, 739–746 (2016).
Peng, S. et al. Direct detection of local electric polarization in the interfacial region in ferroelectric polymer nanocomposites. Adv. Mater. 31, 1807722 (2019).
Liu, M. et al. Compact hole-selective self-assembled monolayers enabled by disassembling micelles in solution for efficient perovskite solar cells. Adv. Mater. 35, 2304415 (2023).
Mirle, C. R., Raja, M., Vasudevarao, P., Sankararaman, S. & Kothandaraman, R. Functionalised carbazole as a cathode for high voltage non-aqueous organic redox flow batteries. N. J. Chem. 44, 14401–14410 (2020).
Ciobanu, M. et al. High contrast hybrid electrochromic film based on cross-linked phosphonated triarylamine on mesoporous antimony doped tin oxide. Sol. Energ. Mat. Sol. C 203, 110186 (2019).
Chen, L. et al. Exploring the influence of the contact resistance on perovskite phototransistors. Appl. Phys. Lett. 124, 162202 (2024).
Liang, J. et al. Origins and influences of metallic lead in perovskite solar cells. Joule 6, 816–833 (2022).
Xu, J. et al. Triple-halide wide-band gap perovskites with suppressed phase segregation for efficient tandems. Science 367, 1097–1104 (2020).
Mariotti, S. et al. Interface engineering for high-performance, triple-halide perovskite–silicon tandem solar cells. Science 381, 63–69 (2023).
Zhao, X. et al. Accelerated aging of all-inorganic, interface-stabilized perovskite solar cells. Science 377, 307–310 (2022).
Hao, M. et al. Flattening grain-boundary grooves for perovskite solar cells with high optomechanical reliability. Adv. Mater. 35, 2211155 (2023).
Ahmad, T., Dasgupta, S., Almosni, S., Dudkowiak, A. & Wojciechowski, K. Encapsulation protocol for flexible perovskite solar cells enabling stability in accelerated aging tests. Energy Environ. Mater. 6, e12434 (2023).
Heo, J. H., Choi, Y. K., Koh, C. W., Woo, H. Y. & Im, S. H. Semitransparent FAPbI3−xBrx perovskite solar cells stable under simultaneous damp heat (85 °C/85%) and 1 sun light soaking. Adv. Mater. Technol. 4, 1800390 (2019).
Lin, X. et al. In situ growth of graphene on both sides of a Cu–Ni alloy electrode for perovskite solar cells with improved stability. Nat. Energy 7, 520–527 (2022).
Li, H. et al. 2D/3D heterojunction engineering at the buried interface towards high-performance inverted methylammonium-free perovskite solar cells. Nat. Energy 8, 946–955 (2023).
Liang, Z. et al. Homogenizing out-of-plane cation composition in perovskite solar cells. Nature 624, 557–563 (2023).
Zhu, P. et al. Aqueous synthesis of perovskite precursors for highly efficient perovskite solar cells. Science 383, 524–531 (2024).
Gaulding, E. A. et al. Package development for reliability testing of perovskites. ACS Energy Lett. 7, 2641–2645 (2022).
Xia, C., Advincula, R. C., Baba, A. & Knoll, W. Electrochemical patterning of a polyfluorene precursor polymer from a microcontact printed (μCP) monolayer. Chem. Mater. 16, 2852–2856 (2004).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 132, 154104 (2010).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192 (1976).
Guo, S., Gregory, G., Gabor, A. M., Schoenfeld, W. V. & Davis, K. O. Detailed investigation of TLM contact resistance measurements on crystalline silicon solar cells. Sol. Energy 151, 163–172 (2017).
Syed, A. M., Iqbal, A. K., Waheed, A. Y. & Khasan, S. K. in Conducting Polymers (ed. Faris, Y.) Ch. 5 (IntechOpen, 2016).
Acknowledgements
The research was made possible by the start-up funding from Xi’an Jiaotong University under the framework of the ‘Young Talent Support Plan’ (71211223010709). The work was supported in part by National Natural Science Foundation of China (GYKP038, Y. Liu.; 52403248, Y. Liu; 12175298, Y.Y.; 22279039, X.L.). This work was also supported under Qin Chuang Yuan grant no. QCYRCXM-2023–067. The work was support by the National Key Research and Development Program of China (2022YFE0132400, W.M., 2022YFB4200305, X.L., 2024YFE0103700, X.L.). M.W. acknowledges the support of the National University of Singapore Presidential Young Professorship (A-0010046-00-00; A-0010046-01-00). A.H. acknowledges support by the Swedish Energy Agency (P2020-90215) and Swedish Research Council (2019-05591, 2023-05244). M.G. and S.M.Z. acknowledge support by Günes Perovskite Solar Cell corporation located in Adana, Turkey. We thank H. Qiu at Orient KOJI Scientific and D. He at the Instrument Analysis Center of Xi’an Jiaotong University for assistance with TRPL measurement; P. Zhou at the Instrument Analysis Center of Xi’an Jiaotong University for NMR analysis; and F. Tan for ATR-FTIR analysis. This work made use of beamlines BL01B1, BL02U2, BL17B1, BL06B, BL16U1 and BL19U2 at SSRF and received support through User Experiment Assist System of SSRF. We thank G. Gao and N. Gao at School of Chemistry of Xi’an Jiaotong University for their assistance with the SEM measurements; and eceshi for the XPS and UV photoelectron spectroscopy analyses.
Author information
Authors and Affiliations
Contributions
Y. Liu conceived the idea and supervised the project. Y. Liu, M.W., S.Y., X.L. and M.G. designed this project. B.D., S.Y., Y. Li and Y.Z. fabricated solar cell devices and conducted characterization of the devices. B.D. conducted XPS, FTIR, TRPL, conductance, stability and CV measurement. Y.Y. conducted GIWAXS measurements and analysis. Y.Z. conducted KPFM measurements. Y. Liu and Y.R. synthesized TATPA. M.C. conducted NMR measurements. Z.S. conducted SEM measurements. Z.B. assisted with GIWAXS analysis. Z.D. and H.J. conducted TOF-SIMS measurements. Y. Liu, M.W. and S.Y. analysed the data. Y. Liu and M.W. wrote the original draft. Y. Liu, M.W., S.Y., W.M., S.M.Z., A.H., T.E., X.L. and M.G. contributed to the editing of the manuscript. All authors discussed the results and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Y. Liu has filed a patent based on the concept developed in this manuscript. The other authors declare no competing interests.
Peer review
Peer review information
Nature Energy thanks Yongzhen Wu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–53, Notes 1–7 and Tables 1–4.
Supplementary Data 1
Source data for Supplementary Figs. 43, 44, 45 and 50.
Supplementary Data 2
Chemical file for the structure in the main text.
Source data
Source Data Fig. 4
The individual data behind the average values and the full photovoltaic parameters in Fig. 4h,i.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dong, B., Wei, M., Li, Y. et al. Self-assembled bilayer for perovskite solar cells with improved tolerance against thermal stresses. Nat Energy 10, 342–353 (2025). https://doi.org/10.1038/s41560-024-01689-2
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41560-024-01689-2
This article is cited by
-
Low-temperature sequential deposition for efficient inverted perovskite solar cells
Nature Communications (2025)
-
Bio-inspired multiscale design for perovskite solar cells
Nature Reviews Clean Technology (2025)
-
Non-volatile solid-state 4-(N-carbazolyl)pyridine additive for perovskite solar cells with improved thermal and operational stability
Nature Energy (2025)
-
Environmentally friendly solvents
Nature Energy (2025)
-
Enhancing the efficiency and stability of perovskite solar cells via a polymer heterointerface bridge
Nature Photonics (2025)
