
Abstract
Earth’s first rise in atmospheric oxygen between about 2.43 billion and 2.1 billion years ago fundamentally transformed the atmosphere and oceans, setting the foundation for the evolution of complex life. However, geochemical evidence reveals intermittent oceanic oxygen oases before the rise of atmospheric oxygen, although the mechanisms that drove the production and accumulation of oxygen remain poorly constrained. Here we present redox-sensitive trace metal and iron speciation data, and phosphorus phase partitioning results, for a 2.93-billion-year-old drill core from the Red Lake area, Canada, to reconstruct oceanic phosphorus cycling and links to oxygen production in the dominantly anoxic, iron-rich Archaean ocean. Our data document one of the earliest known intervals of surface water oxygen accumulation, predating the first accumulation of atmospheric oxygen by about 500 Ma. These intervals were preceded by ferruginous intervals and intervals of enhanced sulfide availability, which led to pulsed increases in oceanic phosphorus bioavailability via anoxic recycling from sediments. Enhanced phosphorus bioavailability would have helped stimulate photosynthetic primary productivity and organic carbon burial, probably exerting a major control on the episodic development of oxygen oases in the late Archaean ocean. This, in turn, led to a critical transitional phase in the development of an oxygenated surface environment.
Similar content being viewed by others


Main
The transition to a persistently oxygenated atmosphere during the Great Oxidation Event (GOE) ~2.43‒2.1 billion years ago (Ga)1 has ultimately been attributed to the evolution of oxygenic photosynthesis2. Cyanobacterial ancestors were among the first organisms to perform oxygenic photosynthesis, and initially, the oxygen they produced would have reacted with reduced species in the ocean and atmosphere, thereby preventing atmospheric accumulation. However, local oxygen accumulation in oceanic oxygen oases and transient ‘whiffs’3,4,5 of atmospheric oxygen before the GOE, representing oxygen production but not atmospheric accumulation6, appear to have been a crucial transitional step in the progression towards persistent Earth surface oxygenation7.
Evidence of pre-GOE whiffs of oxygen is substantiated by multiple independent geochemical proxies, including molybdenum and rhenium enrichments at ~2.5–2.6 Ga in South Africa8 and ~2.5 Ga in Western Australia3, manganese oxides burial and molybdenum stable isotope compositions at ~2.5 Ga (ref. 9) in Australia and ~2.95 Ga (refs. 10,11) in South Africa, mobilization of selenium by free oxygen at ~2.5 Ga (ref. 12) in Australia and Ce abundance anomalies at ~2.6 Ga (ref. 13) in South Africa and Canada. In the Red Lake area (Canada), the presence of whiffs of oxygen has been inferred in 2.93-Ga deposits based on trace element enrichments and rare earth elements4,5. However, the mechanisms that promoted the production and accumulation of oxygen have scarcely been explored, reflecting a critical knowledge gap in our understanding of oxygen evolution on the early Earth. Furthermore, the factors causing the delay in pervasive oxygenation of the atmosphere–ocean system after the establishment of oxygenic photosynthesis in the Archaean remain poorly constrained, particularly regarding the role of bio-limiting nutrients such as nitrogen and phosphorus (P)14.
On geologic timescales, P is generally considered the ultimate limiting nutrient for primary productivity, which is intrinsically linked to organic carbon (Corg) production and burial, and therefore to atmospheric oxygen production15. Multiple studies point to persistent low seawater P concentrations throughout the Archaean16,17 and Proterozoic18,19, with the depletion in P primarily attributed to scavenging by iron minerals in the water column17,18,19. However, recent studies have challenged the view of low P in the Archaean ocean20,21, and while there is evidence for P–Corg–O2 coupling in late Neoarchaean marine sediments deposited immediately before the GOE7, the behaviour of the P cycle in earlier oxygen oasis settings remains entirely unconstrained.
Here we seek to bridge this knowledge gap by conducting an in-depth analysis of P cycling in Mesoarchaean (~2.93 Ga) open marine sediments from the Ball Assemblage, Canada, to evaluate potential links to periodic oxygenation of the early Earth’s surface. We combine P phase partitioning with Fe speciation and redox-sensitive trace metal data to evaluate local redox controls on P cycling and its availability. This constitutes the oldest examination of coupled redox and P cycling in marine sediments, reflecting one of the earliest known examples of oasis-style oceanic oxygenation.
Geological setting
To investigate local redox conditions, P bioavailability and its role in primary productivity, Corg burial and subsequent oxygen production, we studied a Mesoarchaean drill core (NGI10-31) from the Red Lake Greenstone Belt, Canada (Supplementary Fig. 1). The Red Lake area represents the oldest carbonate platform on Earth4,5,22. It was deposited in an open marine setting and contains some of the earliest geochemical evidence for oxygenic photosynthesis and whiffs of oxygen5,23. The NGI10-31 core consists of alternating siliciclastics and offshore chemical sediments, which are comprised of chert, alongside oxide-rich and sulfide-rich iron formations from the basinal facies of the Red Lake carbonate platform24 (Supplementary Information provides full details of the geologic setting, age constraints, core location and evaluation of metamorphism).
Ocean redox conditions
To constrain water column redox variability through the NGI10-31 core, we combine Fe speciation with redox-sensitive trace metal (Mo, V and U) systematics (Supplementary Information provides detailed analytical procedures and Methods provides the interpretational framework). To evaluate trace metal data, we calculate enrichment factors (EFs), utilizing a modified approach25,26 to allow for artificially inflated EF values in chemical sediments (iron formations and cherts)25, due to their low Al content (Methods).
Generally elevated FeHR/FeT (>0.38) and FeT/Al (>0.66) ratios, combined with enrichments in Mo and V, indicate deposition under dominantly anoxic bottom water conditions27,28, but with better oxygenated intervals potentially being indicated by lower values for these proxies during deposition of siltstones (Fig. 1; below). However, the U record is relatively stable, with little evidence for sediment enrichment. In the Red Lake area, redox-sensitive metals were delivered to seawater largely via oxidative continental weathering5. We thus attribute the limited enrichment in U to a particularly low oxidative weathering influx, relative to Mo and V, due to the high oxidation requirements of minerals such as uraninite, the main mineral host for crustal U, relative to Mo and V host phases29. Intervals of water column anoxia are further supported by moderate EF–Mo values (>1 to <10; Fig. 1), which commonly occur via a particulate shuttle mechanism following Mo uptake by Fe minerals precipitating in a ferruginous water column26,30.

a, Stratigraphy and lithology of the NGI core. b, Total organic carbon contents. c, Total iron versus aluminium ratio reported as wt%/wt%. d, Highly reactive iron versus total iron. e, Pyrite–Fe versus highly reactive Fe. f–h, Enrichment factors (EFs) for molybdenum (f), vanadium (g) and uranium (h). i, Total phosphorus content. j, Phosphorus versus aluminium ratio reported as wt%/wt%. k, Proportion of Pdet, Pauth, Porg and PFe within the total P pool. The yellow dots and light-yellow areas indicate the oxic water column conditions. In f–h, circle symbols correspond to EFs calculated with the standard Al normalization equation, and square symbols represent revised EFs calculations for chemical samples (Methods). Dashed lines: in c, upper boundary for recognition of anoxic deposition; in d, boundaries for identifying oxic (<0.22) and anoxic deposition (>0.38), with equivocal samples falling between these lines; in e, boundaries for identifying euxinic (>0.8) from ferruginous (<0.6) deposition with possibly euxinic conditions in between 0.6 and 0.8; in c, i and j, represent average shale values (ref. 46). Note that samples NGI 26 and NGI 29 have been excluded from i, j and k (see ‘Phosphorus phase partitioning’ section in the Supplementary Information).
We note, however, that because the apparent development of better oxygenated water column conditions is restricted to siltstone intervals, this may represent rapid sedimentation due to relocation of the river system, which would potentially mask water column FeHR/FeT and FeT/Al enrichments even under anoxic conditions31. Conversely, it is well documented that in modern alluvial systems (for example, estuaries or river input systems), high FeHR inputs can lead to high FeHR/FeT ratios, giving an apparent anoxic depositional condition32,33, especially if eroded sediments are sourced from highly weatherable mafic volcanic terrains, such as in the Red Lake area. However, the low FeHR/FeT and FeT/Al ratios during deposition of the siltstone horizons (Fig. 1) rule out this second possibility.
In assessing the possibility that FeHR/FeT and FeT/Al ratios were masked by rapid sedimentation, we note that EFs for Mo and V are similarly depleted across these intervals (Fig. 1f,g). The relative depletion in Mo may also be an expectation during rapid sedimentation under ferruginous conditions because this would potentially mask the drawdown of Mo delivered to the sediment via the particulate shuttle mechanism. However, the geochemical behaviour of V contrasts with that of Mo in that it is primarily affected by diagenetic transformations close to the sediment–water interface (Methods). Indeed, under partially oxygenated (dysoxic) conditions, V may be mobilized and lost from the sediment34. Rapid sedimentation would probably minimize this loss of V to the overlying water column, which may have occurred across the middle siltstone horizon, where V is not depleted (Fig. 1g). However, while the redox interpretation for the siltstone layers is complicated by the potential for rapid sedimentation, the depletion in V observed across two of these horizons is consistent with loss of V from the sediments under dysoxic conditions. This is similar to the redox state proposed for the 2.6–2.5-Ga Campbellrand–Malmani carbonate platform in South Africa8, and we note that partially oxygenated surface waters may have dominated during the early stages of biospheric oxygenation.
Indeed, the presence of at least partially oxygenated surface waters in the vicinity of a redox boundary separating the stromatolite-rich shallow shelf from further offshore lithofacies has been invoked as an oxidative mechanism for the deposition of the oxide iron formations (IFs) in the Red Lake5,23 and nearby areas4,22. Similarly, oxidation and removal of Ce and Mn from seawater and subsequent enrichment of these elements in chemical sediments near the carbonate platform required oxygen in the depositional environment5. Thus, independent lines of evidence suggest that the water column periodically became partially oxygenated in the Red Lake area, with the extent of oxygenation progressively increasing in time and space in the run-up to the GOE9,35.
In addition, episodes of enhanced sulfide availability, at least in shallow pore waters but also potentially in the water column, are implied for three intervals (at 67–75 m, 88–93 m and 118 m) by elevated FeHR/FeT (>0.38) and high FePy/FeHR (>0.6) ratios, along with elevated EF–Mo (>6) and EF–V (>1) values. The consensus for Precambrian sedimentary sulfide production invokes microbial reduction of sulfate, itself derived from oxidative weathering and continental run-off36. Therefore, sulfide production in our ~2.93-Ga sediments implies oxidative weathering of Archaean37 continents and supply of riverine sulfate. Alternatively, as supported by rare earth element patterns12,27, seawater sulfate could have been sourced from hydrothermally derived upwelling waters.
Phosphorus cycling in the dominantly iron-rich Archaean ocean
The transfer of P to marine sediments primarily occurs through the deposition of detrital apatite, organic matter and Fe minerals. In the modern ocean, rivers provide the dominant source of dissolved P (ref. 38), and concentrations can be sustained in the water column by the remineralization of sinking organic matter. Indeed, under oxic conditions, most organic P is remineralized during deposition, whereas a considerable fraction of organic and Fe-bound P is released to the pore waters of anoxic sediments during diagenesis39. Under oxic water column conditions, P released to pore waters during early diagenesis is dominantly trapped in the sediment via ‘sink switching’, either forming carbonate fluorapatite or re-adsorbing to Fe minerals near the sediment–water interface40,41. However, under euxinic water column conditions or under ferruginous conditions where pore waters are sulfidic close to the sediment–water interface, a major proportion of the P released during diagenesis may be recycled back to the water column, potentially promoting a positive productivity feedback7,42. By contrast, under oligotrophic ferruginous conditions, P adsorbed to Fe (oxyhydr)oxide minerals formed in the water column may be efficiently retained in the sediment7,18. In addition, under anoxic conditions, P is preferentially released from organic matter during remineralization, particularly during microbial sulfate reduction42,43. This results in elevated molar Corg/Porg ratios, surpassing the canonical Redfield ratio of 106/1 (ref. 44). In the Archaean, therefore, the nature of local redox conditions, both in the water column and in the sediment, would have exerted a strong control on the fate of P.
To evaluate the P cycle on the Red Lake Platform, we determined the phase partitioning of P via a sequential extraction scheme adapted to ancient sediments45 (Methods). The technique targets four operationally defined P pools, including iron-bound P (PFe), authigenic P (Pauth), organic-bound P (Porg) and detrital P (Pdet), with the sum of PFe, Pauth and Porg defining a reactive P pool (Preac), representing P that is potentially bioavailable during deposition and early diagenesis. The correlation between Pdet and unreactive detrital elements such as Ti in our samples (Supplementary Fig. 3) implies that Pdet is dominantly derived from detrital sources and not from diagenetic recrystallization of Pauth (refs. 18,45) (Methods and Supplementary Information provide further discussion).
Total P contents in the NGI10-31 core are generally below the average shale value46 (Fig. 1i), which is consistent with a low supply of P from the water column due to global depletion of P via widespread removal in association with Fe minerals under ferruginous conditions18,19. Accordingly, Pdet is the largest P pool, due to the dominance of detrital riverine inputs over bioavailable forms (Fig. 1k). The two sections of the core that capture deposition under oxic water column conditions exhibit P contents close to average shale (Fig. 1i,j). These intervals also record the highest Preac values, dominantly characterized by PFe and Pauth phases (Fig. 1k), with the latter resulting from diagenetic sink switching from phases (PFe and Porg) that dominantly reflect drawdown of dissolved water column phosphate47. At the bottom of the core (75 to 90 m) and immediately after the lower oxic interval (108 to 112 m), P/Al surpasses the average shale value, but this is probably due to the very low Al contents in iron formations and ferruginous chert, and in both cases, Pdet represents ~70% of the total P budget.
Archaean oxygen oases within a P-limited ferruginous ocean
We next explore C/P ratios through the NGI10-31 core to address potential P limitation on primary productivity. Most of the core exhibits elevated Corg/Porg ratios, approaching ~2,000 (Fig. 2a), considerably above the Redfield ratio of 106/1 (ref. 44). Such elevated Corg/Porg ratios demonstrate pronounced preferential release of P during anaerobic remineralization of organic matter under anoxic conditions39,43. This P release occurred during intervals of enhanced sulfide availability (high FeHR/FeT and FePy/FeHR ratios; Fig. 1d,e and Supplementary Fig. 4), but also during ferruginous intervals (that is, deposition of oxide IFs and, to a lesser extent, dolostone and ferruginous cherts; Fig. 2a), where there was probably limited sulfide generation (as evidenced by low FePy/FeHR ratios; Fig. 1e). This highlights that while microbial sulfate reduction is important for driving preferential release of P during organic matter remineralization42,43, other microbial pathways (such as dissimilatory Fe reduction) may also promote preferential P release. Indeed, the organic matter preserved in these ferruginous, low-sulfide samples is relatively high (between 0.54 wt% and 1.05 wt%; Fig. 1b), suggesting abundant availability to fuel microbial remineralization pathways.

a, Corg versus Porg in moles per 100 g of sediment. b, Corg versus Preac in moles per 100 g of sediment. c, Depth (m) versus Corg/Preac ratios. Black lines represent the Redfield ratio (106/1).
To address whether the preferential release of P from organic matter during diagenesis resulted in P recycling to the water column, we next consider Corg/Preac ratios (Fig. 2b). Except for two samples, sulfidic slates and oxide-rich IFs mostly plot well above the Redfield ratio, confirming efficient recycling of P to the water column during early diagenesis. Samples showing no evidence for P recycling (hence plotting below the Redfield ratio) are less provisioned in organic matter, with total organic carbon (TOC) contents <0.5 wt%, suggesting that the supply of organic carbon exerted a positive feedback on P recycling under anoxic conditions. Contrasting with relatively TOC-rich samples (that is, >0.5 wt% TOC), ferruginous cherts and dolostone also mostly yield Corg/Preac plotting near or below the Redfield ratio. This supports limited P recycling to the water column, with P fixation as Pauth and PFe for the less productive, carbonate- or chert-dominated facies. Samples deposited under oxic water conditions have Corg/Preac ratios below the Redfield ratio (Fig. 2b). This is consistent with efficient P fixation in the sediment after drawdown in association with organic matter and, to a lesser extent, Fe minerals, coupled with extensive release of P during aerobic organic matter oxidation and trapping of that P by microbial biomass48. If organic matter was the main carrier of reactive phosphorus under oxic water column conditions, this suggests that P released from organic decay was effectively trapped, ultimately as Pauth or PFe (Fig. 1k). This process is reflected by the increase in Preac (Fig. 1k) and P/Al ratios close to the average shale41 value (Fig. 1j).
Archaean oxygen oases were probably spatially and temporally limited, and their development was largely dependent on local redox conditions and nutrient cycling. A comparison between our Corg/Preac results and data from Neoarchaean (2.65 to 2.43 Ga) S- and Fe-rich samples from South Africa7 suggests that oxygen oases were less extensive and less intense ~2.93 Ga, with average Neoarchaean Corg/Preac values of 3,900 ± 5,764, indicating particularly active P recycling7, in sharp contrast to average values of 128 ± 160 in our earlier Archaean samples (Fig. 2c). This aligns with a period of increased sulfate delivery from 2.8 to 2.4 Ga (refs. 49,50), which would have enhanced sulfide production and, consequently, the extent of P recycling. Enhanced P recycling probably created a positive productivity feedback that progressively increased oxygen production in these oasis-style settings. Over time, oxygenation expanded and intensified, progressing from limited areas in the Archaean to well-oxygenated continental shelves by ~2.5 Ga (ref. 9). Ultimately, this led to coupled atmosphere–ocean oxygenation by 2.32 Ga (ref. 35), when oxygen sources began to surpass oxygen sinks, driving Earth’s surface oxygenation1.
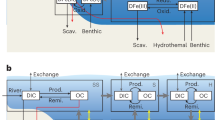
Our results collectively depict a dynamic redox scenario that promoted P recycling into the water column. Figure 3 illustrates how P recycling was primarily driven by sulfide production, initially triggered by oxygen ‘whiffs’ that facilitated an influx of sulfate, promoting sulfide production. This process, in turn, helped sustain productivity and oxygen generation. Specifically, during periods of enhanced sulfide availability, sulfidic slates were deposited, enhancing P recycling back to the water column. The subsequent increase in organic matter generation triggered additional P recycling from the IFs deposited deeper in the water column (note that these IFs are relatively enriched in TOC, up to 1 wt%), beneath the sulfidic wedge. The extent of the sulfidic zone probably fluctuated over time, with its spatial distribution waxing and waning, leading to the alternating deposition of sulfidic slates, IF and ferruginous chert observed in the NGI10-31 core. Although Archaean seawater was generally P limited, and ferruginous sediments were typically P depleted and dominated by detrital inputs, microbially mediated P recycling to the water column appears to have been sufficient to stimulate photosynthetic primary production in the oxygen oasis of the Red Lake area, as evidenced by diverse stromatolitic assemblages5,23.

The development of oxygen oases was strongly localized and limited by nutrient availability, particularly phosphorus. Ferruginous settings typically acted as phosphorus sinks under oligotrophic conditions, restricting productivity and the potential for oxygen production. However, during enhanced microbial iron reduction or when sulfidic intervals developed near the sediment–water interface, efficient phosphorus recycling could fuel localized primary production and drive oxygen generation. P recycling during periods of enhanced sulfide availability triggered additional P recycling from the IFs deposited deeper in the water column. P commonly underwent efficient recycling to the water column through anaerobic remineralization of organic matter and reduction of iron (oxyhydr)oxides, resulting in Corg/Preac ratios close to or higher than the Redfield ratio (106/1). The increased bioavailability of P promoted enhanced primary productivity, increased Corg burial and periodic accumulation of oxygen in surface waters.
The cycling and recycling of P played a critical role in shaping Earth’s early biosphere and the evolution of life, particularly during the Archaean. Our findings highlight the dynamic redox conditions of the Archaean ocean, where specific redox conditions at the bottom of the water column—such as organic-rich ferruginous or euxinic environments—enabled the recycling of P from the sediments into the water column. In these relatively shallow water settings, recycled P would have provided an additional source of bioavailable phosphorus to fuel primary productivity, particularly in localized oxygen oases. These oases, though spatially and temporally limited, probably facilitated the development of early microbial life, including stromatolite-forming cyanobacteria, by maintaining a supply of bioavailable P. In turn, enhanced local organic carbon burial would have allowed for oxygen accumulation and the establishment of intermittent oxygen oases in shallow waters, marking a crucial transitional phase in Earth’s oxygenation history.
The interplay between redox conditions and nutrient cycling may have created feedback loops that promoted oxygen production in these localized environments, contributing to gradual increases in atmospheric and oceanic oxygen levels. This process, which could have extended over millions of years, set the stage for the eventual rise of more complex life forms and the evolution of Earth’s biogeochemical cycles, influencing the planet’s capacity to support life in the long term. Thus, our data support a biogeochemical coupling between bioavailable P, organic carbon and O2 production some 500 million years before the first accumulation of atmospheric O2. This suggests that the mechanisms linking the cycling of bioavailable nutrients to coevolving organisms, which led to the modern stoichiometry of life, may trace their origins back to the Archaean.
Methods
Iron speciation
The iron speciation method evaluates local redox conditions51. It targets four operationally defined iron fractions, including carbonate-associated iron (FeCarb), ferric oxides (FeOx), magnetite (FeMag) and pyrite-associated Fe (FePy), which together comprise highly reactive iron (FeHR)31. Ratios of FeHR to total Fe (FeT) above 0.38 suggest deposition from anoxic bottom waters52. By contrast, FeHR/FeT ratios below 0.22 suggest deposition from oxic bottom waters, whereas intermediate values are considered equivocal52. Shales and iron-formation samples deposited in ferruginous settings can be impacted by the transfer of non-sulfidized FeHR to poorly reactive sheet silicates during early diagenesis, which lowers FeHR/FeT ratios53,54. To account for this possibility, we also considered FeT/Al ratios, whereby values >0.66 provide a robust indication of anoxic depositional conditions55, and thus we identify anoxic water column deposition by a combination of FeHR/FeT > 0.38 and/or FeT/Al > 0.66 (Fig. 1).
Redox-sensitive trace metals
Redox-sensitive elements such as V, Mo and U tend to be more soluble under oxidizing conditions and less soluble under reducing conditions, resulting in authigenic enrichments in oxygen-depleted sedimentary facies27. The removal of these metals from seawater under anoxic conditions may result in sediment enrichments several orders of magnitude higher than detrital values27.
Under oxic conditions, Mo is transported as the molybdate anion (MoO42−) and remains largely unreactive, with water column drawdown primarily occurring via uptake to Fe–Mn (oxyhydr)oxide minerals56. However, particle-reactive thiomolybdate forms under sulfidic conditions, commonly resulting in extensive sequestration in the sediments57,58. Under oxic–suboxic conditions, U is present mainly in the form of uranyl carbonate (UO2(CO3)34−) and is largely chemically unreactive59. However, under anoxic conditions in the sediments, U(VI) is reduced to U(IV), which may result in enrichments in the sediment, regardless of whether the water column is euxinic or ferruginous60. Vanadium is commonly transported to sediments as the vanadate ion (H2V(VI)O−4) adsorbed onto Mn oxides. Under mildly reducing conditions, where Mn oxides are reduced to Mn2+, V is commonly released from the sediment34, resulting in V depletion. Under anoxic conditions, vanadate is reduced to the vanadyl ion (V(IV)O2+), which is highly surface reactive and tends to be retained in the sediment57.
Redox-sensitive trace metal enrichment factors
Redox-sensitive trace metals may be controlled by intrinsic basinal factors, such as provenance and thus they are generally normalized to aluminium to account for terrigenous detrital inputs. A common way to approach this normalization is via the calculation of enrichment factors (EFs) for a specific element relative to average continental crust61. It is important to highlight that the primary control on elemental ratios in siliciclastics is usually the source area composition62. Therefore, the composition of the source rocks must be taken into consideration when calculating trace metal EFs. In this case, we normalized to lower continental crust (LCC) average values63, which better represent the mafic and ultramafic composition of the Red Lake area rocks, via the formula:
Whereas this equation is valid for siliciclastic sediments (and is used for siliciclastic sediments in the present study), the application of EF values to chemical sediments is problematic, as elevated values are commonly obtained relative to siliciclastic sediments27 due to the low detrital Al component characteristic of chemical sediments. An alternative approach is to calculate excess trace metal contents64:
However, this approach lacks the utility of EF values as they provide no information on the relative degree of enrichment, and consideration alongside well-calibrated siliciclastic EF values is not possible. To address this, we adopt a recently modified approach25,26, which utilizes ‘excess’ trace metal concentrations and recasts these data as EF* values:
With this approach, EF* values calculated for chemical sediments (including carbonates, cherts and IFs) can be directly compared to EF values calculated for siliciclastic sediments (Supplementary Table 1).
Phosphorus phase partitioning
We performed a sequential P extraction scheme adapted for ancient sedimentary rocks45. The method targets four operationally defined P pools, including iron-bound P (PFe), authigenic P (Paut), organic-bound P (Porg) and crystalline apatite P (dominantly detrital P; Pdet). Reactive P (Preac) is considered to be potentially available to organisms and is calculated as the sum of PFe + Pauth + Porg. Pdet is considered unreactive during early diagenesis and is buried in the sediment without biogeochemical interactions.
Data availability
All data generated or analysed during this study are available via Figshare at https://doi.org/10.6084/m9.figshare.28359224 (ref. 65) and included within the published article and its Supplementary Information files.
References
Poulton, S. W. et al. A 200-million-year delay in permanent atmospheric oxygenation. Nature 592, 232–236 (2021).
Canfield, D. E. The early history of atmospheric oxygen: homage to Robert M. Garrels. Annu. Rev. Earth Planet. Sci. 33, 1–36 (2005).
Anbar, A. D. et al. A whiff of oxygen before the Great Oxidation Event? Science 317, 1903–1906 (2007).
Riding, R., Fralick, P. & Liang, L. Identification of an Archean marine oxygen oasis. Precambrian Res. 251, 232–237 (2014).
Afroz, M., Fralick, P. W. & Lalonde, S. V. Sedimentology and geochemistry of basinal lithofacies in the Mesoarchean (2.93 Ga) Red Lake carbonate platform, northwest Ontario, Canada. Precambrian Res. 388, 106996 (2023).
Kaufman, A. J. et al. Late Archean biospheric oxygenation and atmospheric evolution. Science 317, 1900–1903 (2007).
Alcott, L. J., Mills, B. J. W., Bekker, A. & Poulton, S. W. Earth’s Great Oxidation Event facilitated by the rise of sedimentary phosphorus recycling. Nat. Geosci. 15, 210–215 (2022).
Kendall, B. et al. Pervasive oxygenation along late Archaean ocean margins. Nat. Geosci. 3, 647–652 (2010).
Ostrander, C. M. et al. Fully oxygenated water columns over continental shelves before the Great Oxidation Event. Nat. Geosci. 12, 186–191 (2019).
Albut, G. et al. Modern weathering in outcrop samples versus ancient paleoredox information in drill core samples from a Mesoarchaean marine oxygen oasis in Pongola Supergroup, South Africa. Geochim. Cosmochim. Acta 265, 330–353 (2019).
Planavsky, N. J. et al. Evidence for oxygenic photosynthesis half a billion years before the Great Oxidation Event. Nat. Geosci. 7, 283–286 (2014).
Stüeken, E. E., Buick, R. & Anbar, A. D. Selenium isotopes support free O2 in the latest Archean. Geology 43, 259–262 (2015).
Thoby, M. et al. Global importance of oxic molybdenum sinks prior to 2.6 Ga revealed by the Mo isotope composition of Precambrian carbonates. Geology 47, 559–562 (2019).
Ossa, F. O. et al. Limited oxygen production in the Mesoarchean ocean. Proc. Natl Acad. Sci. USA 116, 6647–6652 (2019).
Tyrrell, T. The relative influences of nitrogen and phosphorus on oceanic primary production. Nature 400, 525–531 (1999).
Bjerrum, C. J. & Canfield, D. E. Ocean productivity before about 1.9 Gyr ago limited by phosphorus adsorption onto iron oxides. Nature 417, 159–162 (2002).
Rego, E. S. et al. Low-phosphorus concentrations and important ferric hydroxide scavenging in Archean seawater. Proc. Natl Acad. Sci. Nexus 2, pgad025 (2023).
Guilbaud, R. et al. Phosphorus-limited conditions in the early Neoproterozoic ocean maintained low levels of atmospheric oxygen. Nat. Geosci. 13, 296–301 (2020).
Reinhard, C. T. et al. Evolution of the global phosphorus cycle. Nature 541, 386–389 (2017).
Ingalls, M., Grotzinger, J. P., Present, T., Rasmussen, B. & Fischer, W. W. Carbonate-associated phosphate (CAP) indicates elevated phosphate availability in Neoarchean shallow marine environments. Geophys. Res. Lett. 49, e2022GL098100 (2022).
Planavsky, N. J. et al. The evolution of the marine phosphate reservoir. Nature 467, 1088–1090 (2010).
Fralick, P. & Riding, R. Steep Rock Lake: sedimentology and geochemistry of an Archean carbonate platform. Earth Sci. Rev. 151, 132–175 (2015).
McIntyre, T. & Fralick, P. Sedimentology and geochemistry of the 2930 Ma Red Lake-Wallace Lake carbonate platform, Western Superior Province, Canada. Depositional Rec. 3, 258–287 (2017).
Fralick, P. & Pufahl, P. K. Iron formation in Neoarchean deltaic successions and the microbially mediated deposition of transgressive systems tracts. J. Sediment. Res. 76, 1057–1066 (2006).
Krewer, C. et al. Controls on the termination of Cretaceous oceanic anoxic event 2 in the Tarfaya Basin, Morocco. Am. J. Sci. 324, 11 (2024).
Li, S., Wignall, P. B. & Poulton, S. W. Co-application of rhenium, vanadium, uranium and molybdenum as paleo-redox proxies: insight from modern and ancient environments. Chem. Geol. 674, 122565 (2025).
Tribovillard, N., Algeo, T. J., Lyons, T. & Riboulleau, A. Trace metals as paleoredox and paleoproductivity proxies: an update. Chem. Geol. 232, 12–32 (2006).
Tribovillard, N., Algeo, T. J., Baudin, F. & Riboulleau, A. Analysis of marine environmental conditions based on molybdenum-uranium covariation—applications to Mesozoic paleoceanography. Chem. Geol. 324–325, 46–58 (2012).
Sverjensky, D. A. & Lee, N. The Great Oxidation Event and mineral diversification. Elements 6, 31–36 (2010).
Algeo, T. J. & Tribovillard, N. Environmental analysis of paleoceanographic systems based on molybdenum–uranium covariation. Chem. Geol. 268, 211–225 (2009).
Poulton, S. W. & Canfield, D. E. Ferruginous conditions: a dominant feature of the ocean through Earth’s history. Elements 7, 107–112 (2011).
Poulton, S. W. & Raiswell, R. The low-temperature geochemical cycle of iron: from continental fluxes to marine sediment deposition. Am. J. Sci. 302, 774–805 (2002).
Wei, G. Y. et al. A chemical weathering control on the delivery of particulate iron to the continental shelf. Geochim. Cosmochim. Acta 308, 204–216 (2021).
Morford, J. L. & Emerson, S. The geochemistry of redox sensitive trace metals in sediments. Geochim. Cosmochim. Acta 63, 1735–1750 (1999).
Ostrander, C. M. et al. Onset of coupled atmosphere—ocean oxygenation 2. 3 billion years ago. Nature 631, 335–339 (2024).
Canfield, D. E. A new model for Proterozoic ocean chemistry. Nature 396, 450–453 (1998).
Johnson, A. C. et al. Reconciling evidence of oxidative weathering and atmospheric anoxia on Archean Earth. Sci. Adv. 7, eabj0108 (2021).
Filippelli, G. M. The global phosphorus cycle: past, present, and future. Elements 4, 89–95 (2008).
Krom, M. D. & Berner, R. A. The diagenesis of phosphorus in a nearshore marine sediment. Geochim. Cosmochim. Acta 45, 207–216 (1981).
Slomp, C. P., Van Der Gaast, S. J. & Van Raaphorst, W. Phosphorus binding by poorly crystalline iron oxides in North Sea sediments. Mar. Chem. 52, 55–73 (1996).
Ruttenberg, K. C. & Berner, R. A. Authigenic apatite formation and burial in sediments from non-upwelling, continental margin environments. Geochim. Cosmochim. Acta 57, 991–1007 (1993).
Slomp, C. P., Thomson, J. & De Lange, G. J. Controls on phosphorus regeneration and burial during formation of eastern Mediterranean sapropels. Mar. Geol. 203, 141–159 (2004).
Van Cappellen, P. & Ingall, E. D. Benthic phosphorus regeneration, net primary production, and ocean anoxia: a model of the coupled marine biogeochemical cycles of carbon and phosphorus. Paleoceanography 9, 677–692 (1994).
Redfield, A. C. The processes determining the concentration of oxygen, phosphate and other organic derivatives within the depths of the Atlantic Ocean. Pap. Phys. Oceanogr. Meteorol. 9, 1–22 (1942).
Thompson, J. et al. Development of a modified SEDEX phosphorus speciation method for ancient rocks and modern iron-rich sediments. Chem. Geol. 524, 383–393 (2019).
Wedepohl, K. H. in Physics and Chemistry of the Earth (eds Ahrens, L. H. et al.) 305–333 (Pergamon, 1971).
Anderson, L. D., Delaney, M. L. & Faul, K. L. Carbon to phosphorus ratios in sediments: implications for nutrient cycling. Glob. Biogeochem. Cycles 15, 65–79 (2001).
Ingall, E. D., Bustin, R. M. & Van Cappellen, P. Influence of water column anoxia on the burial and preservation of carbon and phosphorus in marine shales. Geochim. Cosmochim. Acta 57, 303–316 (1993).
Fakhraee, M., Hancisse, O., Canfield, D. E., Crowe, S. A. & Katsev, S. Proterozoic seawater sulfate scarcity and the evolution of ocean–atmosphere chemistry. Nat. Geosci. 12, 375–380 (2019).
Fakhraee, M., Crowe, S. A. & Katsev, S. Sedimentary sulfur isotopes and Neoarchean ocean oxygenation. Sci. Adv. 4, e1701835 (2018).
Poulton, S. W. & Canfield, D. E. Development of a sequential extraction procedure for iron: implications for iron partitioning in continentally derived particulates. Chem. Geol. 214, 209–221 (2005).
Poulton, S. W. The Iron Speciation Paleoredox Proxy (Cambridge Univ. Press, 2021).
Poulton, S. W., Fralick, P. W. & Canfield, D. E. Spatial variability in oceanic redox structure 1.8 billion years ago. Nat. Geosci. 3, 486–490 (2010).
Tosca, N. J., Guggenheim, S. & Pufahl, P. K. An authigenic origin for Precambrian greenalite: implications for iron formation and the chemistry of ancient seawater. Geol. Soc. Am. Bull. 128, 511–530 (2016).
Clarkson, M. O., Poulton, S. W., Guilbaud, R. & Wood, R. A. Assessing the utility of Fe/Al and Fe-speciation to record water column redox conditions in carbonate-rich sediments. Chem. Geol. 382, 111–122 (2014).
Bertine, K. K. & Turekian, K. K. Molybdenum in marine deposits. Geochim. Cosmochim. Acta 37, 1415–1434 (1973).
Emerson, S. R. & Huested, S. S. Ocean anoxia and the concentrations of molybdenum and vanadium in seawater. Mar. Chem. 34, 177–196 (1991).
Scholz, F., McManus, J. & Sommer, S. The manganese and iron shuttle in a modern euxinic basin and implications for molybdenum cycling at euxinic ocean margins. Chem. Geol. 355, 56–68 (2013).
Calvert, S. E. & Pedersen, T. F. Geochemistry of recent oxic and anoxic marine sediments: implications for the geological record. Mar. Geol. 113, 67–88 (1993).
Anderson, R. F., Lehuray, A. P., Fleisher, M. Q. & Murray, J. W. Uranium deposition in Saanich Inlet sediments, Vancouver Island. Geochim. Cosmochim. Acta 53, 2205–2213 (1989).
McLennan, S. M. Relationships between the trace element composition of sedimentary rocks and upper continental crust. Geochem. Geophys. Geosyst. 2, 1021 (2001).
Fralick, P. W. & Kronberg, B. I. Geochemical discrimination of clastic sedimentary rock sources. Sediment. Geol. 113, 111–124 (1997).
Wedepohl, K. H. The composition of the continental crust. Geochim. Cosmochim. Acta 59, 1217–1232 (1995).
Brumsack, H.-J. The trace metal content of recent organic carbon-rich sediments: implications for Cretaceous black shale formation. Palaeogeogr. Palaeoclimatol. Palaeoecol. 232, 344–361 (2006).
Cañadas, F. et al. Archean oxygen oases driven by pulses of enhanced phosphorus recycling in the ocean. Figshare https://doi.org/10.6084/m9.figshare.28359224 (2025).
Acknowledgements
F.C. acknowledges support from Marie Skłodowska-Curie Actions (MSCA) grant agreement H2020-MSCA-IF-EF-ST/101022397. R.G. acknowledges support from L’Agence Nationale de la Recherche (ANR) grant agreement ANR-21-CE49-0007-01 and help from C. Causserand for inductively coupled plasma analysis. A.G.F. was supported by the European Research Council Consolidator grant number 818602. P.F. is supported by a Natural Science and Engineering Research Council of Canada Discovery Grant.
Author information
Authors and Affiliations
Contributions
Conceptualization: F.C., R.G.; Methodology: F.C., R.G., M.-P.M.-R.; Supervision: F.C., R.G.; Writing–original draft: F.C.; Writing–review and editing: F.C., R.G., P.F., Y.X., S.W.P., A.G.F.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Geoscience thanks Dan Asael and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Alison Hunt, in collaboration with the Nature Geoscience team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Geological context, methods, data quality checks, Supplementary Fig. 4 and references.
Supplementary Table 1
Supplementary Table 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cañadas, F., Guilbaud, R., Fralick, P. et al. Archaean oxygen oases driven by pulses of enhanced phosphorus recycling in the ocean. Nat. Geosci. 18, 430–435 (2025). https://doi.org/10.1038/s41561-025-01678-4
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41561-025-01678-4
This article is cited by
-
Enhanced phosphorus regeneration linked to Ediacaran ocean oxygenation
Communications Earth & Environment (2025)
-
Oscillating Archean oxygen oases
Nature Geoscience (2025)
