
Abstract
To ensure sustainable aquaculture, it is essential to understand the path ‘from feed to fish’, whereby the gut microbiome plays an important role in digestion and metabolism, ultimately influencing host health and growth. Previous work has reported the taxonomic composition of the Atlantic salmon (Salmo salar) gut microbiome; however, functional insights are lacking. Here we present the Salmon Microbial Genome Atlas consisting of 211 high-quality bacterial genomes, recovered by cultivation (n = 131) and gut metagenomics (n = 80) from wild and farmed fish both in freshwater and seawater. Bacterial genomes were taxonomically assigned to 14 different orders, including 35 distinctive genera and 29 previously undescribed species. Using metatranscriptomics, we functionally characterized key bacterial populations, across five phyla, in the salmon gut. This included the ability to degrade diet-derived fibres and release vitamins and other exometabolites with known beneficial effects, which was supported by genome-scale metabolic modelling and in vitro cultivation of selected bacterial species coupled with untargeted metabolomic studies. Together, the Salmon Microbial Genome Atlas provides a genomic and functional resource to enable future studies on salmon nutrition and health.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
The Norwegian Atlantic Salmon Gut Bacteria Culture Collection is maintained and stored at the Norwegian University of Life Sciences. Bacterial strains are available from the lead contacts upon request by email to S.L.L.R. (sabina.leantilarosa@nmbu.no) or P.B.P. (phil.pope@nmbu.no). A five-working-day response time should be expected. Isolates will be sent as freeze stock cultures and shipped in dry ice via courier. Shipping costs will be charged to the person or institution requesting them. Isolates are for research purposes only; a declaration that the isolates will not be used for commercial purposes is required before sample shipment. Oxford Nanopore sequencing reads have been deposited in the SRA with project numbers PRJEB45024 and PRJEB61648. Publicly available 16S amplicon datasets of Atlantic salmon gut samples were downloaded from NCBI (bioprojects: PRJEB39298, PRJNA498084, PRJNA555355, PRJNA590084, PRJNA594310, PRJNA650141, PRJNA730696, PRJNA733893, PRJNA824235, PRJNA824256, PRJNA866155) together with data derived from two in-house trials with salmons feeding on a commercial standard diet (ImpTrial2: PRJEB60544 and ImpTrial1: PRJEB60545). Shotgun metagenomic reads have been deposited at SRA bioProjects PRJEB60591 and PRJNA947914. RNA sequencing reads can be found in SRA under accession number PRJEB60552. Mass spectrometry data for this study can be found on the Mass Spectrometry Interactive Virtual Environment (MassIVE) repository (massive.ucsd.edu) with accession number MSV000089895. The SMGA is publicly available via Figshare (Genomes_fasta, https://figshare.com/s/049af70f3d15d05e690d; Genes_nuc_fna, https://figshare.com/s/e56cdce3cd422e43a1e6; Genes_prot_faa, https://figshare.com/s/517b6e611fd896314dd0) and by the National e-Infrastructure for Research Data (NIRD) of Norway (https://ns9864k.web.sigma2.no/TheMEMOLab/projects/SMGA_v1.0/). Metabolic models can be inspected via the html files supplied in Supplementary Dataset 1. The complete list of detected pathogenicity factors is available at https://ns9864k.web.sigma2.no/TheMEMOLab/projects/SMGA_v1.0/annotation/SMGA_PathoFact_Annotation.csv.
Code availability
Scripts used to generate the genome-scale metabolic models, phylogenetic trees, the GIFTs and MCI can be found at https://github.com/TheMEMOLab/SalmonMicrobialGenomeAtlas_SMGA.
References
La Rosa, S. L. et al. Glycan processing in gut microbiomes. Curr. Opin. Microbiol. 67, 102143 (2022).
Louis, P. & Flint, H. J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 19, 29–41 (2017).
Egerton, S., Culloty, S., Whooley, J., Stanton, C. & Ross, R. P. The gut microbiota of marine fish. Front. Microbiol. 9, 873 (2018).
Legrand, T. P. R. A., Wynne, J. W., Weyrich, L. S. & Oxley, A. P. A. A microbial sea of possibilities: current knowledge and prospects for an improved understanding of the fish microbiome. Rev. Aquac. 12, 1101–1134 (2020).
Llewellyn, M. S. et al. The biogeography of the Atlantic salmon (Salmo salar) gut microbiome. ISME J. 10, 1280–1284 (2016).
Zhao, R. et al. Salinity and fish age affect the gut microbiota of farmed Chinook salmon (Oncorhynchus tshawytscha). Aquaculture 528, 735539 (2020).
Wang, J. et al. Microbiota in intestinal digesta of Atlantic salmon (Salmo salar), observed from late freshwater stage until one year in seawater, and effects of functional ingredients: a case study from a commercial sized research site in the Arctic region. Anim. Microbiome 3, 14 (2021).
Rasmussen, J. A. et al. Genome-resolved metagenomics suggests a mutualistic relationship between Mycoplasma and salmonid hosts. Commun. Biol. 4, 579 (2021).
Rasmussen, J. A. et al. Co-diversification of an intestinal Mycoplasma and its salmonid host. ISME J. https://doi.org/10.1038/s41396-023-01379-z (2023).
Li, Y., Gajardo, K., Jaramillo-Torres, A., Kortner, T. M. & Krogdahl, Å. Consistent changes in the intestinal microbiota of Atlantic salmon fed insect meal diets. Anim. Microbiome 4, 8 (2022).
Agboola, J. O. et al. Effect of yeast species and processing on intestinal microbiota of Atlantic salmon (Salmo salar) fed soybean meal-based diets in seawater. Anim. Microbiome 5, 21 (2023).
Weththasinghe, P. et al. Modulation of Atlantic salmon (Salmo salar) gut microbiota composition and predicted metabolic capacity by feeding diets with processed black soldier fly (Hermetia illucens) larvae meals and fractions. Anim. Microbiome 4, 9 (2022).
Ringø, E. et al. Lactic acid bacteria associated with the digestive tract of Atlantic salmon (Salmo salar L.). J. Appl. Microbiol. 89, 317–322 (2000).
Wylensek, D. et al. A collection of bacterial isolates from the pig intestine reveals functional and taxonomic diversity. Nat. Commun. 11, 6389 (2020).
Beresford-Jones, B. S. et al. The mouse gastrointestinal bacteria catalogue enables translation between the mouse and human gut microbiotas via functional mapping. Cell Host Microbe 30, 124–138.e128 (2022).
Wong Erin, O.-Y. et al. The CIAMIB: a large and metabolically diverse collection of inflammation-associated bacteria from the murine gut. mBio 13, e02949–02921 (2022).
Saheb Kashaf, S. et al. Integrating cultivation and metagenomics for a multi-kingdom view of skin microbiome diversity and functions. Nat. Microbiol. 7, 169–179 (2022).
Browne, H. P. et al. Culturing of ‘unculturable’ human microbiota reveals novel taxa and extensive sporulation. Nature 533, 543–546 (2016).
Parks, D. H. et al. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38, 1079–1086 (2020).
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114 (2018).
Yarza, P. et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat. Rev. Microbiol. 12, 635–645 (2014).
Cathers, H. S. et al. In silico, in vitro and in vivo characterization of host-associated Latilactobacillus curvatus strains for potential probiotic applications in farmed Atlantic salmon (Salmo salar). Sci. Rep. 12, 18417 (2022).
Jin, Y. et al. Atlantic salmon raised with diets low in long-chain polyunsaturated n-3 fatty acids in freshwater have a Mycoplasma-dominated gut microbiota at sea. Aquac. Environ. Interact. 11, 31–39 (2019).
Bowers, R. M. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725–731 (2017).
Whitman, W. B. (ed.) Bergey’s Manual of Systematics of Archaea and Bacteria (Wiley Online Library, 2015).
Stefanovic, E., Fitzgerald, G. & McAuliffe, O. Advances in the genomics and metabolomics of dairy lactobacilli: a review. Food Microbiol 61, 33–49 (2017).
Li, F. et al. A phylogenomic analysis of Limosilactobacillus reuteri reveals ancient and stable evolutionary relationships with rodents and birds and zoonotic transmission to humans. BMC Biol. 21, 53 (2023).
Parata, L. et al. Age, gut location and diet impact the gut microbiome of a tropical herbivorous surgeonfish. FEMS Microbiol Ecol. 96, fiz179 (2020).
Dvergedal, H., Sandve, S. R., Angell, I. L., Klemetsdal, G. & Rudi, K. Association of gut microbiota with metabolism in juvenile Atlantic salmon. Microbiome 8, 160 (2020).
Fogarty, C. et al. Diversity and composition of the gut microbiota of Atlantic salmon (Salmo salar) farmed in Irish waters. J. Appl. Microbiol. 127, 648–657 (2019).
Heys, C. et al. Neutral processes dominate microbial community assembly in Atlantic salmon, Salmo salar. Appl. Environ. Microbiol. 86, e02283–02219 (2020).
Huyben, D., Roehe, B. K., Bekaert, M., Ruyter, B. & Glencross, B. Dietary lipid:protein ratio and n-3 long-chain polyunsaturated fatty acids alters the gut microbiome of Atlantic salmon under hypoxic and normoxic conditions. Front. Microbiol. 11, 589898 (2020).
Kazlauskaite, R. et al. Deploying an in vitro gut model to assay the impact of the mannan–oligosaccharide prebiotic bio-mos on the Atlantic salmon (Salmo salar) gut microbiome. Microbiol. Spectr. 10, e01953-21 (2022).
Kazlauskaite, R. et al. SalmoSim: the development of a three-compartment in vitro simulator of the Atlantic salmon GI tract and associated microbial communities. Microbiome 9, 179 (2021).
Leeper, A. et al. Feeding black soldier fly larvae (Hermetia illucens) reared on organic rest streams alters gut characteristics of Atlantic salmon (Salmo salar). J. Insects Food Feed 8, 1355–1372 (2022).
Li, Y. et al. Differential response of digesta- and mucosa-associated intestinal microbiota to dietary insect meal during the seawater phase of Atlantic salmon. Anim. Microbiome 3, 8 (2021).
Schaal, P. et al. Links between host genetics, metabolism, gut microbiome and amoebic gill disease (AGD) in Atlantic salmon. Anim. Microbiome 4, 53 (2022).
Villasante, A. et al. Dietary carbohydrate-to-protein ratio influences growth performance, hepatic health and dynamic of gut microbiota in atlantic salmon (Salmo salar). Anim. Nutr. 10, 261–279 (2022).
Drula, E. et al. The carbohydrate-active enzyme database: functions and literature. Nucleic Acids Res. 50, D571–D577 (2022).
Cabello, F. C., Godfrey, H. P., Buschmann, A. H. & Dolz, H. J. Aquaculture as yet another environmental gateway to the development and globalisation of antimicrobial resistance. Lancet Infect. Dis. 16, e127–e133 (2016).
Burridge, L., Weis, J. S., Cabello, F., Pizarro, J. & Bostick, K. Chemical use in salmon aquaculture: a review of current practices and possible environmental effects. Aquaculture 306, 7–23 (2010).
Tofalo, R., Cocchi, S. & Suzzi, G. Polyamines and gut microbiota. Front. Nutr. https://doi.org/10.3389/fnut.2019.00016 (2019).
Wu, G. et al. Arginine metabolism and nutrition in growth, health and disease. Amino Acids 37, 153–168 (2009).
Strandwitz, P. et al. GABA-modulating bacteria of the human gut microbiota. Nat. Microbiol. 4, 396–403 (2019).
Magnúsdóttir, S., Ravcheev, D., de Crécy-Lagard, V. & Thiele, I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front. Genet. 6, 148 (2015).
Rudi, K. et al. Stable core gut microbiota across the freshwater-to-saltwater transition for farmed Atlantic salmon. Appl. Environ. Microbiol. 84, e01974-17 (2018).
Serrato-Salas, J. & Gendrin, M. Involvement of microbiota in insect physiology: focus on B vitamins. mBio 14, e02225–02222 (2022).
Saito, T. et al. Micronutrient supplementation affects transcriptional and epigenetic regulation of lipid metabolism in a dose-dependent manner. Epigenetics 16, 1217–1234 (2021).
Akhtar, M. S. & Ciji, A. Pyridoxine and its biological functions in fish: current knowledge and perspectives in aquaculture. Rev. Fish. Sci. Aquac. 29, 260–278 (2021).
Andersen, S. M. et al. Dietary arginine affects energy metabolism through polyamine turnover in juvenile Atlantic salmon (Salmo salar). Br. J. Nutr. 110, 1968–1977 (2013).
Berge, G. E., Sveier, H. & Lied, E. Effects of feeding Atlantic salmon (Salmo salar L.) imbalanced levels of lysine and arginine. Aquac. Nutr. 8, 239–248 (2002).
Roager, H. M. & Licht, T. R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 9, 3294 (2018).
Huang, D., Alexander, P. B., Li, Q.-J. & Wang, X.-F. GABAergic signaling beyond synapses: an emerging target for cancer therapy. Trends Cell Biol. 33, 403–412 (2023).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Herlemann, D. P. R. et al. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5, 1571–1579 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience 10, giab008 (2021).
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Wu, Y.-W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607 (2016).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316 (2022).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T. T.-Y. ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2017).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Shaffer, M. et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 48, 8883–8900 (2020).
de Nies, L. et al. PathoFact: a pipeline for the prediction of virulence factors and antimicrobial resistance genes in metagenomic data. Microbiome 9, 49 (2021).
Ginestet, C. ggplot2: elegant graphics for data analysis. J. R. Stat. Soc. Ser. A 174, 245–246 (2011).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2022); https://www.r-project.org/
Buck, M., Mehrshad, M. & Bertilsson, S. mOTUpan: a robust Bayesian approach to leverage metagenome-assembled genomes for core-genome estimation. NAR Genom. Bioinform. 4, lqac060 (2022).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Arkin, A. P. et al. KBase: the United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 36, 566–569 (2018).
Heinken, A. et al. Genome-scale metabolic reconstruction of 7,302 human microorganisms for personalized medicine. Nat. Biotechnol. 41, 1320–1331 (2023).
Heinken, A., Magnúsdóttir, S., Fleming, R. M. T. & Thiele, I. DEMETER: efficient simultaneous curation of genome-scale reconstructions guided by experimental data and refined gene annotations. Bioinformatics 37, 3974–3975 (2021).
Noronha, A. et al. The Virtual Metabolic Human database: integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res 47, D614–D624 (2019).
Heirendt, L. et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 14, 639–702 (2019).
Lieven, C. et al. MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 38, 272–276 (2020).
Orth, J. D., Thiele, I. & Palsson, B. Ø. What is flux balance analysis? Nat. Biotechnol. 28, 245–248 (2010).
Lewis, N. E. et al. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol. Syst. Biol. 6, 390 (2010).
Thiele, I., Vlassis, N. & Fleming, R. M. T. fastGapFill: efficient gap filling in metabolic networks. Bioinformatics 30, 2529–2531 (2014).
Kanehisa, M. et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484 (2008).
Heirendt, L., Thiele, I. & Fleming, R. M. T. DistributedFBA.jl: high-level, high-performance flux balance analysis in Julia. Bioinformatics 33, 1421–1423 (2017).
Koziol, A. et al. Mammals show distinct functional gut microbiome dynamics to identical series of environmental stressors. mBio 14, e0160623 (2023).
Leinonen, R., Sugawara, H., Shumway, M. & On behalf of the International Nucleotide Sequence Database CollaborationThe Sequence Read Archive. Nucleic Acids Res. 39, D19–D21 (2011).
Callahan, B. J. et al. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Bozzi, D. et al. Salmon gut microbiota correlates with disease infection status: potential for monitoring health in farmed animals. Anim. Microbiome 3, 30 (2021).
Kopylova, E., Noe, L. & Touzet, H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217 (2012).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
La Rosa, S. L. et al. The human gut firmicute Roseburia intestinalis is a primary degrader of dietary beta-mannans. Nat. Commun. 10, 905 (2019).
La Rosa, S. L. et al. Wood-derived dietary fibers promote beneficial human gut microbiota. mSphere 4, 00554-18 (2019).
Acknowledgements
This work was supported by the Research Council of Norway (project number 300846), the Swedish Research Council Formas (grant number 2019-02336), the European Union’s Horizon 2020 research and innovation programme under the ERA-Net Cofund project BlueBio (grant agreement number 311913) and the Danish National Research Foundation grant CEH-DNRF143. I.T. received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement number 757922) and from the Science Foundation Ireland under grant number 12/RC/2273-P2. Sequencing was performed by the SNP&SEQ Technology Platform in Uppsala, part of the National Genomics Infrastructure (NGI) Sweden and SciLifeLab. Cell sorting and whole-genome amplification was performed at the Microbial Single Cell Genomics Facility (MSCG) at SciLifeLab. Computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under projects SNIC 2021/5-51 and SNIC 2021/22-602. The Orion High Performance Computing Cluster at the Norwegian University of Life Sciences and Saga HPC Sigma2—the National Infrastructure for High Performance Computing and Data Storage in Norway are acknowledged for providing computational resources that have contributed to meta-omics analyses described in this study. We acknowledge Claudia Bergin at the SciLifeLab Microbial Single Cell Genomics Facility for support with cell sorting, genome amplification and library preparation.
Author information
Authors and Affiliations
Contributions
P.B.P., S.L.L.R., A.V.-P.L., S.R.S., S.B. and T.R.H. designed the study. Isolation experiments and genomics analysis of the cultured microorganisms were carried out by A.V.-P.L., S.L.L.R. and S.M.J. Metagenomic analyses were performed by A.V.-P.L. and M.H. Culture experiments and untargeted metabolomic analyses were carried out by S.L.L.R. Constraint-based metabolic models were generated by T.H., B.W. and I.T. M.H. and S.G. conducted amplicon sequencing analyses. C.R.K., K.R., L.S., J.A.R., M.T.L. and S.B. obtained isolates and generated shotgun sequencing data. The draft of this paper was written by S.L.L.R., P.B.P., M.H., A.V.-P.L. and T.H. All authors contributed to the editing of the text and content and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Microbiology thanks Hilary Browne, Matthew Olm and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Strategy for the generation of the SMGA.
Digesta samples were collected from 107 farmed and 70 wild fish either at the freshwater or seawater stage. Genomic and metagenomic datasets were combined to generate a collection of 211 salmon gut microbial genomes. Green boxes indicate the number of genomes from bacterial isolates or bacterial MAGs obtained using two different approaches in this study. Turquoise boxes indicate the number of genomes for cultured isolates or bacterial MAGs from publicly available studies. For a description of the different assembly strategies, see the Methods section.
Extended Data Fig. 2 16S rRNA gene-based phylogenetic tree illustrating diversity and origin of SMGA’s bacterial genomes as well as identified close relatives in the Silva database.
The cladogram depicts the taxonomical position of all the sequences coloured by genus (outer ring). The sequence ID of each organism (in the SMGA and Silva database) used in the phylogeny are printed in the top of the outer ring. Grey dots in the cladogram indicates a Bootstrap support higher than 70 %. The genome of Prochlorococcus marinus subsp. marinus CCMP1375 (Silva ID: AE017126) was used as an outgroup. Scale bar indicates 5% estimated sequence divergence.
Extended Data Fig. 3 Coverage of the SMGA in shotgun metagenomic datasets generated from salmon digesta samples and mOTUs.
a) Percentage of metagenomic reads mapped against all reconstructed genomes and MAGs in the SMGA examined across 562 datasets (represented by a data point) from four publicly available Bioprojects. Mean values for each Bioproject are indicated on top of each boxplot. The boxes represent the interquartile range, with the central bars being the medians, and the whiskers denoting the lowest and highest values. Coverage % for each dataset can be found in Supplementary Table 1. The SMGA recruited 60.52% of metagenomic reads from a seawater dataset, 61.03% metagenomic reads from a flow-through system dataset, as well as 2.83% and 7.22% reads from a recirculated aquaculture system and a seawater land cages dataset, respectively. b) Pan- genome sizes of species-like mOTUs. The 211 SMGA genomes were clustered into 62 mOTUs (x-axis) based on 95% ANI. Bar heights indicate the number of protein clusters within the core and accessory genome of each mOTU.
Extended Data Fig. 4 Functional ordination of bacterial genomes from the SMGA and genome-inferred functional traits (GIFTs) abundance.
a) Ordination using a t-Distributed Stochastic Neighbor Embedding (t-SNE) analysis for clustering GIFTs values of the SMGA’s bacterial genomes. GIFTs values indicate functional capabilities of bacteria based on their genome content. These metrics are computed for individual functions within a genome, representing the proportion of biochemical reactions mediated by the genes to fulfill specific metabolic tasks. Genomes are colored by taxonomic Order. b) Identical ordination but genomes colored by their Metabolic Capacity Index (MIC). Color gradient indicates the increase of MIC; increasing MIC can be observed from bottom to top. Low-MCI: MCI < 0.15; mid-MCI: 0.15 < MCI < 0.3; high-MCI: MCI > 0.3. A PERMANOVA (Permutational Multivariate Analysis of Variance) was conducted to analyse differences in the MCI across genomes of bacteria collected from fish in fresh and seawater. PERMANOVA results are reported under the MCI legend, ** indicates a significance of p < 0.01. The PERMANOVA was two-sided, and no adjustments were made for multiple comparisons. c) Heatmap showing the abundance of GIFTs associated with a particular metabolic function (columns) in each bacterial genome from the SMGA (rows). Bacterial genomes were hierarchically clustered according to their GIFTs abundance (left dendrogram).
Extended Data Fig. 5 Detecting genomes from the SMGA in publicly available datasets.
The detection of isolate genomes and MAGs from the SMGA (y-axis) in selected publicly available 16S rRNA gene amplicon datasets (x-axis) based on alignment of 16S rRNA gene sequences. 16S rRNA gene detection is coloured based on the % identity of the gene alignment. At a 97% identity level to amplicon sequence variants (ASVs), 144 out of 146 SMGA bacteria were detected in publicly available 16S rRNA gene datasets from either in vivo trials or in vitro models with salmon gut microbial communities. This included prevalent genera such as Carnobacterium, Lactobacillus, Flavobacteria, Photobacterium, Shewanella, Vibrio and Aliivibrio that are routinely observed in salmon microbiome research1,2,3,4.
Extended Data Fig. 6 pFBA model predictions on a complex medium.
a) Heatmap of the normalised mean subsystem fluxes in a complex medium across all 211 strain models. The distributions of the mean subsystem fluxes have been averaged for each microbial genus. In addition, the metabolic subsystems have been ordered from most active (top) to least active (bottom). The heatmap cell colours represent the relative subsystem-level metabolic activity, which is defined as the mean of absolute flux values scaled by the sum of predicted flux values in the models. The values in each column sum up to 100%. Subsystems without flux in any of the models were further removed for this visualisation. This panel shows that the most active subsystems were related to energy metabolism and nucleotide synthesis and that substantial heterogeneity exists between the genera. Note that high fluxes in a subsystem do not mean that a subsystem is more important for cellular growth as all growth-associated reactions fluxes are needed to Panels b) and c) show heatmaps of the pFBA predicted exchange fluxes for the 25 metabolites with the highest predicted consumption rates (b) and 25 metabolites with the highest predicted secretion rates (c) across all 211 models. The shown fluxes represent the mean exchange flux for each microbial genus. Exchange fluxes below zero indicate a net metabolic uptake, while fluxes above zero indicate a predicted net secretion of a metabolite. Metabolites that were consumed the most on average include water, nitrate, L-malate, and D-glucose. Hydrogen, acetate, and formate were among the most secreted metabolites on average. Together, these simulations show that the metabolic models reflected expected metabolic properties, for example, higher metabolic fluxes in energy metabolism and nucleotide synthesis, of growing microbes. Note that these predictions are not necessarily representative of metabolic features of these microbes when directly in the salmon gut environment.
Extended Data Fig. 7 Heatmaps on agreements between measured consumed (a) and secreted (b) metabolites and predicted model capabilities in an unlimited aerobe medium.
The yellow cells show metabolites that were not consumed or not secreted in vitro. Turquoise cells indicate metabolites that were consumed or secreted in vitro but could not be consumed or secreted by the metabolic models, that is, the predictions were false negatives. Finally, the blue cells show which metabolites were also consumed or secreted in vitro and could be consumed or secreted by the metabolic models, that is, the true positives. The Photobacterium phosphoreum S39_bc34 metabolic model captured 78% of the 55 measured in vitro metabolic exchanges. Pseudomonas E_sp_S3_bc03 captured 75% of the 65 in vitro metabolic exchanges, while Serratia liquefaciens S38_bc38 captured 79% of the 68 in vitro exchanged metabolic exchanges. The false negative predictions were likely due to incomplete pathways in the respective strain-specific metabolic models and require further refinement.
Extended Data Fig. 8 CAZyme profiles of the 211 salmon microbial genomes and MAGs in the SMGA.
Heatmap showing the presence of CAZy families (listed on the righthand y-axis) arranged into the different glycan substrate categories (listed on lefthand y-axis) found in each genome that are arranged in taxonomic orders (x-axis). The presence of CAZy genes is denoted by grey-black boxes that are weighted for copy number. CAZy families that are not detected are represented by a white box. GH: glycoside hydrolase, PL: polysaccharide lyase, AA: auxiliary activity. While the dominant CAZy family within the SMGA was GH13, prevalent enzymes in Enterobacterales, Lactobacillales and Pseudomonadales were GH18 and GH19 (followed by GH20), which enable microorganisms to depolymerize chitin through the hydrolytic utilization pathway5. Intriguingly, AA10 LPMOs, that have been shown to be involved in an alternative (oxidative) chitin utilization pathway6, were detected in the genomes of Enterobacterales, Lactobacillales and a few Pseudomonadales. CAZymes involved in utilization of terrestrial and marine plant-derived carbohydrates (for example beta-mannan, beta-glucans, xylans, cello-oligosaccharides, manno-oligosaccharides and algal polysaccharides) included GH1, GH2, GH3, GH5, GH8, GH9, GH10, GH16, GH26, GH36, GH43 and GH94 among others5. In addition, CAZymes belonging to the families GH28, GH35, GH78, GH105, GH147, PL2, PL9 and PL22 for deconstruction of the plant pectic polysaccharide rhamnogalacturonan-I were detected in some Enterobacterales and Lactobacillales genomes5. A few Pseudomonadota and Bacteroidota genomes harboured genes encoding CAZymes for depolymerization of host mucin-derived oligosaccharides, including GH29, GH33, GH109, GH112 and GH1295.
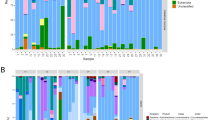
Extended Data Fig. 9 Heatmap illustrating the application of the SMGA as a database to map metatranscriptomes from gut samples.
a) Variation in gene expression of all bacterial genes in the SMGA database (x-axis) and b) a subset of three Enterobacterales (Photobacterium phosphoreum S39_bc34, Pseudomonas_E sp. S3_bc03 and Serratia liquefaciens S38_bc38) in metatranscriptomes generated from gut samples obtained from 33 growing fish fed a standard commercial diet and collected at different life stages (y-axis). T0: 30 g fish (parr), freshwater; T1, 90 g fish (pre-smolt), freshwater; T2, 130 g fish (smolt), freshwater; T3, 300 g fish (adult), seawater.
Extended Data Fig. 10 Examples of the gating strategy used for flow cytometric data.
Examples of the gating strategy used for flow cytometric cell sorting. Cells were stained with SYBR Green I, gated based on forward scatter (x-axes) and fluorescence intensity at 488/530 nm excitation/emission (y-axes) and sorted into 384-well plates by collecting 1 - 200 events per well. Samples were further processed to obtain host-depleted mini-metagenomes, as described in the Method section.
Supplementary information
Supplementary Information
Extended Data Figures 1–10.
Supplementary Table 1
Overview of the SMGA content and its presence in other metagenomic data.
Supplementary Table 2
Metabolomic predictions and models of the SMGA.
Supplementary Table 3
Overview of the in vitro metabolomic reactions of the SMGA.
Supplementary Table 4
CAZy genes encoded in the SMGA.
Supplementary Table 5
Number of expressed genes in terms of TPM in different genomes across different phyla in the SMGA.
Supplementary Table 6
Particularly expressed genes, in terms of TPM, from members of the Lactobacillales, Photobacterium, Serratia and Pseudomonas genomes of the SMGA.
Supplementary Table 7
Untargeted metabolomic values of the SMGA.
Supplementary Table 8
Details about the sequencing depth and mapping rates to non-host and host genome (Salmo salar) for each salmon gut metagenome samples.
Supplementary Dataset 1
Full reports on the benchmarking results on the draft and refined reconstructions from all 211 genome assemblies in the SMGA.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vera-Ponce de León, A., Hensen, T., Hoetzinger, M. et al. Genomic and functional characterization of the Atlantic salmon gut microbiome in relation to nutrition and health. Nat Microbiol 9, 3059–3074 (2024). https://doi.org/10.1038/s41564-024-01830-7
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41564-024-01830-7
This article is cited by
-
A bacterial and viral genome catalogue from Atlantic salmon highlights diverse gut microbiome compositions at pre- and post-smolt life stages
Animal Microbiome (2025)
-
The need for high-resolution gut microbiome characterization to design efficient strategies for sustainable aquaculture production
Communications Biology (2024)
