
Abstract
Advancements in single-atom-based catalysts are crucial for enhancing oxygen evolution reaction (OER) performance while reducing precious metal usage. A comprehensive understanding of underlying mechanisms will expedite this progress further. Here we report Ir single atoms coordinated out-of-plane with dimethylimidazole (MI) on CoFe hydroxide (Ir1/(Co,Fe)-OH/MI). This Ir1/(Co,Fe)-OH/MI catalyst, which was prepared using a simple immersion method, delivers ultralow overpotentials of 179 mV at a current density of 10 mA cm−2 and 257 mV at 600 mA cm−2 as well as an ultra-small Tafel slope of 24 mV dec−1. Furthermore, Ir1/(Co,Fe)-OH/MI has a total mass activity exceeding that of commercial IrO2 by a factor of 58.4. Ab initio simulations indicate that the coordination of MI leads to electron redistribution around the Ir sites. This causes a positive shift in the d-band centre at adjacent Ir and Co sites, facilitating an optimal energy pathway for OER.
Similar content being viewed by others

Main
Electrochemical water splitting powered by renewable electricity is a promising green pathway to realizing large-scale hydrogen production1,2. One of the major bottlenecks of water splitting is the oxygen evolution reaction (OER), which is a sluggish reaction involving the transfer of four electrons3. As the breaking of O–H bonds and the formation of subsequent O–O bonds are required, significant overpotentials are needed to achieve sufficiently high current densities4. OER can be speeded up if effective electrocatalysts containing precious metals are used. For instance, commercial OER catalysts are materials based on IrO2 or RuO2 (refs. 5,6). However, these catalysts are far from ideal owing to their high costs linked to high precious material loading, limited stability and significant overpotentials. These factors collectively constrain the commercial viability of this technology6,7.
Over the past two decades, researchers have extensively studied high-performance OER catalysts, focusing on materials containing early-transition metals such as metal oxyhydroxides/hydroxides8,9, perovskite oxides10, metal phosphides11, metal nitrides12, metal sulfides13, metal oxides14, metal borides15 and metal carbides4. Co-, Fe- and Ni-based metal oxyhydroxides/hydroxides have emerged as particularly promising candidates owing to their cost-effectiveness and strong OER activity16,17,18,19. For instance, NiFe, CoFe and NiCo oxyhydroxides/hydroxides show promising OER overpotentials; the reported values in the 220–350 mV (at 10 mA cm−2) range19,20,21 are comparable to those of commercial IrO2/RuO2 (300–380 mV at 10 mA cm−2)20,21. Although these Co/Fe/Ni-based oxyhydroxides/hydroxides have relativity low OER overpotentials, they are still far from the desired target (that is, overpotentials below 200 mV at 10 mA cm−2)22. Several strategies have been used to lower the overpotentials of metal oxyhydroxides/hydroxides, including morphology regulation23, defect engineering24, creation of heterogeneous interfaces25, doping with multiple elements26 and single-atom loading27,28,29.
Loading noble-metal single atoms onto metal oxyhydroxides/hydroxides has proven effective in reducing OER overpotentials to values below 200 mV (refs. 27,28,29). For example, Li et al. developed CoFe hydroxides loaded with atomically dispersed Ru atoms using a simple chemical precipitation method, achieving an OER overpotential of only 198 mV (ref. 27). Zhai et al. incorporated Ru single atoms into defect-rich NiFe hydroxides via an electrodeposition-etching method, further reducing the OER overpotential to 189 mV (ref. 28). Mu et al. used a hydrothermal-soaking process to create hole-rich CoFe hydroxides with atomically dispersed Ru single atoms, achieving OER overpotentials as low as 194 mV (ref. 29). Further research is needed to fully exploit the interplay between single-atom coordination, bonding and catalytic performance.
Researchers in the field of single-atom catalysts have used out-of-plane ligand coordination (axial coordination) to enhance the electrocatalytic activity of various electrochemical reactions30,31,32. For instance, Zhang et al. designed a Pt single-atom catalyst on NiFe hydroxide with chloride coordination using an irradiation-impregnation procedure, which improved water dissociation and increased hydrogen production in alkaline media30. Similarly, Liu et al. enhanced OER performance by coordinating out-of-plane, a phosphate group to a CoN4 site (the CoN4 site is a single Co atom surrounded by four planar nitrogen atoms)32. These studies demonstrate the power of out-of-plane ligand coordination engineering to optimize the performance of single-atom catalysts. Applying this technique to noble-metal single atoms on metal oxyhydroxides/hydroxides can significantly improve OER performance. However, no studies have explored this promising avenue of research in the context of OER.
Herein we prepared Ir single atoms out-of-plane coordinated with dimethylimidazole (MI) on CoFe hydroxide nanosheets (denoted as Ir1/(Co,Fe)-OH/MI). As an OER catalyst, the as-prepared Ir1/(Co,Fe)-OH/MI showed impressively low overpotential, small Tafel slope, high areal activity (based on electrochemically active surface area (ECSA)) and mass activity (based on total catalyst mass). First-principle simulations suggest that this excellent OER performance stems from the unique coordination between the Ir single atoms and MI. This coordination effectively redistributed charge around the Ir sites, which favourably shifted the d-band centres of both Ir and adjacent Co atoms and reduced the reaction energy barrier. When used as overall water splitting electrodes, the Ir1/(Co,Fe)-OH/MI exhibited exciting application potential.
Synthesis and characterization
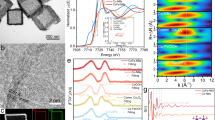
Ir1/(Co,Fe)-OH/MI was prepared using a process that converted the Co-based complex (Co-MI) into CoFe hydroxide. As illustrated in Supplementary Scheme 1, this synthesis procedure required two steps. The synthesis of the Ir1/(Co,Fe)-OH/MI sample started with the growth of Co-MI on nickel foam using an aqueous solution containing Co2+ ions and MI molecules. The resulting Co-MI featured petal-shaped sheets characterized by nanometre-sized pores (Supplementary Fig. 1). As a second step, Co-MI was converted into Ir1/(Co,Fe)-OH/MI following immersion in an ethylene glycol/water solution containing Co2+, Fe3+ and Ir3+ (Supplementary Figs. 2−7 and Table 1). For a more detailed description of the synthesis procedure, refer to ‘Synthesis of Ir1/(Co,Fe)-OH/MI’ in Methods and ‘Synthesis process for Ir1/(Co,Fe)-OH/MI’ section in Supplementary Information. The morphology of Ir1/(Co,Fe)-OH/MI was characterized using transmission electron microscopy (TEM) combined with selected area electron diffraction (SAED) and high-resolution TEM (HR-TEM) (Fig. 1). The TEM images indicate that Ir1/(Co,Fe)-OH/MI has a nanosheet morphology characterized by the presence of many holes, similar to that of Co-MI (Fig. 1a,b and Supplementary Fig. 1). Additional SAED image indicates that Ir1/(Co,Fe)-OH/MI is polycrystalline (Fig. 1a). The HR-TEM image indicates a 0.23 nm lattice fringe of CoFe hydroxide in Ir1/(Co,Fe)-OH/MI (Fig. 1b). Atomic force microscopy (AFM) analysis suggests that the thickness of the Ir1/(Co,Fe)-OH/MI nanosheet is 3–4 nm (Fig. 1c). Aberration-corrected high angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) images highlight the presence of isolated Ir atoms (bright dots marked by pink circles in Fig. 1e), which were dispersed on the surface of (Co,Fe)-OH (Fig. 1d,e and Supplementary Fig. 8). Elemental mapping images show that the dispersion of Co, Fe, Ir, O, C and N atoms (with C and N originating from MI) was uniform across the entire Ir1/(Co,Fe)-OH/MI sample (Fig. 1f,g and Supplementary Figs. 9 and 10). Analysis of atom-overlapping intensity values further confirms atomic dispersion of Ir on (Co,Fe)-OH (Fig. 1h and Supplementary Fig. 11 with the red regions indicating Ir)14. To confirm isolated Ir atoms, the intensity profile in Fig. 1i (from the dashed rectangle in Fig. 1e) shows a prominent Ir peak, visually supporting atomic dispersion. Inductively coupled plasma mass spectrometry (ICP-MS) confirmed 0.33 wt% Ir loading (Supplementary Table 2). Electrodeposited Ir1/(Co,Fe)-OH without MI also showed similar atomically dispersed Ir within porous nanosheets (Supplementary Fig. 12), with 0.30 wt% Ir loading (Supplementary Table 3). For comparison, Ir-free (Co,Fe)-OH/MI and (Co,Fe)-OH were prepared and characterized by TEM (Supplementary Figs. 13 and 14).

a, TEM micrograph with a corresponding SAED image. b, Integrated TEM and HR-TEM images. c, AFM characterization. d,e, HAADF-STEM images. Holes (d). Ir single atoms (e). f, TEM image with corresponding elemental maps. g, High-resolution elemental map. h, Three-dimensional atom-overlapping intensity value representation. i, Intensity profile along the dashed rectangle in e. Note: M in i represents either Co or Fe.
Further structural characterizations are presented in Fig. 2. The X-ray powder diffraction (XRD) patterns of Ir-containing catalysts closely resemble their Ir-free counterparts, confirming the absence of Ir nanoparticles or bulk Ir metal (Fig. 2a and Supplementary Fig. 15). Characteristic layered double hydroxide peaks in the XRD patterns identify CoFe hydroxides as the supports for Ir single atoms33,34,35. X-ray absorption spectroscopy (XAS) was used to probe the electronic structure and local atomic environment of Ir. The Ir L3-edge X-ray absorption near edge structure (XANES) spectra for both Ir1/(Co,Fe)-OH/MI and Ir1/(Co,Fe)-OH show similar peaks, shifted ~0.4 eV higher than Ir foil but lower than IrO2, suggesting an Ir oxidation state between 0 and +4 (refs. 14,36) (Fig. 2b and Supplementary Fig. 16). Linear regression analysis of the first-order derivative zero further refines the Ir oxidation states to +1.02 and +0.91 for Ir1/(Co,Fe)-OH/MI and Ir1/(Co,Fe)-OH, respectively (Fig. 2c and Supplementary Fig. 17). The slightly higher Ir oxidation state in Ir1/(Co,Fe)-OH/MI may be due to partial coordination of Ir with MI.

a, XRD patterns. b, XANES spectra. c, Valence of various Ir species obtained from Ir L3-edge XANES. d, Normalized Ir L3-edge FT-EXAFS spectra. e, Wavelet-transformed spectra of Ir L3-edge FT-EXAFS. f,g, Fitting results of the FT-EXAFS spectra of Ir1/(Co,Fe)-OH/MI. Real part (f). Imaginary part (g).
The Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectra reveal the nature of Ir bonds in Ir1/(Co,Fe)-OH/MI, Ir1/(Co,Fe)-OH, IrO2 and Ir foil (Fig. 2d). In R-space, IrO2 and Ir foil exhibit single peaks at 1.60 Å and 2.60 Å, corresponding to Ir–O and Ir–Ir bonds, respectively. Ir1/(Co,Fe)-OH shows two peaks at 1.65 Å and 2.63 Å, attributed to Ir–O and Ir–Co/Ir–Fe. By contrast, Ir1/(Co,Fe)-OH/MI shows a slightly shifted profile with peaks at 1.68 Å (Ir–O or Ir–N) and 2.68 Å (Ir–Co or Ir–Fe), implying an impact of MI coordination. The wavelet-transformed Ir L3-edge EXAFS spectra of Ir1/(Co,Fe)-OH/MI and Ir1/(Co,Fe)-OH exhibit distinct differences compared with IrO2 and Ir foil (Fig. 2e), further supporting the atomic dispersion of Ir species on CoFe hydroxides, with no evidence of Ir clusters or nanoparticles. EXAFS analysis and structural simulations reveal the coordination structure of Ir to be Ir–CoFe for Ir1/(Co,Fe)-OH and Ir(N)–CoFe for Ir1/(Co,Fe)-OH/MI (Fig. 2f,g and Supplementary Figs. 18−20). Notably, in Ir1/(Co,Fe)-OH/MI, the MI ligand coordinates out-of-plane with the Ir single atom.
OER performance
To investigate the impact of MI–Ir coordination on OER performance, we evaluated the prepared samples electrochemically in 1 M KOH (Fig. 3). The linear sweep voltammetry (LSV) results demonstrate high current densities (up to 700 mA cm−2) and excellent OER activity across all catalysts. Notably, Ir1/(Co,Fe)-OH/MI exhibits the highest activity, followed by (Co,Fe)-OH/MI, Ir1/(Co,Fe)-OH and (Co,Fe)-OH, all outperforming IrO2 and Ni foam (Fig. 3a). At 10 mA cm−2, 100 mA cm−2, 300 mA cm−2 and 600 mA cm−2, the overpotentials of Ir1/(Co,Fe)-OH/MI are 179 mV, 241 mV, 252 mV and 257 mV, respectively. These values are notably lower than those observed for (Co,Fe)-OH/MI, Ir1/(Co,Fe)-OH, (Co,Fe)-OH and IrO2 at the same current densities (Fig. 3b). The Tafel slope of Ir1/(Co,Fe)-OH/MI is 24 mV dec−1, significantly lower than the other catalysts (Fig. 3c). Moreover, the low overpotential (179 mV at 10 mA cm−2) and Tafel slope (24 mV dec−1) of Ir1/(Co,Fe)-OH/MI outperform many reported hydroxide-based noble-metal single-atom electrocatalysts and other state-of-the-art OER catalysts27,28,29,37,38,39,40,41,42,43,44,45 (Fig. 3d and Supplementary Tables 4 and 5). The Faraday efficiency exceeds 98%, indicating near-theoretical O2 production during OER (Supplementary Fig. 21).

a, LSV curves in 1 M KOH at a scanning rate of 5 mV s−1. b, Overpotentials at 10 mA cm−2, 100 mA cm−2, 300 mA cm−2 and 600 mA cm−2. c, Tafel plots. d, Comparison of overpotentials and Tafel plots in reported hydroxide-based noble-metal single-atom OER electrocatalysts. e, Areal activity based on ECSA. f, Activity normalized relative to the catalyst mass. g, TOF from ICP-MS characterization. h, Electrochemical impedance spectroscopy. i, Chronoamperometry measurements of Ir1/(Co,Fe)-OH/MI at 10 mA cm−2, 100 mA cm−2 and 300 mA cm−2.
The catalyst showcased exceptional performance in both areal activity (based on ECSA) and mass activity (based on total catalyst mass) (Fig. 3e,f, Supplementary Figs. 22 and 23, and Table 6). At an overpotential of 250 mV, Ir1/(Co,Fe)-OH/MI achieved an areal activity of 8.20 mA cm−2, surpassing Ir1/(Co,Fe)-OH and commercial IrO2 by factors of 15.5 and 16.4, respectively (Fig. 3e). Furthermore, Ir1/(Co,Fe)-OH/MI showed a mass activity of 329.89 A g−1, exceeding Ir1/(Co,Fe)-OH and IrO2 by factors of 9.8 and 58.4, respectively (Fig. 3f). When normalized to Ir mass, Ir1/(Co,Fe)-OH/MI delivered an impressive 105 A gIr−1, outperforming Ir1/(Co,Fe)-OH by a factor of 8.9 and IrO2 by a remarkable 15,000-fold margin (Supplementary Fig. 24 and Table 7). The turnover frequency (TOF) of Ir1/(Co,Fe)-OH/MI significantly surpassed that of other catalysts, outperforming Ir1/(Co,Fe)-OH and IrO2 by factors of 9.8 and 35.8, respectively (Fig. 3g and Supplementary Tables 2, 3, 8 and 9). In addition, Ir1/(Co,Fe)-OH/MI exhibited the lowest charge transfer resistance among the prepared catalysts (Fig. 3h and Supplementary Table 10). Chronoamperometry tests confirmed the catalyst’s stability, with no significant degradation observed over 10 h at various current densities (10 mA cm−2, 100 mA cm−2 and 300 mA cm−2) (Fig. 3i). Multistep chronopotentiometry consistently supported this stability data across a broad current density range (50–400 mA cm−2) (Supplementary Fig. 25). Remarkably, Ir1/(Co,Fe)-OH/MI maintained stable operation for over 120 h as evidenced by chronoamperometric and chronopotentiometric measurements (Supplementary Fig. 26) as well as post-mortem morphological, chemical and structural characterizations (Supplementary Figs. 27−31).
Density functional theory calculations
To elucidate the OER mechanisms in Ir1/(Co,Fe)-OH and Ir1/(Co,Fe)-OH/MI, we constructed Ir–CoFe and Ir(N)–CoFe atomic models, respectively, based on modified Co hydroxide single-crystal structures46 (Supplementary Fig. 20). Density functional theory (DFT) simulations on these models (Supplementary Figs. 32−37 and ‘DFT software, modules and functions’ in Supplementary Information) revealed distinct OER pathways. In the Ir–CoFe model, only the Ir site showed stable adsorption of OER intermediates (*OH, *O and *OOH), suggesting that OER primarily occurs at Ir, not adjacent Co or Fe sites (Supplementary Figs. 32−34). Conversely, in the Ir(N)–CoFe model, favourable OER energetics were observed at both Ir and adjacent Co sites, while Fe sites were unfavourable owing to unstable intermediate formation (Supplementary Figs. 35−37).
We analysed differential charge density, Bader charge, partial density of states (PDOS) and Gibbs free energies for both Ir–CoFe and Ir(N)–CoFe models (Fig. 4 and Supplementary Figs. 38−41). The Ir(N)–CoFe model exhibited significant charge redistribution around the Ir site owing to out-of-plane coordination with MI (Fig. 4a). Bader charge analysis revealed a greater electron loss at the Ir site for Ir(N)–CoFe (−0.53 e) compared with Ir–CoFe (−0.22 e), indicating a higher Ir valence in Ir(N)–CoFe. This aligns with the XANES-estimated Ir valence of +1.02 for Ir1/(Co,Fe)-OH/MI and +0.91 for Ir1/(Co,Fe)-OH (Fig. 2c and Supplementary Fig. 41). Similarly, Co or Fe sites adjacent to Ir in Ir(N)–CoFe showed greater electron loss than in Ir–CoFe, suggesting increased Co or Fe valence in Ir1/(Co,Fe)-OH/MI compared with Ir1/(Co,Fe)-OH (Supplementary Figs. 41−44). To examine the impact of the electronic structure on the adsorption of OER intermediates, we calculated the corresponding PDOS of Ir, Co and Fe sites. The Ir(N)–CoFe model showed higher d-band centres for both Ir (−1.78 eV) and its adjacent Co (−1.17 eV) compared with their counterparts in the Ir–CoFe model (−2.01 eV at Ir and −1.22 eV at Co). Conversely, the adjacent Fe site in Ir(N)–CoFe exhibited a lower d-band centre (−0.91 eV) than in Ir–CoFe (−0.82 eV) (Fig. 4b−d). On the basis of the d-band centre model, the elevated d-band centres at Ir and adjacent Co in Ir(N)–CoFe suggest more unoccupied orbitals, implying stronger adsorption of OER intermediates at these sites compared with the Ir–CoFe model47,48.

a, Differential charge density of catalysts, where the yellow and green regions represent electronic accumulation and depletion, respectively. b, PDOS of Ir sites. c, PDOS of Co sites. d, PDOS of Fe sites. e, Gibbs free energy diagram of the Ir site in Ir1/(Co,Fe)-OH. f, Gibbs free energy diagram of the Ir site in Ir1/(Co,Fe)-OH/MI. g, Gibbs free energy diagram of the Co site adjacent to Ir in Ir1/(Co,Fe)-OH/MI. h, Proposed 4e− mechanism of OER at the Co site adjacent to Ir in Ir1/(Co,Fe)-OH/MI. Note: the asterisk denotes the active site for adsorption.
To understand the OER process at Ir and adjacent Co sites in Ir1/(Co,Fe)-OH/MI and Ir1/(Co,Fe)-OH, the Gibbs free energies (G) of adsorbed OER intermediates for the Ir(N)–CoFe and Ir–CoFe models were calculated49 (Fig. 4e−g). In alkaline electrolytes, OER follows four steps28: * → *OH, *OH → *O, *O → *OOH and *OOH → * + ½O2. At Ir sites, only the first step (* → *OH) was calculated to be spontaneous (ΔG < 0) at 0 V for both models. However, Ir(N)–CoFe exhibits a more negative ΔG for the first step than Ir–CoFe, indicating stronger OH adsorption on Ir1/(Co,Fe)-OH/MI (Fig. 4e,f). At 1.23 V, both models exhibit two spontaneous (that is, * → *OH and *OH → *O) and two unfavourable (that is, *O → *OOH and *OOH → * + ½O2) steps. The energy barriers for the unfavourable steps are lower for Ir(N)–CoFe (2.35 eV) compared with Ir–CoFe (2.76 eV), suggesting higher electrocatalytic activity for the former (Fig. 4e,f). Increasing the applied potential drives the transition of non-spontaneous steps (*O → *OOH and OOH → * + ½O2) towards spontaneity. Specifically, in Ir–CoFe, the *O → *OOH step becomes thermodynamically favourable at 2.43 V (Fig. 4e). By contrast, Ir(N)–CoFe exhibits favourable energetics for both *O → OOH and OOH → * + ½O2 at 2.88 V (Fig. 4f). These simulations indicate that the Ir site of Ir1/(Co,Fe)-OH/MI has enhanced OER activity compared with that of Ir1/(Co,Fe)-OH.
At the Co site adjacent to Ir in the Ir(N)–CoFe model, the OER process is non-spontaneous at 0 V (Fig. 4g). At 1.23 V, one step (that is, * → *OH) is spontaneous, while three steps (that is, *OH → *O, *O → *OOH and *OOH → * + ½O2) are not (Fig. 4g). Importantly, the total energy barrier at this Co site adjacent to Ir is only 0.78 eV, a value lower than that computed for the Ir site (2.35 eV for Ir(N)–CoFe and 2.76 eV for Ir–CoFe), indicating a more efficient OER pathway around Co than the Ir site (Fig. 4e−g). Upon increasing the potential to 1.59 V, the previously three non-spontaneous steps at the Co site become spontaneous, implying greater OER reactivity (lower overpotentials) at this site relative to the Ir site (Fig. 4f,g). Correspondingly, the 4e− mechanism for OER at the Co site is outlined in Fig. 4h. The lower computed overpotential for the model corresponding to Ir1/(Co,Fe)-OH/MI relative to the one for Ir1/(Co,Fe)-OH is consistent with the trend observed experimentally (Fig. 3b and Supplementary Fig. 45).
In summary, out-of-plane coordination of Ir atoms with MI molecules increases the oxidation states of Ir (+1.02) and Co (+2.11) compared with Ir1/(Co,Fe)-OH (+0.91 for Ir and +2.05 for Co) (Fig. 2c and Supplementary Fig. 42). This increase aligns with the reduced charge density around Ir and Co sites, as evidenced by the analysis of the differential and Bader charges (Fig. 4a and Supplementary Figs. 38−41). These changes induce a positive shift in the d-band centres of Ir and adjacent Co sites (Fig. 4b,c), promoting adsorption of OER intermediates (Fig. 4e–g) and enhancing catalytic performance. For instance, the Ir1/(Co,Fe)-OH/MI catalyst exhibits lower overpotentials and Tafel slope (Fig. 3a–d) relative to Ir1/(Co,Fe)-OH. Moreover, its activity normalized by Ir mass exceeds that of commercial IrO2 by more than four orders of magnitude (Supplementary Fig. 24). These findings indicate that out-of-plane coordination with MI molecules is a suitable approach for tuning electronic structure and improving OER activity in noble-metal single-atom catalysts on metal oxyhydroxide/hydroxide supports.
Electrochemical performance of overall water splitting
To assess the practical feasibility of the Ir1/(Co,Fe)-OH/MI catalyst, we evaluated its overall water splitting performance in a two-electrode cell (Fig. 5). The asymmetric Ir1/(Co,Fe)-OH/MI||20% Pt/C cell exhibited superior performance below 500 mA cm−2 compared with both the symmetric Ir1/(Co,Fe)-OH/MI||Ir1/(Co,Fe)-OH/MI cell and the asymmetric IrO2||20% Pt/C cell (Fig. 5a,b and Supplementary Fig. 46). However, the symmetric cell excelled at current densities above 500 mA cm−2 (Fig. 5b). Specifically, at 10 mA cm−2, 300 mA cm−2 and 500 mA cm−2, the asymmetric Ir1/(Co,Fe)-OH/MI||20% Pt/C cell achieved water splitting voltages of 1.44 V, 1.70 V and 1.76 V, respectively, outperforming the symmetric Ir1/(Co,Fe)-OH/MI||Ir1/(Co,Fe)-OH/MI cell (Fig. 5c). At higher current densities (600 mA cm−2 and 800 mA cm−2), the asymmetric Ir1/(Co,Fe)-OH/MI||20% Pt/C cell reached 1.79 V and 1.80 V, while the symmetric Ir1/(Co,Fe)-OH/MI||Ir1/(Co,Fe)-OH/MI cell showed slightly lower voltages of 1.78 V and 1.79 V. This Ir1/(Co,Fe)-OH/MI catalyst shows high overall water splitting performance compared with reported OER electrocatalysts (Fig. 5d, Supplementary Fig. 47 and Tables 11 and 12). The asymmetric Ir1/(Co,Fe)-OH/MI||20% Pt/C cell operated stably for 120 h at 300 mA cm−2, and the symmetric Ir1/(Co,Fe)-OH/MI||Ir1/(Co,Fe)-OH/MI cell functioned for 120 h at 700 mA cm−2 (Fig. 5e). Even in a two-electrode flow cell, the symmetric Ir1/(Co,Fe)-OH/MI||Ir1/(Co,Fe)-OH/MI cell maintained stability for 100 h at 800 mA cm−2 (Supplementary Fig. 48). To further demonstrate practical applicability, an anion exchange membrane (AEM) water electrolyser using Ir1/(Co,Fe)-OH/MI as both anode and cathode exhibited a lower voltage at 500 mA cm−2 than an IrO2||20% Pt/C electrolyser (Supplementary Fig. 49 and Table 13). Impressively, this electrolyser operated stably for over 150 h at 500 mA cm−2 with negligible degradation.

a, Schematic diagram of water splitting. b, LSV curves in 1 M KOH at a scanning rate of 5 mV s−1. c, Water splitting voltage at 10 mA cm−2, 300 mA cm−2, 500 mA cm−2, 600 mA cm−2 and 800 mA cm−2. d, Comparison of water splitting voltage (10 mA cm−2) in reported hydroxide-based OER electrocatalysts. e, Chronoamperometry measurements at 300 mA cm−2 and 700 mA cm−2 for 120 h.
Generalizing the preparation method
Our results show that coordinating MI improves the OER performance of Ir single atoms supported on CoFe hydroxide. We achieved this coordination using a simple, two-step method conducted under mild conditions (Supplementary Scheme 1). Extending our approach, we successfully synthesized Pt1/(Co,Fe)-OH/MI (Fig. 6a−c and Supplementary Figs. 50 and 51), Pd1/(Co,Fe)-OH/MI (Fig. 6d−f and Supplementary Figs. 52 and 53) and Ru1/(Co,Fe)-OH/MI (Fig. 6g−i and Supplementary Figs. 54 and 55), all of which exhibited the desired morphology: atomically dispersed single atoms on porous hydroxide supports (Figs. 1 and 6). This versatile approach opens a new platform of MI-coordinated, noble-metal, single-atom catalysts for exploring a wide range of catalytic reactions.

a–c, Pt1/(Co,Fe)-OH/MI. d–f, Pd1/(Co,Fe)-OH/MI. g–i, Ru1/(Co,Fe)-OH/MI. TEM images (a,d,g). HR-TEM images (b,e,h). HAADF-STEM images (c,f,i).
Conclusion
This article introduces a straightforward two-step method to prepare Ir single atoms on CoFe hydroxide supports, coordinated with the MI molecule under mild conditions. The resulting catalyst, Ir1/(Co,Fe)-OH/MI, features atomically dispersed Ir atoms with enhanced valence, confirmed by HAADF-STEM and XANES analyses. The catalyst showed exceptional OER performance, with an ultralow overpotential of 179 mV at 10 mA cm−2 and superior stability even at high current densities. The superior performance compared with commercial IrO2 and similar catalysts is attributed to the precise coordination between Ir and MI, which optimizes charge distribution and adjusts d-band centres, thereby enhancing the adsorption of OER intermediates. When used in water splitting systems, Ir1/(Co,Fe)-OH/MI exhibited low operating voltages and stable long-term performance across various cell configurations. In addition, this preparation method is versatile, can be extended to other noble metals such as Pt, Pd and Ru, and offers a new platform for studying catalytic mechanisms across multiple applications.
Methods
Synthesis of Co-MI
First, 2-methylimidazole (20 mmol, MI) was dissolved into deionized water (50 ml) to form a solution, denoted as ‘solution A’. Cobalt nitrate (2.5 mmol, Co(NO3)2∙6H2O) was dissolved in deionized water (50 ml) to form another solution, denoted as ‘solution B’. Then, ‘solution B’ was poured into ‘solution A’ to form ‘solution C’. Next, Ni foam (1 × 2 cm2) was put into ‘solution C’. After soaking for 6 h, the Co-MI (that is, Co-MOF) electrode was obtained. Subsequently, the electrode was rinsed with deionized water.
Synthesis of Ir1/(Co,Fe)-OH/MI
Ir1/(Co,Fe)-OH/MI was synthesized using a straightforward immersion technique conducted at ambient temperature and pressure. A solution was prepared by dissolving cobalt nitrate (Co(NO3)2∙6H2O, 1 mmol), iron nitrate (Fe(NO3)3∙6H2O, 1 mmol) and an Ir3+ aqueous solution (7.14 mg ml−1, 350 µl) in a mixture of deionized water (20 ml) and ethylene glycol (10 ml). The pH value of this solution was kept at ~4.5 by adding an appropriate amount of 2 mol l−1 NaOH. Subsequently, the pre-prepared Co-MI electrode was immersed in this solution for 20 h. Following immersion, the Ir1/(Co,Fe)-OH/MI was obtained and washed three times using deionized water. The mass loading of the Ir1/(Co,Fe)-OH/MI catalyst loaded on Ni foam was 0.70 mg cm−2. For comparative purposes, Ir1/(Co,Fe)-OH, a material without MI, was synthesized. This was achieved through a simple electrodeposition method using an aqueous solution of Co2+/Fe3+/Ir3+. During this process, an electrodeposition potential of −1 V (versus a saturated calomel reference electrode) was maintained, and the electrodeposition time was set at 240 s. In addition, samples without Ir atoms, specifically (Co,Fe)-OH/MI and (Co,Fe)-OH, were synthesized using the above two methods. The active material average mass loadings of the (Co,Fe)-OH/MI, Ir1/(Co,Fe)-OH and (Co,Fe)-OH electrodes were recorded as 0.70 mg cm−2, 0.71 mg cm−2 and 0.73 mg cm−2, respectively. The impact of Ni foam thickness, Co/Fe ratio and Ir species’ mass fractions on the OER performance of Ir1/(Co,Fe)-OH/MI were investigated for optimization purposes (see Supplementary Figs. 56 and 57).
Materials characterization
SEM images were obtained using a Thermo Scientific APREO S microscope. TEM, HR-TEM, SAED and elemental mapping images were acquired with a JEOL JEM-F200 transmission electron microscope. AFM images were captured using a DIMENSION ICON CLOSED LOOP SPM. HAADF-STEM images, along with corresponding elemental images, were obtained using a transmission electron microscope (FEI/Thermo Scientific Themis Z) equipped with double spherical aberration correction, operating at 200 kV (the beam current was 44 pA, and the dwell time was 2 μs). The three-dimensional atom-overlapping intensity value representation was obtained by first extracting the grey value of each pixel of the HAADF-STEM image using the Velox software part of the spherical aberration correction STEM software packages, followed by fitting the obtained grey values using intensities ranging from 0 to 100. The two-dimensional atom-overlapping intensity map representation is the top view (or contour plot) of the three-dimensional plot. XRD patterns were recorded using an Ultima IV X-ray powder diffractometer, using Cu Kα radiation with a wavelength of 0.154056 nm. In situ Raman spectra were collected on a laser micro confocal Raman spectrometer (inVia, Renishaw). XAS spectra were used to examine the electronic structures and the local atomic environments of the Ir, Co and Fe elements. More details on the XAS technique are provided in Supplementary Information. The atomic ratios of the as-prepared samples were determined using an Agilent 7900 inductively coupled plasma mass spectrometer.
Electrochemical measurements for OER
The electrochemical measurements of as-prepared catalysts were conducted on an electrochemical workstation (CHI760E) with a three-electrode cell system. The Ir1/(Co,Fe)-OH/MI, (Co,Fe)-OH/MI, Ir1/(Co,Fe)-OH and (Co,Fe)-OH catalysts grown on Ni foam were used as the working electrodes. A graphite rod was utilized as the counter electrode. The Hg/HgO (1 M KOH, 0.098 V at 25 °C) electrode acted as the reference electrode. For comparison purposes, IrO2 and 20% Pt/C inks were dropped on Ni foam to produce the IrO2 and 20% Pt/C electrodes (with a mass loading of 0.7 mg cm−2), respectively. Before the electrochemical measurements, the working electrodes required electrochemical activation using cyclic voltammetry for 50 cycles at a scan rate of 100 mV s−1 in a potential range of 0.2–1.0 V (versus Hg/HgO). The electrochemical activation and working electrode measurements were performed in an oxygen-saturated 1 M KOH aqueous electrolyte at 25 °C. The LSV curves of OER and hydrogen evolution reaction were all measured under a scanning rate of 5 mV s−1 with 100% iR compensation. The electrochemical impedance spectroscopy spectra were tested in the frequency range of 0.1 Hz to 100 kHz. Chronoamperometry was performed on Ir1/(Co,Fe)-OH/MI at 10 mA cm−2, 100 mA cm−2, 300 mA cm−2 and 600 mA cm−2 in a three-electrode cell system. In this paper, the potentials (versus reversible hydrogen electrode, E(RHE)) are given using the formula E(RHE) = E(Hg/HgO) + 0.098 + 0.059 × pH, where pH = 14 in 1 M KOH solution.
Overall water splitting performance was measured in a two-electrode membrane-free cell system and a two-electrode flow cell system at 25 °C. The LSV curves of overall water splitting were measured in an oxygen-saturated 1 M KOH aqueous electrolyte at a scanning rate of 5 mV s−1. The chronoamperometry measurements of overall water splitting at 300 mA cm−2 and 700 mA cm−2 were performed in a two-electrode membrane-free cell system. The chronoamperometry measurement of overall water splitting at 800 mA cm−2 was performed in a two-electrode flow cell system.
First-principle simulations
The specific software, modules and functions used for DFT are detailed in Supplementary Information. The (Co,Fe)-OH models were constructed based on the single-crystal structure of Co(OH)2. The primitive cell model of (Co,Fe)-OH underwent full optimization. The 4 × 2 × 1 supercell from the (001) surface was sliced from the bulk (Co,Fe)-OH. To create a model of the Ir single-atom-anchored (Co,Fe)-OH catalyst, an Ir atom was positioned directly on the catalyst surface, coordinating with various metallic and oxygen atoms. The Ir1/(Co,Fe)-OH/MI model was constructed by introducing an MI group at the apex of the Ir atom. Throughout the structural optimizations, all atoms were permitted to relax. A vacuum layer of 15 Å was used to prevent periodic interactions in the direction perpendicular to the catalyst’s surface. The convergence criterion for geometry optimization was set with the energy and force converging to 1.0 × 10−5 eV per atom and 0.01 eV Å−1, respectively. The differential charge density, projected density of states and Gibbs free energy calculations related to the OER of the constructed models are described in the ‘DFT software, modules and functions’ section of Supplementary Information.
Data availability
The data that support the main findings are available in the main text and Supplementary Information. Source data are provided with this paper.
References
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294–303 (2012).
Zhang, B. et al. Homogeneously dispersed multimetal oxygen-evolving catalysts. Science 352, 333–337 (2016).
Seitz, L. C. et al. A highly active and stable IrOx/SrIrO3 catalyst for the oxygen evolution reaction. Science 353, 1011–1014 (2016).
Li, S. et al. Oxygen-evolving catalytic atoms on metal carbides. Nat. Mater. 20, 1240 (2021).
Jiang, K. et al. Dynamic active-site generation of atomic iridium stabilized on nanoporous metal phosphides for water oxidation. Nat. Commun. 11, 2701 (2020).
Tobias, R., Nhan, N. H., Detre, T., Robert, S. & Peter, S. Electrocatalytic oxygen evolution reaction in acidic environments—reaction mechanisms and catalysts. Adv. Energy Mater. 7, 1601275 (2017).
Cherevko, S. et al. Oxygen and hydrogen evolution reactions on Ru, RuO2, Ir, and IrO2 thin film electrodes in acidic and alkaline electrolytes: a comparative study on activity and stability. Catal. Today 262, 170–180 (2016).
Yan, Z. et al. Anion insertion enhanced electrodeposition of robust metal hydroxide/oxide electrodes for oxygen evolution. Nat. Commun. 9, 2373 (2018).
Huang, W. et al. Ligand modulation of active sites to promote electrocatalytic oxygen evolution. Adv. Mater. 34, 2200270 (2022).
Yang, L. et al. Efficient oxygen evolution electrocatalysis in acid by a perovskite with face-sharing IrO6 octahedral dimers. Nat. Commun. 9, 5236 (2018).
Huang, C. J. et al. A review of modulation strategies for improving catalytic performance of transition metal phosphides for oxygen evolution reaction. Appl. Catal. B Environ. 325, 122313 (2023).
Batool, M., Hameed, A. & Nadeem, M. A. Recent developments on iron and nickel-based transition metal nitrides for overall water splitting: a critical review. Coord. Chem. Rev. 480, 215029 (2023).
Mohili, R., Hemanth, N. R., Jin, H., Lee, K. & Chaudhari, N. Emerging high entropy metal sulphides and phosphides for electrochemical water splitting. J. Mater. Chem. A 11, 10463–10472 (2023).
Zhu, Y. et al. Iridium single atoms incorporated in Co3O4 efficiently catalyze the oxygen evolution in acidic conditions. Nat. Commun. 13, 7754 (2022).
Dincă, M., Surendranath, Y. & Nocera, D. G. Nickel-borate oxygen-evolving catalyst that functions under benign conditions. Proc. Natl Acad. Sci. USA 107, 10337 (2010).
Bauer, J., Buss, D. H., Harms, H.-J. & Glemser, O. The electrochemical behavior of positive cobalt/aluminum and cobalt/iron hydroxide electrodes. J. Electrochem. Soc. 137, 173–178 (1990).
Benson, P., Briggs, G. W. D. & Wynne-Jones, W. F. K. The cobalt hydroxide electrode—I. Structure and phase transitions of the hydroxides. Electrochim. Acta 9, 275–276 (1964).
Benson, P., Briggs, G. W. D. & Wynne-Jones, W. F. K. The cobalt hydroxide electrode—II. Electrochemical behavior. Electrochim. Acta 9, 281–288 (1964).
Babar, P. et al. Cobalt iron hydroxide as a precious metal-free bifunctional electrocatalyst for efficient overall water splitting. Small 14, 1702568 (2018).
Wu, J. et al. Constructing electrocatalysts with composition gradient distribution by solubility product theory: amorphous/crystalline CoNiFe-LDH hollow nanocages. Adv. Funct. Mater. 33, 2300808 (2023).
Enkhtuvshin, E. et al. Surface reconstruction of Ni–Fe layered double hydroxide inducing chloride ion blocking materials for outstanding overall seawater splitting. Adv. Funct. Mater. 33, 2214069 (2023).
Li, Z. et al. High-density cationic defects coupling with local alkaline-enriched environment for efficient and stable water oxidation. Angew. Chem. Int. Ed. 62, e2022178 (2023).
Arshad, F. et al. Microwave-assisted growth of spherical core-shell NiFe LDH@CuxO nanostructures for electrocatalytic water oxidation reaction. Int. J. Hydrog. Energy 48, 4719–4727 (2023).
Li, H. et al. Efficient electrocatalysis for oxygen evolution: W-doped NiFe nanosheets with oxygen vacancies constructed by facile electrodeposition and corrosion. Chem. Eng. J. 452, 139104 (2023).
Cui, H. et al. Synergistic electronic interaction between ruthenium and nickel-iron hydroxide for enhanced oxygen evolution reaction. Rare Met. 41, 2606–2615 (2022).
Kim, S. J. et al. Zn-doped nickel iron (oxy)hydroxide nanocubes passivated by polyanions with high catalytic activity and corrosion resistance for seawater oxidation. J. Energy Chem. 81, 82–92 (2023).
Li, P. et al. Boosting oxygen evolution of single-atomic ruthenium through electronic coupling with cobalt-iron layered double hydroxides. Nat. Commun. 10, 1711 (2019).
Zhai, P. et al. Engineering single-atomic ruthenium catalytic sites on defective nickel-iron layered double hydroxide for overall water splitting. Nat. Commun. 12, 4587 (2021).
Mu, X. et al. Breaking the symmetry of single-atom catalysts enables an extremely low energy barrier and high stability for large-current-density water splitting. Energy Environ. Sci. 15, 4048 (2022).
Zhang, T. et al. Pinpointing the axial ligand effect on platinum single-atom-catalyst towards efficient alkaline hydrogen evolution reaction. Nat. Commun. 13, 6875 (2022).
Li, X. et al. Convergent paired electrosynthesis of dimethyl carbonate from carbon dioxide enabled by designing the superstructure of axial oxygen coordinated nickel single-atom catalysts. Energy Environ. Sci. 16, 502–512 (2023).
Liu, Y. et al. Axial phosphate coordination in Co single atoms boosts electrochemical oxygen evolution. Adv. Sci. 10, 2206107 (2023).
Zhao, J. et al. Sub-nanometer-scale fine regulation of interlayer distance in Ni-Co layered double hydroxides leading to high-rate supercapacitors. Nano Energy 76, 105026 (2020).
Zhao, J. et al. Insight into the decay mechanism of cycling capacitance for layered double hydroxides at subnanometer scale. Chem. Commun. 58, 9124–9127 (2022).
Zhao, J. et al. Balancing loading mass and gravimetric capacitance of NiCo−layered double hydroxides to achieve ultrahigh areal performance for flexible supercapacitors. Adv. Powder Mater. 3, 100151 (2024).
Li, N. et al. Identification of the active-layer structures for acidic oxygen evolution from 9R-BaIrO3 electrocatalyst with enhanced iridium mass activity. J. Am. Chem. Soc. 143, 18001–18009 (2021).
Yang, Y. et al. Enhancing water oxidation of Ru single atoms via oxygen-coordination bonding with NiFe layered double hydroxide. ACS Catal. 13, 2771–2779 (2023).
Hu, Y. et al. Single Ru atoms stabilized by hybrid amorphous/crystalline FeCoNi layered double hydroxide for ultraefficient oxygen evolution. Adv. Energy Mater. 11, 2002816 (2021).
Duan, X. et al. Stabilizing single-atomic ruthenium by ferrous ion doped NiFe-LDH towards highly efficient and sustained water oxidation. Chem. Eng. J. 466, 136962 (2022).
Wang, Y. et al. Interfacial synergy between dispersed Ru sub-nanoclusters and porous NiFe layered double hydroxide on accelerated overall water splitting by intermediate modulation. Nanoscale 12, 9669–9679 (2020).
Jia, C. et al. Ir single atoms modified Ni(OH)2 nanosheets on hierarchical porous nickel foam for efficient oxygen evolution. Nano Res. 15, 10014–10020 (2022).
Xing, Y., Ku, J., Fu, W., Wang, L. & Chen, H. Inductive effect between atomically dispersed iridium and transition-metal hydroxide nanosheets enables highly efficient oxygen evolution reaction. Chem. Eng. J. 395, 125149 (2020).
He, Q. et al. Confining high-valence iridium single sites onto nickel oxyhydroxide for robust oxygen evolution. Nano Lett. 22, 3832–3839 (2022).
Zhang, Z. et al. Selectively anchoring single atoms on specific sites of supports for improved oxygen evolution. Nat. Commun. 13, 2473 (2022).
Zhang, J. et al. Single-atom Au/NiFe layered double hydroxide electrocatalyst: probing the origin of activity for oxygen evolution reaction. J. Am. Chem. Soc. 140, 3876–3879 (2018).
Li, J., Li, Z., Zhan, F. & Shao, M. Phase engineering of cobalt hydroxide toward cation intercalation. Chem. Sci. 12, 1756–1761 (2021).
Nørskov, J. K., Abild-Pedersen, F., Studt, F. & Thomas, B. Density functional theory in surface chemistry and catalysis. PNAS 108, 937–943 (2011).
Hu, Q. et al. Subnanometric Ru clusters with upshifted d band center improve performance for alkaline hydrogen evolution reaction. Nat. Commun. 13, 3958 (2022).
Shi, H. et al. A sodium-ion-conducted asymmetric electrolyzer to lower the operation voltage for direct seawater electrolysis. Nat. Commun. 14, 3934 (2023).
Acknowledgements
The work was supported by funding from the National Natural Science Foundation of China (numbers 52003163 and 52373266 received by X.C., number 22105129 received by D.L. and number 22369020 received by J. Zhao), the Guangdong Basic and Applied Basic Research Foundation (numbers 2022A1515010670 and 2024A1515012334 received by X.C. and number 2022A1515011048 received by D.L.), and the Science and Technology Innovation Commission of Shenzhen (numbers KQTD20170810105439418 and 20200812112006001 received by X.C.). The project was funded by the China Postdoctoral Science Foundation (number 2023M732352 received by J. Zhao), and financial support came from the Start-up Research Fund of Southeast University (number RF1028623161 received by Z.Z.) and the Fundamental Research Funds for the Central Universities (number 2242024K40008 received by Z.Z.). We thank the staff of beamline BL13SSW at Shanghai Synchrotron Radiation Facility and the staff at Taiwan Synchrotron Radiation Research Center for XAS experiments support. We also gratefully acknowledge the support of the Research Grant Council of Hong Kong for support through projects 18201820 and 16201622 received by F.C. This work was partly supported by the Project of Hetao Shenzhen-Hong Kong Science and Technology Innovation Cooperation Zone (HZQB-KCZYB-2020083, received by F.C.). F.C. thanks the University of Bayreuth and the Bavarian Center for Battery Technology (BayBatt) for providing a start-up fund. We gratefully acknowledge the valuable assistance of our laboratory colleagues: Y. Li, Y. Wu, Y. Deng, Z. Xie, G. Luo, W. Liu, C. Li, X. Liang, H. Huang, M. Ahmad, L. N. U. Rehman and M. J. Robson. We thank the HKUST Fok Ying Tung Research Institute and National Supercomputing Center in Guangzhou Nansha Sub-Center for providing high-performance computational resources. We also thank H.-Y. Hsu (City University of Hong Kong) and M. Ellwood (The Hong Kong University of Science and Technology) for their support and assistance.
Funding
Open access funding provided by Universität Bayreuth.
Author information
Authors and Affiliations
Contributions
J. Zhao, X.C. and F.C. conceived the idea. J. Zhao designed the study and performed the catalysts’ syntheses and characterizations, including their morphology, structure and electrochemical performance. J. Zeng and C.C. carried out the XAS measurements. H.Z. and G.H. performed the AEM electrolyser measurements. X.Z. performed the DFT calculations. J. Zhao, Y.G., Z.Z., X.Z., Q.J., Z.S., D.L., E.Z., Y.W., M.A.M., W.R., X.C. and F.C. analysed and discussed the experimental and computational data. J. Zhao, X.C. and F.C. wrote the article. X.C. and F.C. supported the project. All authors reviewed the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Nanotechnology thanks Gonzalo Abellán and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
The Supplementary Information file includes experimental section, DFT calculations, electrochemically active surface area calculation, turnover frequency calculation, synthesis process for Ir1/(Co,Fe)-OH/MI, summary of the research results, Supplementary Scheme 1, Figs. 1−57 and Tables 1−13.
Supplementary Data 1
Source data for the Supplementary figures.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, J., Guo, Y., Zhang, Z. et al. Out-of-plane coordination of iridium single atoms with organic molecules and cobalt–iron hydroxides to boost oxygen evolution reaction. Nat. Nanotechnol. 20, 57–66 (2025). https://doi.org/10.1038/s41565-024-01807-x
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41565-024-01807-x
This article is cited by
-
High-performance ionomer-free gas diffusion cathodes with low Pt loading for proton exchange membrane water electrolysis
Communications Materials (2026)
-
Defect-interface coupling for stable lattice-oxygen-driven oxygen evolution at industrial current densities
Nature Communications (2026)
-
Enhanced oxygen evolution reaction through Gd doping-enabled manipulation of oxygen coupling pathway in CoFe oxyhydroxide
Science China Chemistry (2026)
-
Oxophilic sites activate asymmetric IrNi atomic dimers and clusters for efficient hydrogen oxidation and CO tolerance
Science China Materials (2026)
-
Self-replenishing Ni-rich stainless-steel electrode toward oxygen evolution reaction at ampere-level
Communications Chemistry (2025)
