
Abstract
Collagen IV is a basement membrane component that is encoded by six genes in mammals (COL4Α1–COL4A6). The α-chains encoded by these genes assemble into three known heterotrimers — collagen α1α1α2(IV), α3α4α5(IV) and α5α5α6(IV) — that provide structure and act as multifunctional signalling platforms. The ancestral collagen superfamily members collagen alpha-1(IV) chain (COL4Α1) and collagen alpha-2(IV) chain (COL4Α2) are present throughout the animal kingdom and in all developing and most mature mammalian tissues. Consistent with this broad distribution, variants in COL4A1 and COL4A2 cause a congenital multisystem disorder called Gould syndrome (GS), which is characterized by cerebral, ocular, muscular and kidney defects. The main clinical consequences involve the cerebral vasculature (porencephaly, small-vessel disease, leukoencephalopathy and intracerebral haemorrhage). However, the full clinical spectrum, including the organs affected and acquired phenotypes such as vascular dementia, is still being defined. By contrast, variants in COL4A3, COL4A4 or COL4A5 cause Alport syndrome (AS), a disorder of variable severity that affects the kidney, ear and eye. AS nephropathies often progress from haematuria to proteinuria, renal impairment and kidney failure. The auditory features include sensorineural hearing loss, whereas the ocular features comprise corneal dystrophy, lenticonus, dot-and-fleck retinopathy and maculopathy. Although GS and AS have little clinical resemblance, the high conservation of the genes and proteins suggests common elements of underlying pathophysiology. Conventional therapies that modify haemodynamics have lengthened the time to kidney failure for patients living with AS. However, no curative or mechanism-based interventions exist for GS. Gene-editing approaches hold promise for both disorders.
Key points
-
Collagen IV is component of basement membranes that is encoded by six α-chain genes (COL4Α1–COL4A6) that assemble into three known heterotrimers: collagen α1α1α2(IV), α3α4α5(IV) and α5α5α6(IV).
-
COL4Α1 and COL4Α2 variants cause Gould syndrome (GS), which is characterized by cerebral, ocular, muscular and renal manifestations, whereas COL4Α3, COL4Α4 and COL4Α5 variants cause Alport syndrome (AS), which is characterized by a spectrum of pathologies affecting the kidney, ear and eye.
-
GS nephropathies are variable, include haematuria, proteinuria and the presence of cysts and can progress to kidney failure; AS nephropathies are also variable, begin with haematuria and often progress to proteinuria, renal impairment and kidney failure.
-
As no effective treatment options for GS exist, current interventions are aimed at managing the symptoms.
-
Although no curative treatment for AS is available, interventions that target the renin–angiotensin–aldosterone system are efficient at slowing disease progression, delaying kidney failure and increasing life expectancy.
-
Potential shared therapeutic avenues for GS and AS include promoting secretion of defective collagen IV and attenuating TGFβ signalling; gene therapies and gene-editing approaches are also promising treatment strategies.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


References
Fidler, A. L. et al. Collagen IV and basement membrane at the evolutionary dawn of metazoan tissues. Elife 6, e24176 (2017).
Fidler, A. L., Boudko, S. P., Rokas, A. & Hudson, B. G. The triple helix of collagens - an ancient protein structure that enabled animal multicellularity and tissue evolution. J. Cell Sci. 131, jcs203950 (2018).
Khoshnoodi, J., Pedchenko, V. & Hudson, B. G. Mammalian collagen IV. Microsc. Res. Tech. 71, 357–370 (2008).
Boutaud, A. et al. Type IV collagen of the glomerular basement membrane. Evidence that the chain specificity of network assembly is encoded by the noncollagenous NC1 domains. J. Biol. Chem. 275, 30716–30724 (2000).
Meehan, D. T. et al. Biomechanical strain causes maladaptive gene regulation, contributing to Alport glomerular disease. Kidney Int. 76, 968–976 (2009).
Davis, J. M., Boswell, B. A. & Bächinger, H. P. Thermal stability and folding of type IV procollagen and effect of peptidyl-prolyl cis-trans-isomerase on the folding of the triple helix. J. Biol. Chem. 264, 8956–8962 (1989).
Shoulders, M. D. & Raines, R. T. Collagen structure and stability. Annu. Rev. Biochem. 78, 929–958 (2009).
Ramachandran, G. N. & Kartha, G. Structure of collagen. Nature 176, 593–595 (1955).
Salo, A. M. et al. A connective tissue disorder caused by mutations of the lysyl hydroxylase 3 gene. Am. J. Hum. Genet. 83, 495–503 (2008).
Miyatake, S. et al. Biallelic COLGALT1 variants are associated with cerebral small vessel disease. Ann. Neurol. 84, 843–853 (2018).
Ishikawa, Y. et al. Lysyl hydroxylase 3-mediated post-translational modifications are required for proper biosynthesis of collagen ɑ1ɑ1ɑ2(IV). J. Biol. Chem. 298, 102713 (2022).
Aypek, H. et al. Loss of the collagen IV modifier prolyl 3-hydroxylase 2 causes thin basement membrane nephropathy. J. Clin. Invest. 132, e147253 (2022).
Koide, T., Takahara, Y., Asada, S. & Nagata, K. Xaa-Arg-Gly triplets in the collagen triple helix are dominant binding sites for the molecular chaperone HSP47. J. Biol. Chem. 277, 6178–6182 (2002).
Natsume, T., Koide, T., Yokota, S., Hirayoshi, K. & Nagata, K. Interactions between collagen-binding stress protein HSP47 and collagen. Analysis of kinetic parameters by surface plasmon resonance biosensor. J. Biol. Chem. 269, 31224–31228 (1994).
Ono, T., Miyazaki, T., Ishida, Y., Uehata, M. & Nagata, K. Direct in vitro and in vivo evidence for interaction between Hsp47 protein and collagen triple helix. J. Biol. Chem. 287, 6810–6818 (2012).
Saga, S., Nagata, K., Chen, W. T. & Yamada, K. M. pH-dependent function, purification, and intracellular location of a major collagen-binding glycoprotein. J. Cell Biol. 105, 517–527 (1987).
Tasab, M., Batten, M. R. & Bulleid, N. J. Hsp47: a molecular chaperone that interacts with and stabilizes correctly-folded procollagen. EMBO J. 19, 2204–2211 (2000).
Malhotra, V. & Erlmann, P. Protein export at the ER: loading big collagens into COPII carriers. EMBO J. 30, 3475–3480 (2011).
Malhotra, V., Erlmann, P. & Nogueira, C. Procollagen export from the endoplasmic reticulum. Biochem. Soc. Trans. 43, 104–107 (2015).
Wilson, D. G. et al. Global defects in collagen secretion in a Mia3/TANGO1 knockout mouse. J. Cell Biol. 193, 935–951 (2011).
Christiansen, H. E. et al. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet. 86, 389–398 (2010).
Alanay, Y. et al. Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 86, 551–559 (2010).
Barnes, A. M. et al. Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N. Engl. J. Med. 362, 521–528 (2010).
Fang, M. et al. Lifetime risk and projected burden of dementia. Nat. Med. 31, 772–776 (2025).
Merkuryeva, E. S. et al. Presentation of rare phenotypes associated with the FKBP10 gene. Genes 15, 674 (2024).
Nagai, N. et al. Embryonic lethality of molecular chaperone hsp47 knockout mice is associated with defects in collagen biosynthesis. J. Cell Biol. 150, 1499–1506 (2000).
Poschl, E. et al. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 131, 1619–1628 (2004).
Cummings, C. F. et al. Extracellular chloride signals collagen IV network assembly during basement membrane formation. J. Cell Biol. 213, 479–494 (2016).
Vanacore, R. et al. A sulfilimine bond identified in collagen IV. Science 325, 1230–1234 (2009).
Bhave, G. et al. Peroxidasin forms sulfilimine chemical bonds using hypohalous acids in tissue genesis. Nat. Chem. Biol. 8, 784–790 (2012).
McCall, A. S. et al. Bromine is an essential trace element for assembly of collagen IV scaffolds in tissue development and architecture. Cell 157, 1380–1392 (2014).
Timpl, R., Wiedemann, H., van Delden, V., Furthmayr, H. & Kuhn, K. A network model for the organization of type IV collagen molecules in basement membranes. Eur. J. Biochem. 120, 203–211 (1981).
Anazco, C. et al. Lysyl oxidase-like-2 cross-links collagen IV of glomerular basement membrane. J. Biol. Chem. 291, 25999–26012 (2016).
Lopez-Jimenez, A. J., Basak, T. & Vanacore, R. M. Proteolytic processing of lysyl oxidase-like-2 in the extracellular matrix is required for crosslinking of basement membrane collagen IV. J. Biol. Chem. 292, 16970–16982 (2017).
Hofmann, H., Voss, T., Kuhn, K. & Engel, J. Localization of flexible sites in thread-like molecules from electron micrographs. Comparison of interstitial, basement membrane and intima collagens. J. Mol. Biol. 172, 325–343 (1984).
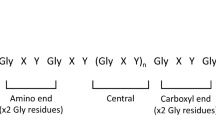
Bella, J., Liu, J., Kramer, R., Brodsky, B. & Berman, H. M. Conformational effects of Gly-X-Gly interruptions in the collagen triple helix. J. Mol. Biol. 362, 298–311 (2006).
Brazel, D. et al. Completion of the amino acid sequence of the ɑ1 chain of human basement membrane collagen (type IV) reveals 21 non-triplet interruptions located within the collagenous domain. Eur. J. Biochem. 168, 529–536 (1987).
Leinonen, A., Mariyama, M., Mochizuki, T., Tryggvason, K. & Reeders, S. T. Complete primary structure of the human type IV collagen ɑ4(IV) chain. Comparison with structure and expression of the other ɑ(IV) chains. J. Biol. Chem. 269, 26172–26177 (1994).
Zhou, J., Ding, M., Zhao, Z. & Reeders, S. T. Complete primary structure of the sixth chain of human basement membrane collagen, ɑ6(IV). Isolation of the cDNAs for ɑ6(IV) and comparison with five other type IV collagen chains. J. Biol. Chem. 269, 13193–13199 (1994).
Abrahamson, D. R., Hudson, B. G., Stroganova, L., Borza, D. B. & St John, P. L. Cellular origins of type IV collagen networks in developing glomeruli. J. Am. Soc. Nephrol. 20, 1471–1479 (2009).
Miner, J. H. & Sanes, J. R. Collagen IV alpha 3, alpha 4, and alpha 5 chains in rodent basal laminae: sequence, distribution, association with laminins, and developmental switches. J. Cell Biol. 127, 879–891 (1994).
Borza, D. B. et al. The NC1 domain of collagen IV encodes a novel network composed of the ɑ1, ɑ2, ɑ5, and ɑ6 chains in smooth muscle basement membranes. J. Biol. Chem. 276, 28532–28540 (2001).
Peissel, B. et al. Comparative distribution of the alpha 1(IV), alpha 5(IV), and alpha 6(IV) collagen chains in normal human adult and fetal tissues and in kidneys from X-linked Alport syndrome patients. J. Clin. Invest. 96, 1948–1957 (1995).
Harvey, S. J. et al. Role of distinct type IV collagen networks in glomerular development and function. Kidney Int. 54, 1857–1866 (1998).
Ninomiya, Y. et al. Differential expression of two basement membrane collagen genes, COL4A6 and COL4A5, demonstrated by immunofluorescence staining using peptide-specific monoclonal antibodies. J. Cell Biol. 130, 1219–1229 (1995).
Kuo, D. S., Labelle-Dumais, C. & Gould, D. B. COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum. Mol. Genet. 21, R97–R110 (2012).
Mao, M., Alavi, M. V., Labelle-Dumais, C. & Gould, D. B. Type IV collagens and basement membrane diseases: cell biology and pathogenic mechanisms. Curr. Top. Membr. 76, 61–116 (2015).
Jeanne, M. & Gould, D. B. Genotype-phenotype correlations in pathology caused by collagen type IV alpha 1 and 2 mutations. Matrix Biol. 57-58, 29–44 (2017).
Labelle-Dumais, C. et al. COL4A1 mutations cause neuromuscular disease with tissue-specific mechanistic heterogeneity. Am. J. Hum. Genet. 104, 847–860 (2019).
Yoneda, Y. et al. Phenotypic spectrum of COL4A1 mutations: porencephaly to schizencephaly. Ann. Neurol. 73, 48–57 (2013).
Meuwissen, M. E. et al. The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature. Genet. Med. 17, 843–853 (2015).
Zagaglia, S. et al. Neurologic phenotypes associated with COL4A1/2 mutations: expanding the spectrum of disease. Neurology 91, e2078–e2088 (2018).
Gasparini, S. et al. Multiorgan manifestations of COL4A1 and COL4A2 variants and proposal for a clinical management protocol. Am. J. Med. Genet. C. Semin. Med. Genet. 196, e32099 (2024).
Whittaker, E. et al. Systematic review of cerebral phenotypes associated with monogenic cerebral small-vessel disease. J. Am. Heart Assoc. 11, e025629 (2022).
Thaung, C. et al. Novel ENU-induced eye mutations in the mouse: models for human eye disease. Hum. Mol. Genet. 11, 755–767 (2002).
Favor, J. et al. Type IV procollagen missense mutations associated with defects of the eye, vascular stability, the brain, kidney function and embryonic or postnatal viability in the mouse, Mus musculus: an extension of the Col4a1 allelic series and the identification of the first two Col4a2 mutant alleles. Genetics 175, 725–736 (2007).
Gould, D. B. et al. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science 308, 1167–1171 (2005).
Gould, D. B. et al. Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N. Engl. J. Med. 354, 1489–1496 (2006).
Breedveld, G. et al. Novel mutations in three families confirm a major role of COL4A1 in hereditary porencephaly. J. Med. Genet. 43, 490–495 (2006).
Vahedi, K. et al. Clinical and brain MRI follow-up study of a family with COL4A1 mutation. Neurology 69, 1564–1568 (2007).
Van Agtmael, T. et al. Dominant mutations of Col4a1 result in basement membrane defects which lead to anterior segment dysgenesis and glomerulopathy. Hum. Mol. Genet. 14, 3161–3168 (2005).
Chen, Z. et al. HANAC syndrome Col4a1 mutation causes neonate glomerular hyperpermeability and adult glomerulocystic kidney disease. J. Am. Soc. Nephrol. 27, 1042–1054 (2016).
Gupta, M. C., Graham, P. L. & Kramer, J. M. Characterization of ɑ1(IV) collagen mutations in Caenorhabditis elegans and the effects of ɑ1 and ɑ2(IV) mutations on type IV collagen distribution. J. Cell Biol. 137, 1185–1196 (1997).
Sibley, M. H., Graham, P. L., von Mende, N. & Kramer, J. M. Mutations in the alpha 2(IV) basement membrane collagen gene of Caenorhabditis elegans produce phenotypes of differing severities. EMBO J. 13, 3278–3285 (1994).
Verdura, E. et al. Disruption of a miR-29 binding site leading to COL4A1 upregulation causes pontine autosomal dominant microangiopathy with leukoencephalopathy. Ann. Neurol. 80, 741–753 (2016).
Yoneda, Y. et al. De novo and inherited mutations in COL4A2, encoding the type IV collagen ɑ2 chain cause porencephaly. Am. J. Hum. Genet. 90, 86–90 (2012).
Verbeek, E. et al. COL4A2 mutation associated with familial porencephaly and small-vessel disease. Eur. J. Hum. Genet. 20, 844–851 (2012).
Gunda, B. et al. COL4A2 mutation causing adult onset recurrent intracerebral hemorrhage and leukoencephalopathy. J. Neurol. 261, 500–503 (2014).
Maurice, P. et al. Prevalence of COL4A1 and COL4A2 mutations in severe fetal multifocal hemorrhagic and/or ischemic cerebral lesions. Ultrasound Obstet. Gynecol. 57, 783–789 (2021).
Coste, T. et al. COL4A1/COL4A2 and inherited platelet disorder gene variants in fetuses showing intracranial hemorrhage. Prenat. Diagn. 42, 601–610 (2022).
George, E. et al. Spectrum of fetal intraparenchymal hemorrhage in COL4A1/A2-related disorders. Pediatr. Neurol. 147, 63–67 (2023).
Gubana, F. et al. Prenatal diagnosis of COL4A1 mutations in eight cases: further delineation of the neurohistopathological phenotype. Pediatr. Dev. Pathol. 25, 435–446 (2022).
Boyce, D., McGee, S., Shank, L., Pathak, S. & Gould, D. Epilepsy and related challenges in children with COL4A1 and COL4A2 mutations: a Gould syndrome patient registry. Epilepsy Behav. 125, 108365 (2021).
Fehlings, D. L. et al. Comprehensive whole-genome sequence analyses provide insights into the genomic architecture of cerebral palsy. Nat. Genet. 56, 585–594 (2024).
Corriveau, R. A. et al. The science of vascular contributions to cognitive impairment and dementia (VCID): a framework for advancing research priorities in the cerebrovascular biology of cognitive decline. Cell Mol. Neurobiol. 36, 281–288 (2016).
Iadecola, C. The pathobiology of vascular dementia. Neuron 80, 844–866 (2013).
Goodman, R. A. et al. Prevalence of dementia subtypes in United States Medicare fee-for-service beneficiaries, 2011–2013. Alzheimers Dement. 13, 28–37 (2017).
Uemura, M., Tanaka, N., Ando, S., Yanagihara, T. & Onodera, O. Missense variants in COL4A1/2 are associated with cerebral aneurysms: a case report and literature review. Neurol. Int. 16, 226–238 (2024).
Aloui, C. et al. An AluYa5 insertion in the 3’UTR of COL4A1 and cerebral small vessel disease. JAMA Netw. Open. 7, e247034 (2024).
Ruigrok, Y. M. et al. Evidence in favor of the contribution of genes involved in the maintenance of the extracellular matrix of the arterial wall to the development of intracranial aneurysms. Hum. Mol. Genet. 15, 3361–3368 (2006).
Livingston, J. et al. COL4A1 mutations associated with a characteristic pattern of intracranial calcification. Neuropediatrics 42, 227–233 (2011).
O’Donnell, C. J. et al. Genome-wide association study for coronary artery calcification with follow-up in myocardial infarction. Circulation 124, 2855–2864 (2011).
Zheng, Q., Ma, Y., Chen, S., Che, Q. & Chen, D. The integrated landscape of biological candidate causal genes in coronary artery disease. Front. Genet. 11, 320 (2020).
Adi, D. et al. Polymorphisms of COL4A1 gene are associated with arterial pulse wave velocity in healthy Han Chinese and Uygur subjects. Int. J. Clin. Exp. Med. 8, 2693–2701 (2015).
Tarasov, K. V. et al. COL4A1 is associated with arterial stiffness by genome-wide association scan. Circ. Cardiovasc. Genet. 2, 151–158 (2009).
Rannikmae, K. et al. Common variation in COL4A1/COL4A2 is associated with sporadic cerebral small vessel disease. Neurology 84, 918–926 (2015).
Ayrignac, X. et al. Adult-onset genetic leukoencephalopathies: a MRI pattern-based approach in a comprehensive study of 154 patients. Brain 138, 284–292 (2015).
Di Donato, I., Dotti, M. T. & Federico, A. Update on several/certain adult-onset genetic leukoencephalopathies: clinical signs and molecular confirmation. J. Alzheimers Dis. 42, S27–S35 (2014).
Traylor, M. et al. Genome-wide meta-analysis of cerebral white matter hyperintensities in patients with stroke. Neurology 86, 146–153 (2016).
Lanfranconi, S. & Markus, H. S. COL4A1 mutations as a monogenic cause of cerebral small vessel disease: a systematic review. Stroke 41, e513–e518 (2010).
Persyn, E. et al. Genome-wide association study of MRI markers of cerebral small vessel disease in 42,310 participants. Nat. Commun. 11, 2175 (2020).
Sargurupremraj, M. et al. Cerebral small vessel disease genomics and its implications across the lifespan. Nat. Commun. 11, 6285 (2020).
Taylor-Bateman, V. et al. Cardiovascular risk factors and MRI markers of cerebral small vessel disease: a Mendelian randomization study. Neurology 98, e343–e351 (2022).
Jeanne, M., Jorgensen, J. & Gould, D. B. Molecular and genetic analyses of collagen type IV mutant mouse models of spontaneous intracerebral hemorrhage identify mechanisms for stroke prevention. Circulation 131, 1555–1565 (2015).
Cozzitorto, C. et al. Evaluating neural crest cell migration in a Col4a1 mutant mouse model of ocular anterior segment dysgenesis. Cell Dev. 179, 203926 (2024).
Ratelade, J. et al. Reducing hypermuscularization of the transitional segment between arterioles and capillaries protects against spontaneous intracerebral hemorrhage. Circulation 141, 2078–2094 (2020).
Branyan, K. et al. Elevated TGFβ signaling contributes to cerebral small vessel disease in mouse models of Gould syndrome. Matrix Biol. 115, 48–70 (2023).
Yamasaki, E. et al. Impaired intracellular Ca2+ signaling contributes to age-related cerebral small vessel disease in Col4a1 mutant mice. Sci. Signal. 16, eadi3966 (2023).
Yamasaki, E. et al. Faulty TRPM4 channels underlie age-dependent cerebral vascular dysfunction in Gould syndrome. Proc. Natl Acad. Sci. USA 120, e2217327120 (2023).
Thakore, P. et al. PI3K block restores age-dependent neurovascular coupling defects associated with cerebral small vessel disease. Proc. Natl Acad. Sci. USA 120, e2306479120 (2023).
Gould, D. B., Marchant, J. K., Savinova, O. V., Smith, R. S. & John, S. W. Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum. Mol. Genet. 16, 798–807 (2007).
Sibon, I. et al. COL4A1 mutation in Axenfeld–Rieger anomaly with leukoencephalopathy and stroke. Ann. Neurol. 62, 177–184 (2007).
Coupry, I. et al. Ophthalmological features associated with COL4A1 mutations. Arch. Ophthalmol. 128, 483–489 (2010).
Rodahl, E. et al. Variants of anterior segment dysgenesis and cerebral involvement in a large family with a novel COL4A1 mutation. Am. J. Ophthalmol. 155, 946–953 (2013).
Mao, M., Kiss, M., Ou, Y. & Gould, D. B. Genetic dissection of anterior segment dysgenesis caused by a Col4a1 mutation in mouse. Dis. Model. Mech. 10, 475–485 (2017).
Labelle-Dumais, C. et al. COL4A1 mutations cause ocular dysgenesis, neuronal localization defects, and myopathy in mice and Walker-Warburg syndrome in humans. PLoS Genet. 7, e1002062 (2011).
Rannikmae, K. et al. Beyond the brain: systematic review of extracerebral phenotypes associated with monogenic cerebral small vessel disease. Stroke 51, 3007–3017 (2020).
Ma, A. et al. Revealing hidden genetic diagnoses in the ocular anterior segment disorders. Genet. Med. 22, 1623–1632 (2020).
Gould, D. B., Smith, R. S. & John, S. W. Anterior segment development relevant to glaucoma. Int. J. Dev. Biol. 48, 1015–1029 (2004).
Gould, D. B. & John, S. W. Anterior segment dysgenesis and the developmental glaucomas are complex traits. Hum. Mol. Genet. 11, 1185–1193 (2002).
Kupfer, C. & Kaiser-Kupfer, M. I. New hypothesis of developmental anomalies of the anterior chamber associated with glaucoma. Trans. Ophthalmol. Soc. U K. 98, 213–215 (1978).
Kupfer, C. & Kaiser-Kupfer, M. I. Observations on the development of the anterior chamber angle with reference to the pathogenesis of congenital glaucomas. Am. J. Ophthalmol. 88, 424–426 (1979).
Noden, D. M. An analysis of migratory behavior of avian cephalic neural crest cells. Dev. Biol. 42, 106–130 (1975).
Trainor, P. A. & Tam, P. P. Cranial paraxial mesoderm and neural crest cells of the mouse embryo: co-distribution in the craniofacial mesenchyme but distinct segregation in branchial arches. Development 121, 2569–2582 (1995).
Mao, M., Labelle-Dumais, C., Tufa, S. F., Keene, D. R. & Gould, D. B. Elevated TGFβ signaling contributes to ocular anterior segment dysgenesis in Col4a1 mutant mice. Matrix Biol. 110, 151–173 (2022).
Mao, M. et al. TGFβ signaling dysregulation may contribute to COL4A1-related glaucomatous optic nerve damage. Invest. Ophthalmol. Vis. Sci. 65, 15 (2024).
Plaisier, E. et al. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N. Engl. J. Med. 357, 2687–2695 (2007).
Guiraud, S. et al. HANAC Col4a1 mutation in mice leads to skeletal muscle alterations due to a primary vascular defect. Am. J. Pathol. 187, 505–516 (2017).
Fox, M. A. et al. Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell 129, 179–193 (2007).
Kuo, D. S. et al. Allelic heterogeneity contributes to variability in ocular dysgenesis, myopathy and brain malformations caused by Col4a1 and Col4a2 mutations. Hum. Mol. Genet. 23, 1709–1722 (2014).
Kelemen-Valkony, I. et al. Drosophila basement membrane collagen col4a1 mutations cause severe myopathy. Matrix Biol. 31, 29–37 (2012).
Kiss, A. A. et al. Type IV collagen is essential for proper function of integrin-mediated adhesion in Drosophila muscle fibers. Int. J. Mol. Sci. 20, 5124 (2019).
Lv, J. C. & Zhang, L. X. Prevalence and disease burden of chronic kidney disease. Adv. Exp. Med. Biol. 1165, 3–15 (2019).
Canadas-Garre, M. et al. Genetic susceptibility to chronic kidney disease — some more pieces for the heritability puzzle. Front. Genet. 10, 453 (2019).
Plaisier, E. et al. Novel COL4A1 mutations associated with HANAC syndrome: a role for the triple helical CB3[IV] domain. Am. J. Med. Genet. A 152A, 2550–2555 (2010).
Meuwissen, M. E. et al. Sporadic COL4A1 mutations with extensive prenatal porencephaly resembling hydranencephaly. Neurology 76, 844–846 (2011).
Gale, D. P. et al. A novel COL4A1 frameshift mutation in familial kidney disease: the importance of the C-terminal NC1 domain of type IV collagen. Nephrol. Dial. Transpl. 31, 1908–1914 (2016).
Cornec-Le Gall, E. et al. The value of genetic testing in polycystic kidney diseases illustrated by a family with PKD2 and COL4A1 mutations. Am. J. Kidney Dis. 72, 302–308 (2018).
Mao, M. et al. Strain-dependent anterior segment dysgenesis and progression to glaucoma in Col4a1 mutant mice. Invest. Ophthalmol. Vis. Sci. 56, 6823–6831 (2015).
Mao, M. et al. Identification of fibronectin 1 as a candidate genetic modifier in a Col4a1 mutant mouse model of Gould syndrome. Dis. Model. Mech. 14, dmm048231 (2021).
Jeanne, M. et al. COL4A2 mutations impair COL4A1 and COL4A2 secretion and cause hemorrhagic stroke. Am. J. Hum. Genet. 90, 91–101 (2012).
Jones, F. E. et al. ER stress and basement membrane defects combine to cause glomerular and tubular renal disease resulting from Col4a1 mutations in mice. Dis. Model. Mech. 9, 165–176 (2016).
Jones, F. E. et al. 4-Sodium phenyl butyric acid has both efficacy and counter-indicative effects in the treatment of Col4a1 disease. Hum. Mol. Genet. 28, 628–638 (2019).
Hudson, B. G., Tryggvason, K., Sundaramoorthy, M. & Neilson, E. G. Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N. Engl. J. Med. 348, 2543–2556 (2003).
Barker, D. F. et al. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248, 1224–1227 (1990).
Alport, A. C. Hereditary familial congenital haemorrhagic nephritis. Br. Med. J. 1, 504–506 (1927).
Spear, G. S. Pathology of the kidney in Alport’s syndrome. Pathol. Annu. 9, 93–138 (1974).
Williamson, D. A. Alport’s syndrome of hereditary nephritis with deafness. Lancet 2, 1321–1323 (1961).
Grunfeld, J. P. Contemporary diagnostic approach in Alport’s syndrome. Ren. Fail. 22, 759–763 (2000).
Kashtan, C. E. Familial hematuric syndromes — Alport syndrome, thin glomerular basement membrane disease and Fechtner/Epstein syndromes. Contrib. Nephrol. 79–99 (2001).
Daga, S. et al. The 2019 and 2021 international workshops on Alport syndrome. Eur. J. Hum. Genet. 30, 507–516 (2022).
Gibson, J. et al. Prevalence estimates of predicted pathogenic COL4A3–COL4A5 variants in a population sequencing database and their implications for Alport syndrome. J. Am. Soc. Nephrol. 32, 2273–2290 (2021).
Malone, A. F. et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 86, 1253–1259 (2014).
Voskarides, K. et al. COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J. Amer. Soc. Nephrol. 18, 3004–3016 (2007).
Furlano, M. et al. Clinical and genetic features of autosomal dominant Alport syndrome: a cohort study. Am. J. Kidney Dis. 78, 560–570.e561 (2021).
Matthaiou, A., Poulli, T. & Deltas, C. Prevalence of clinical, pathological and molecular features of glomerular basement membrane nephropathy caused by COL4A3 or COL4A4 mutations: a systematic review. Clin. Kidney J. 13, 1025–1036 (2020).
Tryggvason, K. Complex genetics of Alport and Goodpasture syndromes. Nat. Rev. Nephrol. 17, 635–636 (2021).
Savige, J., Huang, M., Croos Dabrera, M. S., Shukla, K. & Gibson, J. Genotype-phenotype correlations for pathogenic COL4A3–COL4A5 variants in X-linked, autosomal recessive, and autosomal dominant Alport syndrome. Front. Med. 9, 865034 (2022).
Pieri, M. et al. Evidence for activation of the unfolded protein response in collagen IV nephropathies. J. Am. Soc. Nephrol. 25, 260–275 (2014).
Wang, D. et al. The chemical chaperone, PBA, reduces ER stress and autophagy and increases collagen IV ɑ5 expression in cultured fibroblasts from men with X-linked Alport syndrome and missense mutations. Kidney Int. Rep. 2, 739–748 (2017).
Wickman, L. et al. Podocyte depletion in thin GBM and Alport syndrome. PLoS ONE 11, e0155255 (2016).
Yamamura, T. et al. Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked Alport syndrome. Kidney Int. 98, 1605–1614 (2020).
Said, S. M. et al. Negative staining for COL4A5 correlates with worse prognosis and more severe ultrastructural alterations in males with Alport syndrome. Kidney Int. Rep. 2, 44–52 (2017).
Solanki, K. V. et al. The phenotypic spectrum of COL4A3 heterozygotes. Kidney Int. Rep. 8, 2088–2099 (2023).
Pierides, A. et al. Clinico-pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3/ COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 24, 2721–2729 (2009).
Savige, J. & Harraka, P. Pathogenic variants in the genes affected in Alport syndrome (COL4A3–COL4A5) and their association with other kidney conditions: a review. Am. J. Kidney Dis. 78, 857–864 (2021).
Pagniez, M. S. et al. Challenging the narrative of Alport syndrome spectrum: no link with cystic phenotype. Nephrol. Dial. Transplant. 40, 1408–1415 (2025).
Dufek, B. et al. Endothelin A receptor activation on mesangial cells initiates Alport glomerular disease. Kidney Int. 90, 300–310 (2016).
Clark, S. D., Nabity, M. B., Cianciolo, R. E., Dufek, B. & Cosgrove, D. X-linked Alport dogs demonstrate mesangial filopodial invasion of the capillary tuft as an early event in glomerular damage. PLoS ONE 11, e0168343 (2016).
Randles, M. J. et al. Three-dimensional electron microscopy reveals the evolution of glomerular barrier injury. Sci. Rep. 6, 35068 (2016).
Cosgrove, D. et al. Integrin ɑ1β1 and transforming growth factor-β1 play distinct roles in Alport glomerular pathogenesis and serve as dual targets for metabolic therapy. Am. J. Pathol. 157, 1649–1659 (2000).
Rubel, D. et al. Collagen receptors integrin alpha2beta1 and discoidin domain receptor 1 regulate maturation of the glomerular basement membrane and loss of integrin alpha2beta1 delays kidney fibrosis in COL4A3 knockout mice. Matrix Biol. 34, 13–21 (2014).
Quinlan, C. & Rheault, M. N. Genetic basis of type IV collagen disorders of the kidney. Clin. J. Am. Soc. Nephrol. 16, 1101–1109 (2021).
Gunwar, S. et al. Glomerular basement membrane. Identification of a novel disulfide-cross- linked network of ɑ3, ɑ4, and ɑ5 chains of type IV collagen and its implications for the pathogenesis of Alport syndrome. J. Biol. Chem. 273, 8767–8775 (1998).
Kalluri, R., Gattone, V. H. 2nd & Hudson, B. G. Identification and localization of type IV collagen chains in the inner ear cochlea. Connect. Tissue Res. 37, 143–150 (1998).
Cosgrove, D. et al. Ultrastructural, physiological, and molecular defects in the inner ear of a gene-knockout mouse model for autosomal Alport syndrome. Hear. Res. 121, 84–98 (1998).
Chavez, E., Goncalves, S., Rheault, M. N. & Fornoni, A. Alport syndrome. Adv. Kidney Dis. Health 31, 170–179 (2024).
Ungar, O. J., Nadol, J. B. & Santos, F. Temporal bone histopathology of X-linked inherited Alport syndrome. Laryngoscope Investig. Otolaryngol. 3, 311–314 (2018).
Merchant, S. N. et al. Temporal bone histopathology in Alport syndrome. Laryngoscope 114, 1609–1618 (2004).
Jais, J. P. et al. X-linked Alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J. Am. Soc. Nephrol. 11, 649–657 (2000).
Jais, J. P. et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and women belonging to 195 families: a “European Community Alport Syndrome Concerted Action” study. J. Am. Soc. Nephrol. 14, 2603–2610 (2003).
Bai, X., Dilworth, D. J., Weng, Y. C. & Gould, D. B. Developmental distribution of collagen IV isoforms and relevance to ocular diseases. Matrix Biol. 28, 194–201 (2009).
Savige, J. et al. Ocular features in Alport syndrome: pathogenesis and clinical significance. Clin. J. Am. Soc. Nephrol. 10, 703–709 (2015).
Nozu, K. et al. Characterization of contiguous gene deletions in COL4A6 and COL4A5 in Alport syndrome — diffuse leiomyomatosis. J. Hum. Genet. 62, 733–735 (2017).
Zhou, J. et al. Deletion of the paired ɑ5(IV) and ɑ6(IV) collagen genes in inherited smooth muscle tumors. Science 261, 1167–1169 (1993).
Rost, S. et al. Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6. Eur. J. Hum. Genet. 22, 208–215 (2014).
O’Brien, A. et al. Confirmation of COL4A6 variants in X-linked nonsyndromic hearing loss and its clinical implications. Eur. J. Hum. Genet. 30, 7–12 (2022).
Tang, S. et al. Lack of collagen ɑ6(IV) chain in mice does not cause severe-to-profound hearing loss or cochlear malformation, a distinct phenotype from nonsyndromic hearing loss with COL4A6 missense mutation. PLoS ONE 16, e0249909 (2021).
Meehan, D. T. et al. Endothelin-1 mediated induction of extracellular matrix genes in strial marginal cells underlies strial pathology in Alport mice. Hear. Res. 341, 100–108 (2016).
Mancuso, M. et al. Monogenic cerebral small-vessel diseases: diagnosis and therapy. Consensus recommendations of the European Academy of Neurology. Eur. J. Neurol. 27, 909–927 (2020).
Tambala, D. et al. COL4A1 and COL4A2-related disorders – clinical features, diagnostic guidelines, and management. Genet. Med. (in the press).
Savige, J. et al. Guidelines for genetic testing and management of Alport syndrome. Clin. J. Am. Soc. Nephrol. 17, 143–154 (2022).
Gross, O. et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 81, 494–501 (2012).
Gross, O. et al. A multicenter, randomized, placebo-controlled, double-blind phase 3 trial with open-arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport’s syndrome. Kidney Int. 97, 1275–1286 (2020).
Kashtan, C. E. & Gross, O. Clinical practice recommendations for the diagnosis and management of Alport syndrome in children, adolescents, and young adults — an update for 2020. Pediatr. Nephrol. 36, 711–719 (2021).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/study/NCT04573920 (2025).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/study/NCT05003986 (2025).
Cosgrove, D. et al. Dual inhibition of the endothelin and angiotensin receptor ameliorates renal and inner ear pathologies in Alport mice. J. Pathol. 260, 353–364 (2023).
Tuttle, K. R. et al. SGLT2 inhibition for CKD and cardiovascular disease in type 2 diabetes: report of a scientific workshop sponsored by the National Kidney Foundation. Am. J. Kidney Dis. 77, 94–109 (2021).
Chertow, G. M. et al. Effects of dapagliflozin in chronic kidney disease, with and without other cardiovascular medications: DAPA-CKD trial. J. Am. Heart Assoc. 12, e028739 (2023).
Heerspink, H. J. L. et al. Dapagliflozin in patients with chronic kidney disease. N. Engl. J. Med. 383, 1436–1446 (2020).
Gross, O. et al. Protocol and rationale for a randomized controlled SGLT2 inhibitor trial in paediatric and young adult populations with chronic kidney disease: DOUBLE PRO-TECT Alport. Nephrol. Dial. Transplant. 40, 679–687 (2025).
Zhu, Z. et al. Finerenone added to RAS/SGLT2 blockade for CKD in Alport syndrome. results of a randomized controlled trial with Col4a3−/− mice. J. Am. Soc. Nephrol. 34, 1513–1520 (2023).
Lin, X., Suh, J. H., Go, G. & Miner, J. H. Feasibility of repairing glomerular basement membrane defects in Alport syndrome. J. Am. Soc. Nephrol. 25, 687–692 (2014).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/study/NCT04774536 (2024).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/study/NCT04426669 (2025).
Daga, S. et al. New frontiers to cure Alport syndrome: COL4A3 and COL4A5 gene editing in podocyte-lineage cells. Eur. J. Hum. Genet. 28, 480–490 (2020).
Funk, S. D., Bayer, R. H. & Miner, J. H. Endothelial cell-specific collagen type IV-ɑ3 expression does not rescue Alport syndrome in Col4a3−/− mice. Am. J. Physiol. Renal Physiol. 316, F830–F837 (2019).
Caruso, S. M., Quinn, P. M., da Costa, B. L. & Tsang, S. H. CRISPR/Cas therapeutic strategies for autosomal dominant disorders. J. Clin. Invest. 132, e158287 (2022).
Yamamura, T. et al. Development of an exon skipping therapy for X-linked Alport syndrome with truncating variants in COL4A5. Nat. Commun. 11, 2777 (2020).
Hayashi, G., Labelle-Dumais, C. & Gould, D. B. Use of sodium 4-phenylbutyrate to define therapeutic parameters for reducing intracerebral hemorrhage and myopathy in Col4a1 mutant mice. Dis. Model. Mech. 11, dmm034157 (2018).
Weng, Y. C. et al. COL4A1 mutations in patients with sporadic late-onset intracerebral hemorrhage. Ann. Neurol. 71, 470–477 (2012).
Hartl, F. U., Bracher, A. & Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 (2011).
Elborn, J. S. et al. Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: a pooled analysis. Lancet Respir. Med. 4, 617–626 (2016).
Germain, D. P. et al. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N. Engl. J. Med. 375, 545–555 (2016).
Yu, S. et al. Tauroursodeoxycholic acid ameliorates renal injury induced by COL4A3 mutation. Kidney Int. 106, 433–449 (2024).
Wang, X., Harris, R. E., Bayston, L. J. & Ashe, H. L. Type IV collagens regulate BMP signalling in Drosophila. Nature 455, 72–77 (2008).
Paralkar, V. M., Vukicevic, S. & Reddi, A. H. Transforming growth factor β type 1 binds to collagen IV of basement membrane matrix: implications for development. Dev. Biol. 143, 303–308 (1991).
Ma, M., Cao, X., Dai, J. & Pastor-Pareja, J. C. Basement membrane manipulation in Drosophila wing discs affects Dpp retention but not growth mechanoregulation. Dev. Cell 42, 97–106.e104 (2017).
Bunt, S. et al. Hemocyte-secreted type IV collagen enhances BMP signaling to guide renal tubule morphogenesis in Drosophila. Dev. Cell 19, 296–306 (2010).
Yamamoto, T., Nakamura, T., Noble, N. A., Ruoslahti, E. & Border, W. A. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc. Natl Acad. Sci. USA 90, 1814–1818 (1993).
Yoshioka, K. et al. Transforming growth factor-beta protein and mRNA in glomeruli in normal and diseased human kidneys. Lab. Invest. 68, 154–163 (1993).
Sato, M., Muragaki, Y., Saika, S., Roberts, A. B. & Ooshima, A. Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Invest. 112, 1486–1494 (2003).
Moon, J. A., Kim, H. T., Cho, I. S., Sheen, Y. Y. & Kim, D. K. IN-1130, a novel transforming growth factor-beta type I receptor kinase (ALK5) inhibitor, suppresses renal fibrosis in obstructive nephropathy. Kidney Int. 70, 1234–1243 (2006).
Williams, M. J. et al. The activin receptor is stimulated in the skeleton, vasculature, heart, and kidney during chronic kidney disease. Kidney Int. 93, 147–158 (2018).
Puapatanakul, P. & Miner, J. H. Alport syndrome and Alport kidney diseases — elucidating the disease spectrum. Curr. Opin. Nephrol. Hypertens. 33, 283–290 (2024).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/study/NCT05267262 (2025).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/study/NCT05448755 (2022).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/study/NCT06226896 (2024).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/study/NCT06274489 (2025).
US National Library of Medicine. ClinicalTrials.gov https://www.clinicaltrials.gov/study/NCT06425055 (2025).
Acknowledgements
J.H.M. acknowledges funding from the NIH (R01DK128660, U54DK137332), the Alport Syndrome Foundation and Kidney Research UK under a contract from the University of Manchester. D.B.G. acknowledges funding from the NIH (R33NS115132, RF1NS128217, R21NS133610, R21DK140866). The UCSF Department of Ophthalmology is supported by a Core Grant from the National Eye Institute (P30EY002162) and an unrestricted grant from Research to Prevent Blindness.
Author information
Authors and Affiliations
Contributions
All authors researched data for the article, contributed substantially to writing the article, and reviewed and edited the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
J.H.M. is a member of the Alport Syndrome Foundation’s Scientific Advisory Research Network, has ownership interest in Sintra Therapeutics and has consultancy relationships with Eloxx Pharmaceuticals, Sintra Therapeutics and Bayer AG. The other authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Nephrology thanks Daniel Gale, Rachel Lennon and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Porencephaly
-
A cerebrovascular condition characterized by the presence of cystic cavities, which are caused by destructive vascular lesions in the germinal matrix.
- Schizencephaly
-
A rare congenital cerebral condition characterized by unilateral or bilateral abnormal clefts or splits of the cerebral hemispheres.
- Stria vascularis
-
A capillary loop within the cochlea that produces endolymph for the scala media, one of the three fluid-filled compartments of the cochlea.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Massoudi, D., Miner, J.H. & Gould, D.B. Collagen IV in Gould syndrome and Alport syndrome. Nat Rev Nephrol 21, 778–793 (2025). https://doi.org/10.1038/s41581-025-00982-x
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41581-025-00982-x
