
Abstract
A recent genetic association study1 identified a gene cluster on chromosome 3 as a risk locus for respiratory failure after infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). A separate study (COVID-19 Host Genetics Initiative)2 comprising 3,199 hospitalized patients with coronavirus disease 2019 (COVID-19) and control individuals showed that this cluster is the major genetic risk factor for severe symptoms after SARS-CoV-2 infection and hospitalization. Here we show that the risk is conferred by a genomic segment of around 50 kilobases in size that is inherited from Neanderthals and is carried by around 50% of people in south Asia and around 16% of people in Europe.
Similar content being viewed by others
Main
The COVID-19 pandemic has caused considerable morbidity and mortality, and has resulted in the death of over a million people to date3. The clinical manifestations of the disease caused by the virus, SARS-CoV-2, vary widely in severity, ranging from no or mild symptoms to rapid progression to respiratory failure4. Early in the pandemic, it became clear that advanced age is a major risk factor, as well as being male and some co-morbidities5. These risk factors, however, do not fully explain why some people have no or mild symptoms whereas others have severe symptoms. Thus, genetic risk factors may have a role in disease progression. A previous study1 identified two genomic regions that are associated with severe COVID-19: one region on chromosome 3, which contains six genes, and one region on chromosome 9 that determines ABO blood groups. Recently, a dataset was released by the COVID-19 Host Genetics Initiative in which the region on chromosome 3 is the only region that is significantly associated with severe COVID-19 at the genome-wide level (Fig. 1a). The risk variant in this region confers an odds ratio for requiring hospitalization of 1.6 (95% confidence interval, 1.42–1.79) (Extended Data Fig. 1).

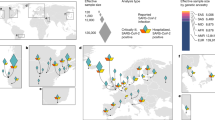
a, Manhattan plot of a genome-wide association study of 3,199 hospitalized patients with COVID-19 and 897,488 population controls. The dashed line indicates genome-wide significance (P = 5 × 10−8). Data were modified from the COVID-19 Host Genetics Initiative2 (https://www.covid19hg.org/). b, Linkage disequilibrium between the index risk variant (rs35044562) and genetic variants in the 1000 Genomes Project. Red circles indicate genetic variants for which the alleles are correlated to the risk variant (r2 > 0.1) and the risk alleles match the Vindija 33.19 Neanderthal genome. The core Neanderthal haplotype (r2 > 0.98) is indicated by a black bar. Some individuals carry longer Neanderthal-like haplotypes. The location of the genes in the region are indicated below using standard gene symbols. The x axis shows hg19 coordinates.
The genetic variants that are most associated with severe COVID-19 on chromosome 3 (45,859,651–45,909,024 (hg19)) are all in high linkage disequilibrium (LD)—that is, they are all strongly associated with each other in the population (r2 > 0.98)—and span 49.4 thousand bases (kb) (Fig. 1b). This ‘core’ haplotype is furthermore in weaker linkage disequilibrium with longer haplotypes of up to 333.8 kb (r2 > 0.32) (Extended Data Fig. 2). Some such long haplotypes have entered the human population by gene flow from Neanderthals or Denisovans, extinct hominins that contributed genetic variants to the ancestors of present-day humans around 40,000–60,000 years ago6,7. We therefore investigated whether the haplotype may have come from Neanderthals or Denisovans.
The index variants of the two studies1,2 are in high linkage disequilibrium (r2 > 0.98) in non-African populations (Extended Data Fig. 3). We found that the risk alleles of both of these variants are present in a homozygous form in the genome of the Vindija 33.19 Neanderthal, an approximately 50,000-year-old Neanderthal from Croatia in southern Europe8. Of the 13 single nucleotides polymorphisms constituting the core haplotype, 11 occur in a homozygous form in the Vindija 33.19 Neanderthal (Fig. 1b). Three of these variants occur in the Altai9 and Chagyrskaya 810 Neanderthals, both of whom come from the Altai Mountains in southern Siberia and are around 120,000 and about 60,000 years old, respectively (Extended Data Table 1), whereas none of the variants occurs in the Denisovan genome11. In the 333.8-kb haplotype, the alleles associated with risk of severe COVID-19 similarly match alleles in the genome of the Vindija 33.19 Neanderthal (Fig. 1b). Thus, the risk haplotype is similar to the corresponding genomic region in the Neanderthal from Croatia and less similar to the Neanderthals from Siberia.
We next investigated whether the core 49.4-kb haplotype might be inherited by both Neanderthals and present-day people from the common ancestors of the two groups that lived about 0.5 million years ago9. The longer a present-day human haplotype shared with Neanderthals is, the less likely it is to originate from the common ancestor, because recombination in each generation will tend to break up haplotypes into smaller segments. Assuming a generational time of 29 years12, the local recombination rate13 (0.53 cM per Mb), a split between Neanderthals and modern humans of 550,000 years9 and interbreeding between the two groups around 50,000 years ago, and using a published equation14, we exclude that the Neanderthal-like haplotype derives from the common ancestor (P = 0.0009). For the 333.8-kb-long Neanderthal-like haplotype, the probability of an origin from the common ancestral population is even lower (P = 1.6 × 10−26). The risk haplotype thus entered the modern human population from Neanderthals. This is in agreement with several previous studies, which have identified gene flow from Neanderthals in this chromosomal region15,16,17,18,19,20,21 (Extended Data Table 2). The close relationship of the risk haplotype to the Vindija 33.19 Neanderthal is compatible with this Neanderthal being closer to the majority of the Neanderthals who contributed DNA to present-day people than the other two Neanderthals10.
A Neanderthal haplotype that is found in the genomes of the present human population is expected to be more similar to a Neanderthal genome than to other haplotypes in the current human population. To investigate the relationships of the 49.4-kb haplotype to Neanderthal and other human haplotypes, we analysed all 5,008 haplotypes in the 1000 Genomes Project22 for this genomic region. We included all positions that are called in the Neanderthal genomes and excluded variants found on only one chromosome and haplotypes seen only once in the 1000 Genomes Project data. This resulted in 253 present-day haplotypes that contained 450 variable positions. Figure 2 shows a phylogeny relating the haplotypes that were found more than 10 times (see Extended Data Fig. 4 for all haplotypes). We find that all risk haplotypes associated with severe COVID-19 form a clade with the three high-coverage Neanderthal genomes. Within this clade, they are most closely related to the Vindija 33.19 Neanderthal.

The coloured area highlights the haplotypes that carry the risk allele at rs35044562—that is, the risk haplotypes for severe COVID-19. Arabic numbers indicate bootstrap support (100 replicates). The phylogeny is rooted with the inferred ancestral sequence of present-day humans. The three Neanderthal genomes carry no heterozygous positions in this region. Scale bar, number of substitutions per nucleotide position.
Among the individuals in the 1000 Genomes Project, the Neanderthal-derived haplotypes are almost completely absent from Africa, consistent with the idea that gene flow from Neanderthals into African populations was limited and probably indirect20. The Neanderthal core haplotype occurs in south Asia at an allele frequency of 30%, in Europe at an allele frequency of 8%, among admixed Americans with an allele frequency of 4% and at lower allele frequencies in east Asia23 (Fig. 3). In terms of carrier frequencies, we find that 50% of people in South Asia carry at least one copy of the risk haplotype, whereas 16% of people in Europe and 9% of admixed American individuals carry at least one copy of the risk haplotype. The highest carrier frequency occurs in Bangladesh, where more than half the population (63%) carries at least one copy of the Neanderthal risk haplotype and 13% is homozygous for the haplotype. The Neanderthal haplotype may thus be a substantial contributor to COVID-19 risk in some populations in addition to other risk factors, including advanced age. In apparent agreement with this, individuals of Bangladeshi origin in the UK have an about two times higher risk of dying from COVID-19 than the general population24 (hazard ratio of 2.0, 95% confidence interval, 1.7–2.4).

It is notable that the Neanderthal risk haplotype occurs at a frequency of 30% in south Asia whereas it is almost absent in east Asia (Fig. 3). This extent of difference in allele frequencies between south and east Asia is unusual (P = 0.006, Extended Data Fig. 5) and indicates that it may have been affected by selection in the past. Indeed, previous studies have suggested that the Neanderthal haplotype has been positively selected in Bangladesh25. At this point, we can only speculate about the reason for this—one possibility is protection against other pathogens. It is also possible that the haplotype has decreased in frequency in east Asia owing to negative selection, perhaps because of coronaviruses or other pathogens. In any case, the COVID-19 risk haplotype on chromosome 3 is similar to some other Neanderthal and Denisovan genetic variants that have reached high frequencies in some populations owing to positive selection or drift14,26,27,28, but it is now under negative selection owing to the COVID-19 pandemic.
It is currently not known what feature in the Neanderthal-derived region confers risk for severe COVID-19 and whether the effects of any such feature are specific to SARS-CoV-2, to other coronaviruses or to other pathogens. Once the functional feature is elucidated, it may be possible to speculate about the susceptibility of Neanderthals to relevant pathogens. However, with respect to the current pandemic, it is clear that gene flow from Neanderthals has tragic consequences.
Methods
Linkage disequilibrium was calculated using LDlink 4.129 and alleles were compared to the archaic genomes8,9,10,11 using tabix30 (HTSlib 1.10). Haplotypes were constructed from the phase 3 release of the 1000 Genomes Project22 as described. Phylogenies were estimated with phyML 3.331 using the Hasegawa–Kishino–Yano-8532 substitution model with a gamma shape parameter and the proportion of invariant sites estimated from the data. The probability of observing a haplotype of a particular length or longer owing to incomplete lineage sorting was calculated as previously described14. The inferred ancestral states at variable positions among present-day humans were taken from Ensembl33. The distribution of frequency differences of Neanderthal haplotypes between east and south Asia was computed by filtering diagnostic Neanderthal variants (fixed positions in the three high-coverage Neanderthal genomes and the Neanderthal allele missing in 108 Yoruba individuals) using a published introgression map20, followed by pruning using PLINK1.9034 (r2 cut-off of 0.5 in a sliding window of 100 variants) and allele frequency assessment in the 1000 Genomes Project. Maps displaying allele frequencies and linkage disequilibrium in different populations were made using Mathematica 11.0 (Wolfram Research) and OpenStreetMap data.
For the meta-analysis carried out by the COVID-19 Host Genetics Initiative2, participants consented and ethical approvals were obtained (https://www.covid19hg.org/partners/). The following eight studies contributed to the meta-analysis of hospitalization versus population controls: Genetic modifiers for COVID-19-related disease ‘BelCovid’ (Université Libre de Bruxelles, Belgium), Genetic determinants of COVID-19 complications in the Brazilian population ‘BRACOVID’ (University of Sao Paulo, Brazil), deCODE (deCODE Genetics, Iceland), FinnGen (Institute for Molecular Medicine Finland, Finland), GEN-COVID (University of Siena, Italy), Genes & Health (Queen Mary University of London, UK), COVID-19-Host(age) (Kiel University and University Hospitals of Oslo and Schleswig-Holstein, Germany and Norway) and the UK Biobank (UK).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The summary statistics of the genome-wide association study that support the finding of this study are available from the COVID-19 Host Genetics Initiative (round 3, ANA_B2_V2: hospitalized patients with COVID-19 compared with population controls; https://www.covid19hg.org/). The genomes used are available from the 1000 Genomes Project (phase 3 release, https://www.internationalgenome.org/) and the Max Planck Institute for Evolutionary Anthropology (Chagyrskaya, Altai and Vindija 33.19, http://cdna.eva.mpg.de/neandertal/). The ancestral alleles are available at Ensembl (release 100, https://www.ensembl.org/). Map data are from OpenStreetMap and available from https://www.openstreetmap.org.
References
Ellinghaus, D. et al. Genomewide association study of severe COVID-19 with respiratory failure. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2020283 (2020).
COVID-19 Host Genetics Initiative. The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur. J. Hum. Genet. 28, 715–718 (2020).
WHO. Coronavirus disease (COVID-19) Weekly Epidemiological Update and Weekly Operational Update: Weekly Epidemiological Update 14 September 2020 https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (2020).
Vetter, P. et al. Clinical features of COVID-19. Br. Med. J. 369, m1470 (2020).
Zhou, F. et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395, 1054–1062 (2020).
Green, R. E. et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010).
Sankararaman, S., Patterson, N., Li, H., Pääbo, S. & Reich, D. The date of interbreeding between Neandertals and modern humans. PLoS Genet. 8, e1002947 (2012).
Prüfer, K. et al. A high-coverage Neandertal genome from Vindija Cave in Croatia. Science 358, 655–658 (2017).
Prüfer, K. et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49 (2014).
Mafessoni, F. et al. A high-coverage Neandertal genome from Chagyrskaya Cave. Proc. Natl Acad. Sci. USA 117, 15132–15136 (2020).
Meyer, M. et al. A high-coverage genome sequence from an archaic Denisovan individual. Science 338, 222–226 (2012).
Langergraber, K. E. et al. Generation times in wild chimpanzees and gorillas suggest earlier divergence times in great ape and human evolution. Proc. Natl Acad. Sci. USA 109, 15716–15721 (2012).
Kong, A. et al. A high-resolution recombination map of the human genome. Nat. Genet. 31, 241–247 (2002).
Huerta-Sánchez, E. et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512, 194–197 (2014).
Sankararaman, S. et al. The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507, 354–357 (2014).
Vernot, B. & Akey, J. M. Resurrecting surviving Neandertal lineages from modern human genomes. Science 343, 1017–1021 (2014).
Vernot, B. et al. Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals. Science 352, 235–239 (2016).
Steinrücken, M., Spence, J. P., Kamm, J. A., Wieczorek, E. & Song, Y. S. Model-based detection and analysis of introgressed Neanderthal ancestry in modern humans. Mol. Ecol. 27, 3873–3888 (2018).
Gittelman, R. M. et al. Archaic hominin admixture facilitated adaptation to out-of-Africa environments. Curr. Biol. 26, 3375–3382 (2016).
Chen, L., Wolf, A. B., Fu, W., Li, L. & Akey, J. M. Identifying and interpreting apparent Neanderthal ancestry in African individuals. Cell 180, 677–687 (2020).
Skov, L. et al. The nature of Neanderthal introgression revealed by 27,566 Icelandic genomes. Nature 582, 78–83 (2020).
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68–74 (2015).
OpenStreetMap. Planet OSM. https://planet.osm.org/ (2017).
Public Health England. COVID-19: Review of Disparities in Risks and Outcomes. https://www.gov.uk/government/publications/covid-19-review-of-disparities-in-risks-and-outcomes (2020).
Browning, S. R., Browning, B. L., Zhou, Y., Tucci, S. & Akey, J. M. Analysis of human sequence data reveals two pulses of archaic Denisovan admixture. Cell 173, 53–61 (2018).
Dannemann, M., Andrés, A. M. & Kelso, J. Introgression of Neandertal- and Denisovan-like haplotypes contributes to adaptive variation in human Toll-like receptors. Am. J. Hum. Genet. 98, 22–33 (2016).
Zeberg, H., Kelso, J. & Pääbo, S. The Neandertal progesterone receptor. Mol. Biol. Evol. 37, 2655–2660 (2020).
Zeberg, H. et al. A Neanderthal sodium channel increases pain sensitivity in present-day humans. Curr. Biol. 30, 3465–3469 (2020).
Machiela, M. J. & Chanock, S. J. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015).
Li, H. Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics 27, 718–719 (2011).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Hasegawa, M., Kishino, H. & Yano, T. Dating of the human–ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 22, 160–174 (1985).
Yates, A. D. et al. Ensembl 2020. Nucleic Acids Res. 48, D682–D688 (2020).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Acknowledgements
We thank the COVID-19 Host Genetics Initiative for making the data from the genome-wide association study available, and the Max Planck Society and the NOMIS Foundation for funding.
Author information
Authors and Affiliations
Contributions
H.Z. performed the haplotype analysis. H.Z. and S.P. jointly wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Tobias Lenz, Yang Luo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Odds ratios for hospitalization owing to COVID-19 for cohorts contributing to the meta-analysis (round 3) of the COVID-19 Host Genetics Initiative (rs35044562).
The odds ratio and the P value for the summary effect are odds ratio = 1.60 (95% confidence interval, 1.42–1.79) and P = 3.1 × 10−15 (two-sided z-test, n = 3,199 patients with COVID-19 and 897,488 controls over 8 independent studies). Data are the odds ratios and 95% confidence intervals. HOST(age), UK Biobank European (EUR), GENCOVID, deCODE and BelCovid use European population controls. BRACOVID, Genes & Health and FinnGen use American, south Asian and Finnish population controls, respectively.
Extended Data Fig. 2 Pairwise linkage disequilibrium between diagnostic Neanderthal variants.
Heat map of linkage disequilibrium between genetic variants in which one allele is shared with three Neanderthal genomes and missing in 108 Yoruba individuals. The black box highlights a haplotype of 333.8 kb between rs17763537 and rs13068572 (chromosome 3: 45,843,315–46,177,096). Red, r2 correlation; blue, D′ correlation.
Extended Data Fig. 3 Linkage disequilibrium between index variant rs11385942 and the index variant of the COVID-19 Host Genetics Initiative (rs35044562).
Shades of red indicate the extent of linkage disequilibrium (r2) in the populations included in the 1000 Genomes Project. Populations labelled ‘n/a’ are monomorphic for the protective allele of rs35044562. The previously described index variant (rs11385942)1 does not have any genetic variants in linkage disequilibrium (r2 > 0.8) in populations from Africa. Map source data from OpenStreetMap23.
Extended Data Fig. 4 Phylogeny of haplotypes in individuals included in the 1000 Genomes Project and Neanderthals covering the genomic region of the core risk haplotype.
The shaded area highlights a monophyletic group that contains all present-day haplotypes carrying the risk allele at rs35044562 and the haplotypes of the three high-coverage Neanderthals. Arabic numbers show bootstrap support (100 replicates). The tree is rooted with the inferred ancestral human sequence. Scale bar, number of substitutions per nucleotide position.
Extended Data Fig. 5 Frequency differences between south and east Asia for haplotypes introgressed from Neanderthals.
The dashed line indicates the frequency difference for the Neanderthal haplotype that confers risk of severe COVID-19.
Supplementary information
Rights and permissions
About this article

Cite this article
Zeberg, H., Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 587, 610–612 (2020). https://doi.org/10.1038/s41586-020-2818-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-020-2818-3
This article is cited by
-
Endometriosis - on the intersection of modern environmental pollutants and ancient genetic regulatory variants
European Journal of Human Genetics (2026)
-
Ancient genomes give insight into 160,000 years of East Asian population dynamics and biological adaptation
Genome Biology (2025)
-
Exploring the genetic landscape of COVID-19 susceptibility and severity among patients in Türkiye
BMC Medical Genomics (2025)
-
Evolution, genetic diversity, and health
Nature Medicine (2025)
-
Severe COVID-19 disease is associated with genetic factors affecting plasma ACE2 receptor and CRP concentrations
Scientific Reports (2025)

Benedikt Buechel
Is nature aware what the political implications of this paper may be, what it could mean for refugees from South Asia? Why is it possible that ap writes an article that is then published in newspapers all over the world (in some already on the 8th of July https://www.wienerzeitung.a... before the final paper is published in nature?
Shalako77 Replied to Benedikt Buechel
I think mainly people just want to stay alive, regardless of whose politics it offends
ciaparker2 Replied to Benedikt Buechel
Truth is important. Political correctness is not, especially when it requires lies or the concealment of the truth.
Harold Morris Replied to ciaparker2
Truth is important. If you are to find it then you must look well beyond this article.
Uwe Holger Replied to Benedikt Buechel
Why don't you show the way and open your own house to the flood of South Asian intruders?
Preferably the women in your family should be in close contact with these creatures.
What a sick bastard!
Laughing_Coyote
Since it is "currently not known what feature in the Neanderthal-derived region confers risk for severe COVID-19," is it possible that other factors are at play in certain immigrant populations in UK that affect the higher rate of more severe forms of the disease? For example, changes in diet, life-style, stress, etc.? For example I understand there is high rate of diabetes among South Asian populations, which also is a risk factor affecting the progress of Corona virus.
Susan Ogg OConnor Replied to Laughing_Coyote
This is a prelim study, there's a lot we don't know yet about implications, and correlation does not always equal causation. But we do know there are genetic traits that impact disease, as well as lifestyle and comorbid disease types.
The neanderthal traits are originating from northern europe and there is a huge Irish diaspora. I'm of Irish/English descent, 2nd generation american. And according to 23 and me, I have a high percentage of neanderthal traits. I am otherwise low risk. While this is prelim, it's enough for me to take my concern for infection up slightly. (Not much, but previously, my assumption would be if I got it, my biggest risk would be spreading it to others. Now there is more potential risk to my own health.)
Thomas Müller
The comment by Uwe Holger is totally inappropriate and personally insulting to Benedikt.
The concern of Benedikt is totally valid, in particular in this time when a lot of preliminary, unconfirmed or simply wrong information is circulated and causes confusion in the population.
mossdale Replied to Thomas Müller
Interesting he missed the point that the gene, as far as we know, originated with Neanderthals, who populated Europe as well as parts of Asia (or that this gene is currently in European populations, albeit at a lesser percentage).
Uwe Holger Replied to Thomas Müller
The truth is insulting to intellectually dishonest people..
Every time is see someone wailing over why something was published in the first place, not because of its truth or falsehood, but because it may offend some PC moron's self esteem, I shudder. Speech codes on college campuses. "Hate" speech, whatever that term means.
"Safe spaces" for braindead imbeciles who should not even be near an institution of higher learning.
Unbelievable degeneracy!
Stalin's Soviet paradise comes to mind. Or the Terror during the French Revolution.
Disgusting!
I guess history repeats itself in cycles.
D
Uwe Holger Replied to Thomas Müller
Benedikt did not say that the claims in the article are untrue or unverified.
His main problem was that the poor millions in South Asia may be rejected at the gates of Europe as a a result of the facts outlined. It would break his tender heart........
"Willkommen Kultur" is the death of Europe. It is crystal clear to anyone with a brain. One does not need the Neanderthal genes angle to see that.
Klampettsen
How do we know for sure this very short stretch (about 50Kb) of DNA is indeed 'Neanderthal'? It could be sapien in origin and ended up in Neanderthal. It could be both Neanderthal and sapien - descended from a common ancestor. Fact is we don't actually know for sure and the claims being presented here in this 'stamp collection' represent little more than an untestable hypothesis. A just-so story. At best it's likely to be abused as 'evidence' Germany has few Neanderthals and Sweden's full of them. I'm really struggling to understand why this idea was even submitted let alone published. It adds nothing of any interest to a better understanding of, and ability to deal with, SARS-CoV-2. It certainly isn't publishable science, frankly.
Bsr Sharma Replied to Klampettsen
Don't you think the following data deserves some genetic research?
Covid-19 Death Statistics: (per million)
Eritrea: 0
Burundi: 0.08
Tanzania: 0.3
Mozambique: 2
Uganda: 2
Rwanda 2
Western Sahara: 2
DRC: 3
Benin: 3
Burkina Faso: 3
Niger: 3
South Sudan: 4
Nigeria: 5
Ivory Coast: 5
Guinea: 5
Chad: 5
Angola: 6
Somalia: 6
Mali: 6
Togo: 6
U.K.: 621
U.S.A. 640
Spain: 684
Belgium: 863
Marlene Toscano Replied to Bsr Sharma
What about Mexico and South America??
Bsr Sharma Replied to Marlene Toscano
Many South/Latin American countries have high death rates. But, I don't know if they have enough Neanderthal ancestry. May be a little bit from the European side and not much from the Amerindian side. But Europe Vs. Africa provides the best contrast in terms of Neanderthal gene set since Africans have none and Europeans have it. It also is interesting in that Europe has the best medical care ("First Word") while Africa has little in terms of medical care. So, the 100:1 death rate stands out even more. My guess is South/Latin American's have an in between healthcare: Better than Africa but worse than Europe.
CovidSurvivor Replied to Bsr Sharma
These statistics overlook the significant difference in rates of Covid-19 testing. If they don't test for it, they don't find it. The United States cannot seem to get to providing one million tests a day (estimates are that 4.4 million per day are needed just to contain the virus), so there's no way poor nations, which often receive their medical supplies from Western nations, would be able to supply enough testing to provide meaningful information about the infections in their borders. The only real way to look at Covid-19 deaths in these areas is to look at deaths in excess of the normal. Of course, even there issues remain that in developing nations some deaths are simply never reported or information perceived as negative is altered.
Bsr Sharma Replied to CovidSurvivor
Agree testing is inadequate/non-existent in many African countries. However, large death statistics (equal or exceeding Europe in proportion) would have attracted attention from medical/scientific community as well as media. The initial crisis in China and later problems in Italy, Spain, Brazil, Mexico, U.S.A. are all widely known. When Ebola epidemic was killing in the hundreds in Africa, it was a big news. Similarly Zika in South America. African nations (except may be South Africa) are hardly mentioned in any news reports of Covid-19 Pandemic. This shows the impact is very low to non-existent in most African countries.
Klampettsen Replied to Bsr Sharma
Africa is infinitely more diverse genetically due to its much, much longer human history. Therefore it's quite possible greater resistance to covid-19-like viruses in African populations explains the much lower death rates. Although we need to be cautious here, because it's early in this pandemic. There are some potentially confounding factors, including - as already mentioned - stage of epidemics; Africans are generally less mobile potentially slowing contagion; and broadly lower population densities in Africa relative to, say, much of Europe.
Bsr Sharma Replied to Klampettsen
Your observation on how a long history and diversity may help increase immunity makes sense intuitively. But, why is it not helpful in other infectious diseases like Malaria, Tuberculosis, HIV/AIDS, Ebola, Yellow Fever etc., that have ravaged Africa? But I agree we have to wait for some time for the Covid-19 disease to reach steady state before inferring genetic differences. If, a year from now, we still see this 1:100 ratio, we may be a lot more confident that something fundamental at genetic level is probably at work.
Klampettsen Replied to Bsr Sharma
But, why is it not helpful in other infectious diseases like Malaria, Tuberculosis, HIV/AIDS, Ebola, Yellow Fever etc? What makes you so sure it isn't? Would we observe the same levels of natural immunity in, say, European populations, if these diseases were endemic in Europe? Relatively speaking, I'd expect a significant difference - less immunity in European populations. In terms of the effects genetics might have, we should consider the epigenome - the interface of genetics and environment - of black males living in Africa vs those living outside of Africa (who are currently assumed to be at a greater risk from covid-19). The genome alone might not be sufficient to shed any light on the matter.
Bsr Sharma Replied to Klampettsen
Would we observe the same levels of natural immunity in, say, European populations, if these diseases were endemic in Europe? Yes, I had not thought about that. Black Plague and 1918 Influenza pandemic show that Europe may have worse outcomes if natural conditions prevail.
Susan Ogg OConnor Replied to Klampettsen
Because we know the genome of neanderthals, we have their genetic samples from ones that froze in ice. We can compare that to sapiens and know which genes came from where.
Klampettsen Replied to Susan Ogg OConnor
Yeah, that's little more than an insufficiently tested hypothesis, based on very limited sampling therefore prone to bias. We really shouldn't assume our knowledge here is sufficiently detailed, because it isn't. Let's be honest, anthropologists/archeologists sequencing genomes is hardly science, is it?
Susan Ogg OConnor Replied to Klampettsen
You don't think sequencing genomes is science?
It's a correlation right now, and it's premature to enact wide-scale policy or treatment based on it, but we're trying to find correlations and then hopefully causations within that.
Klampettsen Replied to Susan Ogg OConnor
Sequencing genomes is technology, like any omics platform. Science is a methodology based on good experimental design mainly. It ceases to amaze me how many scientists these days don't seem to actually know what science is. Education was clearly dumbed down at some point. Don't take it personally, I don't blame you.
golfcrackerjack
Odd, in light of the fact that, outside Africa anyhow, blacks are more likely to die than whites. (Yes, I realize that there are all sorts of factors in play, but if so, why is this important enough to publish in Nature?)
Susan Ogg OConnor Replied to golfcrackerjack
Because the risks that many black people are facing are because of systemic racism, which are potentially modifiable by society, but genetic cannot be modified. Additionally, if that portion of the genome is predisposing you to severe covid disease, that portion can be further studied and provide greater understanding to *all* patients.
Lyngvyst Replied to Susan Ogg OConnor
It has clearly a lot to do with vitamin D levels. The darker our skin is, the longer it takes to create enough vitamin D from the Sun. Meaning it is a bit of disadvantage to be out of Africa (or other sunny regions) with darker skin. White skin is actually an adaptation to the Northern climate with little sun as opposed to sunshine in Africa or around the Equator. So if we filter out ideology or desire to be praised for racism vice versa, we can only find the vitamin D levels and their difference as a significant factor for this.
Klampettsen Replied to Lyngvyst
Outside of peak sunlight levels (the summer months) in the northern hemisphere neither black Africans nor white Europeans produce much vitamin D. Black people live perfectly healthy lives in northern Europe. We should note too that Europeans were thought to be very dark skinned not that long ago and Inuits are not exactly pale. That's not to say vitamin D isn't an important factor, but I'm not aware of any irrefutable evidence suggesting it's not simply a correlation hitching a ride promoting a falsehood and, yes, promoting racism in the dullest minds.
Lyngvyst Replied to Klampettsen
Well, yes, but Inuits eat a lot of vitamin D in fish/seafood and that makes quite a lot of difference in their blood levels of vitamin D. Levels created in summer last many months, decreasing slowly and it is highly probable that levels in people with lighter skin last longer as they are higher in summer (provided they go outside). No racism there, this is just a wave that someone pushes on the left to divide us which in some people works, unfortunately. Not in people who know we all came ultimately from Africa.
Susan Ogg OConnor Replied to Lyngvyst
Vitamin D levels are frequently looked at but they are a consequence of poor health in general, not necessarily an independent factor.
golfcrackerjack Replied to Susan Ogg OConnor
Nonsense, sorry. First of all, it's my guess you mean "systematic" racism; "systemic" racism would be genetic and not modifiable. Second, systematic racism is a trope... and a tiresome one at that.
Cornucopial Replied to golfcrackerjack
No, actually the term is, in fact, "systemic racism," meaning racism affects many systems, like policing, or health care, in the entire country. The term "systematic" has a different meaning, indicating plans or methods.
Dictionary.com says,
"Especially when talking about social institutions like government or healthcare, systemic is used when discussing something that affects the whole—systemic problems, systemic change."
Merriam-Webster says:
"a systematic approach to learning that involves carefully following the program's steps. Systemic describes what relates to or affects an entire system."
And it's very easy to call it a tiresome "trope" when you are privileged to not suffer its effects -- systemically.
golfcrackerjack Replied to Cornucopial
Good response, thanks.
The trope remains tiresome, I know its effects and they too are tiresome... on that we agree.
But systemic as you mean it is a huge overstatement -- one that affects all.
Klampettsen Replied to golfcrackerjack
Arguing semantics - especially when you've clearly misinterpreted the meaning of a phrase - can only get you in 'it' up to your neck.
Cornucopial Replied to golfcrackerjack
I disagree it's a huge overstatement. What's huge is the number of systems that racism does in fact affect. But no point in arguing, as Klampettsen points out below. We clearly have different points of view.
Susan Ogg OConnor Replied to golfcrackerjack
No, at least in the US, it's systemic racism. Maybe your country calls it something different, but that's not a reason to superciliously "correct" me. And it is not simply a trope, it is a major concern. I'm sorry that you as a white man are bored with it.
Klampettsen Replied to Susan Ogg OConnor
But genetics can be modified by the environment. It's called epigenetics and the epigenome. It's possible a potentially protective element in the African genome is silenced in the northern hemisphere. It's difficult to imagine any other factor, especially re data for the UK. Claims it's simply down to racism risk missing real biological causes for what appear to be higher death rates among British black males.
Susan Ogg OConnor Replied to Klampettsen
It's absolutely possible, but are you trying to say there's no racism against black people in the UK?
Klampettsen Replied to Susan Ogg OConnor
No, I believe you typed that, Susan. That 'there's no racism against black people in the UK'. Unfortunately there is, but not as much as elsewhere, e.g., in Europe and the US.
Kim_Lavransdatter Replied to Susan Ogg OConnor
"
It's absolutely possible, but are you trying to say there's no racism against black people in the UK?" That's a specious unscientific strawperson argument. Because "A" may explain some cases, therefore "A" explains all cases.
Mike Ehrmantraut Replied to Susan Ogg OConnor
Shouldn't you be commenting on a political ideological site? Because your comments don't belong here.
John Moore Replied to Susan Ogg OConnor
Since this is a website about science, the political junk term - "systemic racism" - does not belong.
Beyond that, attributing differences to that term is very weak - more a political statement than a reality. Racism in the US is way down from the past, and is lower than in many countries. "Systemic racism" would appear to mean racism by the "system" - which is even weaker.
There are certainly significant average distances in the US black population, but other causes than racism - espeically if you mean current racism - are significant. If you want to talk past racism - hey, that cannot be modified.
www.vintageclothesretro.com Replied to golfcrackerjack
you are ignorant https://www.youtube.com/wat...
golfcrackerjack Replied to www.vintageclothesretro.com
And you would know? Don't make me laugh, I don't want to spew my coffee.
That said, the woman who made this vid seems not only frustrated but friable. But, hey, it is NY -- where Cuomo insisted on sending infectious Covid patients back into nursing homes. Remember who did it -- despite fully set-up sites staffed by competent providers at Javits and on USS Mercy.
The sort of medicine she decries is typical -- esp. in socialized systems -- or in emergencies. And, to her credit, nurses often know more about practical medicine than do many docs. Interns and residents often know close to nothing. But racism? Only insofar as that a lot of patients that they were seeing were minorities.
I find myself wondering when this vid was made. May? That's my guess, 'cause remdesivir had got its EUA then. Happily, what she is describing is not likely applicable any longer.... But remember, Cuomo couldn't give Trump credit for getting COVID capable hospitals set up and staffed pronto -- instead Andy decided on sending patients into what you see here -- back when way, way too many people were getting intubated.
Yet I share your sentiment that it's all very sad. But racist? Sorry....
Mike Ehrmantraut Replied to www.vintageclothesretro.com
Another one with something intelligent to say... what is wrong with you people? Are you all inept, uneducated politically drive children?
Klampettsen Replied to golfcrackerjack
It's not odd at all. As anyone remotely familiar with the field of epigenetics could explain to you. I generally don't waste my time educating fools, so you'll have to ask someone else.
golfcrackerjack Replied to Klampettsen
Thanks for such a polite response. Instead you scream -- from a field of possibilities -- that the differences are the result of epigenetics (in this case heritable epigenetic changes to protein expression, yes?)
Seems possible, but why are you so sure? Occam's razor, if nothing else, would suggest not.
More likely, as some commenters have suggested -- is the "average" black outside of Africa lives longer and I'll add is likely more prone to metabolic syndrome than is the average black in sub-Saharan Africa -- who is both younger and skinnier. And that even if the authors' conjectures are accurate, the effects of metabolic syndrome overwhelm the effect of the Neanderthal insertions among blacks out of Africa, esp. in the U.S.
And one would need to adjust for age to make any such straight-up comparison -- so the rate among black Africans may be lower because they tend to be younger... and skinnier.
My conjecture -- and that's all it is -- would seem a lot more likely than your simplistic overarching that it must be some sort of heritable epigenetic phenomenon -- for reasons unexplained.
And this article doesn't seem weighty enough for Nature, but it's sensational, so maybe Nature's approach to selling its issues on newstands is evolving.
Be well.
Klampettsen Replied to golfcrackerjack
Metabolic syndrome is a socioeconomic disease associated with inequity and poverty. Ethnicity is not a driver of metabolic syndrome. The environment and epigenetics are the drivers. As far as I’m aware the covid-19 data is insufficient to suggest black men are more prone to metabolic syndrome. And it is just one of a number of risk factors now associated with covid-19 severity.
golfcrackerjack Replied to Klampettsen
You failed to answer my observations regarding what would seem to be necessarily heritable epigenetic changes under your theory. Sorry, but it seems you may have recently read an article that decried the old term "junk DNA" and you learned for the first time that it's not so junky after all. Which makes it LOL time -- as you dismiss other thinking as sophomoric.
But the fairly recent field of epigenomics that doesn't mean epigenetics is tightly tied to everything, esp. in a first order sort of way -- as you seem to think.
Sometimes, in fact I'd suggest most often, causality is much more pedestrian, esp. when not considered and adjusted for in what is an interesting"handwaver" paper that hardly helps explain -- rather goes to the contrary -- regarding blacks in America are substantially more prone to Covid morbidity and mortality than others who have the Neanderthal set. (Of course, many if not most blacks in America may have the gene set as the result of generations of mixed parentage, but that's only a thought.)
Klampettsen Replied to golfcrackerjack
One of us has an education and career in molecular genetics. One of us doesn't. Can you guess who's who? Or is that beyond you too?
1bestdog Replied to Klampettsen
Again, what's with the extreme preening arrogance?
Mike Ehrmantraut Replied to Klampettsen
Obviously, that would be golfcrackerjack.
How about justifying your statement instead of ad hominem attacks. Makes you look like a petulant child... and not an educated professional.
1bestdog Replied to Klampettsen
Why must you be rude?
André
Don't forget the study https://www.cnr.it/it/comun...
Dutrás Lali
This may explain why the Hungarians wrote and persecuted the Longobards during the conquest. The peoples found in the Carpathian Basin were usually forced into slavery. However, the Longobards were expelled and settled in northern Italy. The events in Lombardy I think everyone knows ....
Uwe Holger Replied to Dutrás Lali
The Lombards left Western Pannonia and moved to Northern Italy more than 200 years before the Hungarian tribes moved in through what they call the "Vereckei hago" under Arpad. I have no idea how you pull things out of thin air. In fact your entire post makes no sense at all.
Uwe Holger
The censor at Disqus who deletes all my posts almost as soon as I post should find a tall bridge and jump off. Good Riddance! I am done with Disqus.
marymig
A critique here of the the preprint: https://medium.com/@johnhaw...
Harold Morris
CCR5 Δ32: Plague Protection Gene - This article is another example of Euro Psychobabble masquerading as impartial science. CCR5 Δ32, which is most prevalent in Europeans and Han Chinese, is a Neanderthal gene allele, located on chromosome 3. Most Heterozygous carriers often have less severe symptoms than those with the 'complete gene'. Confers resistance to HIV-1, Dengue fever, smallpox, bubonic plague, Ebola, COVID-19 and others. | Galvani AP, Slatkin M (Dec 2003). "Evaluating plague and smallpox as historical selective pressures for the CCR5-Delta 32 HIV-resistance allele". Proceedings of the National Academy of Sciences of the United States of America. | ACE2 Gene Coding Variants and COVID-19 Susceptibility The ACE2 gene encodes the Angiotensin-Converting Enzyme-2. Recent studies proved it to be the receptor for viral entry of SARS-1, SARS-2 (COVID-19) and the human respiratory coronavirus NL63. Several ACE2 variants associated with CCR5 Δ32, could "reduce the association" between ACE2 and S-protein in SARS-C -oV or NL635. | Libert F, Cochaux P, Beckman G, Samson M, Aksenova M, Cao A, et al. (Mar 1998). "The deltaccr5 mutation conferring protection against HIV-1 in Caucasian populations has a single and recent origin in Northeastern Europe". Human Molecular Genetics. 7 (3): 399–406. doi:10.1093/hmg/7.3.399. PMID 9466996 | Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations, Yanan Cao, Lin Li, Zhimin Feng, Shengqing Wan, Peide Huang | Investigating the Likely Association Between Genetic Ancestry and COVID-19 Manifestation - Ranajit Das* and Sudeep Ghate, Yenepoya Research Centre | https://www.medrxiv.org/con... 7, 2020. 04.05.20054627v1.full.pdf | ...To this end we employed 10,215 ancient and modern genomes, obtained from the databank of Dr. David Reich, Harvard Medical School, USA (https://reich.hms.harvard.e... to evaluate the likely correlation between European ancestry and COVID-19. Our findings revealed significant positive correlation between WHG-related ancestry and COVID-19 death/recovery ratio and marginally significant negative correlation between...
FrenchCitizen
Two comments.
1. African people should be less harmed by the virus according to the study and the official death rates are indeed lower in Africa. Knowing the state of public health infrastructure there, the African statistics are to be taken with a pinch of salt. I read actually that African-American and Hispanic people had higher covid mortality rates in the US, which might point to the opposite.
2. Is it possible that another gene or gene combination is the ultimate culprit? Correlation is not causation and one reason for this correlation would e.g. be that the Neanderthal gene and the actual culprit have a correlated prevalence (because they are e.g. incompatible and their joint presence harm the bearer).
Wes Replied to FrenchCitizen
South Africa's data is pretty solid and shows the same pattern of relatively low death rates. Furthermore, many countries in Africa deal with infectious disease, so their infrastructure for recording this is also pretty solid.
Klampettsen Replied to FrenchCitizen
Due to recent epidemics many African countries have adequate networks capable of monitoring the situation. Africans generally express higher tolerance to contagion, most likely due to being much more genetically diverse. Genetic diversity - the most valuable biological currency - accumulates over time. African populations are among the old in human history. Outside Africa, in the northern hemisphere, for instance, epigenetic factors might explain the reported higher death rates among black men
RealDumbTrump
Seemingly in contrast to this analysis is the
higher mortality rate in African–Americans.
Jean Oliveira Replied to RealDumbTrump
GENETIC risk. There is a number of risk factors associated. In US, for example, obesity reaches almost 40% of the population.
Neversleep Replied to Jean Oliveira
And guess who is quite an obesity individual... (answer : Trump ).
Mike Ehrmantraut Replied to Neversleep
And what exactly does Donald Trump have to do with, moron? What? Are you 16 years old?
RealDumbTrump Replied to Jean Oliveira
Does not explain why afroamericans have a three times higher mortality? Or do you want to claim that afroamericans are 3 x more obese than caucasians? Doubt it? General population risk factors do no apply to the context of the paper and my comment.
Jean Oliveira Replied to RealDumbTrump
Obesity is just one among many risk factors.
veggiedude Replied to RealDumbTrump
Most likely due to vitamin D deficiency. Dark skinned people do not get much from the Sun, the melatonin protects them from skin cancer but this advantage has a disadvantage of less Vit D absorption. They have mixed with whites over the centuries and have the Neanderthal DNA. Other reasons still apply; such as likely to have poorer paying jobs which means less healthcare choices, and have jobs that are making them more prone to make contact with others.
RealDumbTrump Replied to veggiedude
Vitamin D deficiency is a symptom, not a cause of severe Covid-19. Most likely the socio-economic situation in a population suffering from severe racism is the reason for the high mortality rate, demonstrating that the Neanderthal genes maybe the most important genetic risk factor, BUT overall just a minor factor wrt mortality.
Margalit Replied to veggiedude
I thought so too. But in the UK they looked at it and it doesn't explain. Race when accounting for Vit D still remains significant, Vit D once accounting for race goes away.
Anna Pin
Any idea of the specific RSID?
Neversleep
Thanks. Now I know why I do NOT prune to infections. I have never a fever.
Johannes van Beek
Advanced age is a major risk factor for severity of COVID-19, as stated in this article. The article proposes that a Neanderthal haplotype (chromosome 3: 45,859,651-45,909,024, hg19) is also a risk factor for COVID-19. I wonder whether correlation between age and frequency of occurrence in the population of this Neanderthal haploptype may explain (part of) the association between the haplotype and hospitalization for COVID-19. Are the controls not substantially younger than the cases? In a previous study showing association of this region with respiratory failure because of COVID-19 the controls were about 20 years younger than the cases (Ellinghaus D. et al., New Eng J Med, June 17, 2020, DOI: 10.1056/NEJMoa2020283; Supplementary Table S1C).
Some genes have been reported to be positively associated with longevity (Deelen J. et al., Nature Communications, 2019, https://doi.org/10.1038/s41... at odds ratios approaching the 1.6 reported for association between the Neanderthal haplotype and COVID-19 in the present study. For someone not an expert in GWAS analysis, it is not easy to find in the present article how correlation between age and occurrence of the Neanderthal haplotype is controlled for.
The article refers to a previous report which suggests that this Neanderthal haplotype has been positively selected in the past, perhaps because of protection against other pathogens. The authors propose that it would have the opposite effect on SARS-CoV-2. Did the analysis prove that correlation between frequency of this Neanderthal haplotype and ageing does not explain (part of) the association with COVID-19 hospitalization? If not, a positive effect of the Neanderthal haplotype on ageing could be mistaken for a negative effect on COVID-19.
Darien Castro
My name is Darien Castro. Biologist
I was fascinated with your research about Neanderthal inheritance with lymphocytes receptors and I'm curious about the following.
The article mentions that HIV immunity is not significant to explain in a time range the positive selection of these genes due to this virus appear so lately, however we need to understand that maybe have existed virus outbreaks or strains in prehistoric periods especially in late Glaciation when some pathogens as Anthrax started to spread around communities because of ice melting (Rayamajhi et al. 2012; Yarzábal et al. 2021, McMichael 2015). The Anthrex Toxin inhibit the Lung B cells receptors and can promote cytokine storm similar to COVID-19 severe pattern (McMichael 2015). Another possibility in paleonthology is to study if epidemic events could promote the extinction rate in Neanderthals and maybe they had continuous pathogen risks as selective pressure in these genes of lymphocytes receptors(Rayamajhi et al. 2012; McMichael 2015).
Best wishes and I would like to know your opinion.
References:
McMichael, A. J., (2015) Extreme weather events and infectious disease outbreaks, Virulence, 6:6, 543-547, DOI: 10.4161/21505594.2014.975022
Rayamajhi M, Delgado C, Condon TV, Riches DW, Lenz LL. Lung B cells promote early pathogen dissemination and hasten death from inhalation anthrax. Mucosal Immunol. 2012 Jul;5(4):444-54. doi: 10.1038/mi.2012.21. Epub 2012 Apr 4. PMID: 22472773; PMCID: PMC5745579.
Yarzábal, L.A., Salazar, L.M.B. & Batista-García, R.A. Climate change, melting cryosphere and frozen pathogens: Should we worry…?. Environmental Sustainability 4, 489–501 (2021). https://doi.org/10.1007/s42...
Zeberg, H., Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 587, 610–612 (2020). https://doi.org/10.1038/s41...