
Abstract
Every generation, the human genome is shuffled during meiosis and a single fertilized egg gives rise to all of the cells of the body1. Meiotic errors leading to chromosomal abnormalities are known causes of pregnancy loss2,3, but genetic aetiologies of euploid pregnancy loss remain largely unexplained4. Here we characterize sequence diversity in early pregnancy loss through whole-genome sequencing of 1,007 fetal samples and 934 parental samples from 467 trios affected by pregnancy loss (fetus, mother and father). Sequenced parental genomes enabled us to determine both the parental and meiotic origins of chromosomal abnormalities, detected in half of our set. It further enabled us to assess de novo mutations on both homologous chromosomes from parents transmitting extra chromosomes, and date them, revealing that 6.6% of maternal mutations occurred before sister chromatid formation in fetal oocytes. We find a similar number of de novo mutations in the trios affected by pregnancy loss as in 9,651 adult trios, but three times the number of pathogenic small (<50 bp) sequence variant genotypes in the loss cases compared with adults. Overall, our findings indicate that around 1 in 136 pregnancies is lost due to a pathogenic small sequence variant genotype in the fetus. Our results highlight the vast sequence diversity that is lost in early pregnancy.
Similar content being viewed by others


Main
Germline mutations are transmitted from parents to their offspring5. They occur before the formation of a fertilized egg, are found in all cells of the offspring and can be assessed by sampling somatic tissues. Considerable knowledge exists on germline mutations that appear de novo in offspring (DNMs). The majority of DNMs are of paternal origin, although there are genomic regions within which parental contributions are almost equal6. DNMs contribute to disease risk7, their frequency correlates with parental age at conception6,7 and they cluster at areas near recombination events8,9. However, little is known about DNMs that prevent a fertilized egg from developing to term and it is unclear why some sites, or genes, lack sequence variants10,11 and why some variants are never observed in homozygous state12.
Recombination shuffles the genome between generations, enabling selection of certain haplotypes in favour of others13. Although this provides an evolutionary advantage, the recombination process comes at a cost, as it is mutagenic8,9. Recombination failure during meiosis can also result in aneuploidies, that is, extra (trisomies) or missing (monosomies) chromosomes1,14. A quarter of all pregnancies ends in pregnancy loss15 and approximately half of all losses are explained by single chromosome aneuploidies or an extra set of chromosomes (triploidies)14. We previously identified that a sequence variant in SYCE2 associates with recombination patterns and pregnancy loss16, with a more pronounced effect on longer chromosomes. Aneuploidies of larger chromosomes are rarer in pregnancy loss than in blastocysts from in vitro fertilization3. Overall, this indicates that there is considerable structural variation present in early pregnancy loss that is only accessible by directly sampling products of conception that are not carried to term.
Finding the genetic causes of chromosomally normal (euploid) pregnancy loss has also proved challenging. Previous genetic studies on euploid pregnancy loss have mainly focused on late losses or on pregnancies complicated by structural anomalies17,18, while studies involving earlier losses have been limited by inaccessibility of fetal material and maternal cell contamination19. The role of small sequence variants (SSVs; small indels and single-nucleotide variants (SNVs)) and copy-number variants (CNVs) has therefore not been directly assessed at scale in early pregnancy loss.
Despite the hefty societal cost, early pregnancy loss is under-researched and there is a paucity of therapeutic interventions15. The Copenhagen Pregnancy Loss (COPL) study was initiated to improve the understanding of pregnancy loss by applying large-scale recruitment of consecutive patients with clinically diagnosed pregnancy loss, detailed phenotyping and comprehensive sampling of fetal and parental material. Here we present the results from our genomic study of 467 trios affected by pregnancy loss (fetus, mother and father), including 1,007 fetal samples and 934 parental samples (Fig. 1a). This enabled us to document the sequence diversity that is present only in early pregnancy loss and describe the complex interplay between meiotic recombination and point mutations.

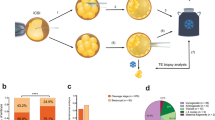
a, Flowchart of the study. b, The gestational ages of fetal samples. The gestational age was estimated as the time difference from the last menstruation to the date of pregnancy loss. c, The paternal fraction of fetal samples. The paternal fraction is shown for intervals of gestational age estimated on the basis of the last menstruation (GA–LM): weeks 0–9, weeks 9–12 and weeks 12–22. d, Kinship between fetal and parental samples. There are 26 (11 pregnancy loss cases) and 93 (59 pregnancy loss cases) fetal samples that had higher than expected kinship with their father and mother, respectively. Furthermore, there are 27 (11 pregnancy loss cases) and 42 (19 pregnancy loss cases) fetal samples from paternal and maternal triploidies, respectively.
The COPL cohort
Most of the pregnancy loss cases occurred in the first 12 weeks of gestation, as measured by time since last menstruation (Fig. 1b). If possible, multiple fetal samples were collected from each pregnancy loss. We sequenced DNA from 1,439 fetal samples from 664 loss cases, along with 1,111 parental samples of predominantly European background (83%), to an average depth of 36.5× (Fig. 1a and Supplementary Figs. 1 and 2). For 467 pregnancy loss cases, we sequenced at least one fetal sample and samples from both parents, with 2.2 samples sequenced on average per fetus (1,007 samples). Collected tissue was dissected and categorized as chorionic villi, fetal and unknown. Chorionic villus samples were the most common (52.4%; Fig. 1b) followed by tissues of unknown (28.1%) and fetal (19.5%) origin.
Tissue samples from pregnancy loss are often maternally contaminated, especially in samples from early loss and in those of unknown origin19,20,21. We found that the paternal fraction (Supplementary Note 1) was less than 25% for 275 out of 1,007 fetal samples, indicating substantial maternal contamination in the cohort (Fig. 1c), comparable to previous studies19.
Triploidies and aneuploidies
Oversharing of alleles between the fetus and mother can indicate either maternal cell contamination or a maternally transmitted triploidy. Triploidies are life-limiting, and known causes of pregnancy loss, particularly in the first trimester2,19,21,22. We found 59 pregnancy loss cases in which at least one fetal sample had higher than expected kinship with the mother, and 11 with the father (Fig. 1d). Maternally transmitted triploidy would present as alternating regions of both homologous chromosomes and of sister chromatids from the mother in the fetal genome (Fig. 2). To discern between maternal oversharing due to contamination and oversharing due to triploidy, we searched these losses for chromosomal crossovers using sequence variants specific to one of the parents (homologous-sister informative alleles; HS alleles; Fig. 2 and Methods). In 19 out of 59 pregnancy loss cases with maternal oversharing, we found numerous crossovers (>20) between sister chromatid state and homologous chromosome state due to meiotic recombination (Figs. 1d and 2). We also found numerous crossovers for all 11 pregnancy loss cases with paternal oversharing. The combined incidence (30 triploidies; 6.4% of the set) is consistent with previous reports2,19,23.

a, Schematic of meiosis II error. Meiosis I error is shown in Extended Data Fig. 1. b, Maternal crossovers at chromosome 2 for a triploid fetus. The phase-informative sequence variants in the fetal genome are informative about transitions between the homologous chromosome and sister chromatid states along the genome. The phase-informative variants are limited to heterozygous (0/1) variants in the mother (1 is the HS allele). In the homologous state, the maternal variants are always present in the fetal genome (0/0/1), as both of the maternal haplotypes are present. In the sister state, the maternal variants are either absent (0/0/0) or have an allelic balance of 2/3 (0/1/1).
We used whole-genome sequencing (WGS) data to search for aneuploidies among the 467 trios affected by pregnancy loss (Methods and Supplementary Note 2), and found them in 206 loss cases (44.1%, 380 fetal samples). Monosomy X (9.9%) and trisomy 16 (9.2%) were the most frequent aneuploidies (Fig. 3a and Supplementary Table 1), comparable with studies using karyotyping or SNP arrays2,19,21,22. WGS also enables the detection of chromosomal structural variants that would not be captured with SNP arrays. We detected 19 large de novo CNVs (>200 kb) in 14 distinct pregnancy loss cases (23 samples) (Methods, Supplementary Fig. 3 and Supplementary Table 2). In five of the loss cases (11 samples), we detected deletion–duplication pairs that were probably part of the same event, three of which represent translocations between two different chromosomes (Fig. 3b and Supplementary Table 2). In two loss cases, we detected a deletion–duplication pair on chromosome 8 (Fig. 3c) that probably represents unbalanced translocations facilitated by non-allelic homologous recombination between the inverted and non-inverted sequence in heterozygous carriers on chromosome 8 (ref. 24).

a, The number and distribution of phased trisomies/monosomies. CNVs are shown in Supplementary Fig. 3. X monosomies are depicted as the loss of the X chromosome (chr.) as we cannot discern between paternal loss of Y and X. b, Translocation of chromosome 15 and chromosome 16. The panels by row are fetal samples from the same pregnancy loss. c, Translocations of 8q. In two pregnancy loss cases, we find pairs of duplications/deletions on chromosome 8 that are probably translocations of 8q. Top, a pregnancy loss in which the mother transmitted the unbalanced translocation. Bottom, a pregnancy loss with a paternally transmitted translocation. The horizontal lines in b and c are the genome-wide averages of the phased coverage. d, Centromere state for trisomies. e, The number of centromeres in a homologous state per triploid case.
We ran structural variant detection25,26 on the flanking regions (±5 kb) of the CNVs to annotate them (Methods and Supplementary Table 2). We found the exact breakpoints for 12 CNVs and read pairs supporting the classification of eight CNVs as part of four translocations (Supplementary Figs. 4–7). Moreover, we found read-alignment patterns of balanced translocations in fathers from two of these losses (Supplementary Figs. 6 and 7), which substantially increases the risk of loss in future pregnancies27. None of the CNVs were near (<100 kb) a common fragile site (Supplementary Table 2). Out of the 14 distinct CNV events, 11 were in euploid losses and 6 of those fulfilled pathogenicity criteria (Methods).
Meiotic origin of the extra chromosomes
At meiosis I, homologous chromosomes segregate to opposite spindle poles, while sister chromatids remain connected at the centromeres28 (Fig. 2a). As crossovers are suppressed at centromeres, the presence of both homologous chromosomes from the same parent at all loci near the centromere (pericentromeric) is indicative of a meiosis I error1 (Extended Data Fig. 1). We found that HS alleles from both homologous chromosomes were generally present at trisomies of chromosomes 15, 16, 21 and 22 (Fig. 3d), pointing to meiosis I errors. Of the 19 maternally transmitted triploidies, 5 and 12 were consistent with meiosis I and II failure, respectively. By contrast, among the paternally transmitted triploidies, we detected homologous chromosomes at the pericentromeres for around half of the chromosomes per fetal sample, pointing to dispermy fertilization29 (Fig. 3e). The two remaining maternally transmitted triploidies showed a similar pattern that might be the product of two independent ova and one spermatid29.
We compared the crossovers identified in the extra chromosomes from triploidies with the sequence-level recombination map from Icelanders30. We found that 27.2% (95% confidence interval (CI) = 23.7–30.6%) and 32.3% (95% CI = 26.6–37.9%; Fig. 4a) of crossovers identified in paternal and maternal triploidies occur within recombination hotspots, respectively (1.6% and 1.8% of the genome). This supports our interpretation that the alternating patterns of homologous-sister segments along the genome represent true crossovers. Although this is substantially less than the fraction of crossovers in recombination hotspots (74.9% and 71.1% for paternal and maternal crossovers, respectively) used to define the recombination map, this could be partly attributed to the window approach used in this study rather than identifying crossovers on the resolution of heterozygous markers.

a, Hotspot use of crossovers in pregnancy loss. The dashed lines are the sex-specific use of hotspots from a previous study8 (male is blue and female is red). The points are the mean values and vertical error bars are the 95% CIs based on a block jackknife procedure. There are 11 paternal triploidies, 16 maternal triploidies and 110 maternal trisomies with detected crossovers. b, Crossover locations in maternal triploidies. The crossover locations in paternal triploidies are shown in Extended Data Fig. 2. c, Crossover locations in trisomies. d, Chromosomes with or without a crossover in maternal trisomies.
To assess whether lost fetuses had a different recombination pattern compared with individuals who made it to term, we tallied the crossovers across the genome for triploidies and trisomies (Fig. 4b,c and Extended Data Fig. 2). We found that only 113 out of the 161 maternal trisomies had a crossover and three of the five paternal ones (Fig. 4c). The rate of crossover absence was higher for trisomies of chromosomes 21 and 22 compared with the rest (Fig. 4d; odds ratio (OR) = 4.5, P = 4.71 × 10−5, Fisher’s exact test). This has also been observed for oocytes derived from fetuses31, supporting that meiotic disjunction of chromosomes 21 and 22 suffers from fewer crossovers established during oocyte arrest. Among autosomes, these chromosomes contribute the fewest number of crossovers to genetic maps8.
Mutation detection
The WGS analysis enabled us to investigate the rate and mutational spectrum of DNMs (SNVs and indels) in the pregnancy loss cases by comparing fetal genomes with parental genomes. In 724 fetal samples with low maternal contamination (paternal fraction > 25%; paternal kinship coefficient > 0.2; 366 distinct pregnancy loss cases; Fig. 1a), we identified 75,345 DNMs, with an average of 103.3 DNMs per loss (95% CI = 100.7–105.9). We estimate a discrepancy rate of 8.0% (95% CI = 7.7–8.2%, binomial test) by comparing multiple fetal samples from the same pregnancy loss (Supplementary Note 3).
Discrepancies between samples can result from artifacts or post-zygotic mutations (Supplementary Note 3 and Extended Data Fig. 3). Using 42 pregnancy loss cases with proper fetal and two villus samples, we classified DNMs as post-zygotic mutations if they were non-constitutional (allelic balance (AB) < 50%) in at least one villus sample, and found 9.7 (P = 1.92 × 10−7, t-test) post-zygotic mutations per villus that were absent from the proper fetus. By contrast, we identified 1.6 (P = 1.07 × 10−6, t-test) post-zygotic mutations in villus samples that were also in the proper fetus, occurring before trophoblast branching in early embryogenesis. This is supported by their high frequency in the proper fetus (Supplementary Fig. 8). Villus samples in general had a lower allelic balance (−2.9%; P = 2.07 × 10−40, t-test) and a higher number of DNMs (11.1 DNMs; P = 9.22 × 10−9, t-test; Fig. 5a and Extended Data Fig. 4) compared with proper fetal samples. We compared the mutational spectrum of the DNMs to mutational signatures identified in cancer samples32 and found that all samples with a detectable mutational signature 18 from COSMIC33 (characterized by C>A mutations) were villus samples (P = 0.0005, Fisher’s exact test), consistent with previous studies of placentas34. Overall, this indicates a considerable accumulation of somatic mutations in early development, confined to the trophoblast lineage.

a, The number of DNMs against the allelic balance per fetal sample. Only fetal samples with low maternal contamination and no detected triploidies were used (Extended Data Fig. 4 includes all samples). b, The paternal fraction of DNMs based on the chromosomal status of the DNM. The estimates are based on 366 (total), 143 (euploid), 337 (chromosome pairs), 11 (triploid father), 18 (triploid mother) and 131 (trisomy mother) unique pregnancy loss cases. c, The number of DNMs per fetal sample against paternal age at conception. The shaded areas represent 95% CIs. See Extended Data Fig. 5 for tissue type colouring. d, A mutation occurring before the formation of sister chromatids (pre-sister) is expected to have an allelic balance of 2/3 in the sister state. A mutation occurring after sister formation (post-sister) is expected to have a 1/3 allelic balance. Paternal triploidies caused by spermatozoa from two distinct spermatogonia fertilizing the oocyte are expected to have mutations in 1/3 allelic balance (Extended Data Fig. 9). e, The allele balance of DNMs conditioned on the sister chromatid state. The vertical line indicates an allelic balance of 50%. Both states are shown in Extended Data Fig. 8. f, Mutation classes of DNMs based on chromosomal status. The asterisk denotes the only significant comparison after multiple-testing correction (P = 0.0002 < 0.05/72) and the block jackknife procedure. The number of DNMs per combination of mutation class and chromosome group are reported above the bars. The numbers of unique pregnancy loss cases for the chromosome sets are 331 (euploid/chromosome pairs), 131 (trisomy–mother), 11 (triploid–father) and 18 (triploid–mother). For b and f, data are mean ± 95% CI based on a block jackknife procedure.
The AB of the DNM allele is a key metric in the quality control of DNMs6. The presence of extra chromosomes, as in the trisomies and triploidies, distorts the expected AB from 1/2 to 1/3 or 2/3, which impacts DNM detection. Correcting for this difference in sensitivity (Supplementary Note 3 and Supplementary Fig. 9), we found that the paternal and maternal triploid fetuses carried 40 (95% CI = 23.0–57.1) and 25.9 (95% CI = 15.8–35.9) more DNMs on average than the euploid fetuses, respectively. This is expected as the triploid fetuses have an extra set of chromosomes.
We phased 15,086 DNMs (71.7%) to the paternal and 5,967 (28.3%) to the maternal chromosomes of the fetus (Supplementary Note 3). The high paternal contribution of DNMs to offspring is well known6,35 and, for a fetus receiving two sets of paternal chromosomes (paternal triploidy), we would expect a corresponding excess of paternal DNMs. Consistent with this expectation, we found that the paternal triploidies showed a higher fraction of paternally phased mutations (85.9%; 95% CI = 82.9–88.9%, block jackknife) compared with the euploid fetuses (71.4%; 95% CI = 69.8–73.0%, block jackknife; Fig. 5b). Furthermore, DNMs identified in maternal triploidies (59.5%; 95% CI = 55.3–63.7%, block jackknife) and trisomic chromosomes (54.5%; 95% CI = 46.4–62.5%, block jackknife) showed a lower paternal fraction than those in euploid fetuses.
The number of DNMs per proband is highly dependent on the parental ages at conception6,8,36,37. We observed a similar age effect in the pregnancy loss cases (Fig. 5c and Extended Data Figs. 5 and 6) as in the Icelandic set6,30. There is a higher number of DNMs (16.3; 95% CI = 14.8–17.8) in the pregnancy loss cases than in the Icelandic set after correcting for parental ages at conception, restricting to non-triploid samples and correcting for villus contribution (Extended Data Fig. 7). The villus contribution is consistent with independence from parental ages at conception, as expected for post-zygotic mutations (Extended Data Figs. 5 and 7). To examine this further, we estimated the mutational spectrum of villus contributions. We found that the highest villus contributions were for C>A (3.4 DNMs; P = 2.11 × 10−13, block jackknife) and indel (4.9 DNMs; P = 1.84 × 10−13, block jackknife) mutations (Extended Data Fig. 7 and Supplementary Table 3). To summarize, there is a greater contribution of post-zygotic mutations and potential false positives compared with the Icelandic set, but the strong parental age effects and paternal fraction indicate that most of the mutations identified in the pregnancy loss cases accumulated before completion of meiosis.
In the female germline, sister chromatids are formed in early development, in contrast to in the male germline, in which they are formed after puberty. If a mutation occurred before sister formation (pre-sister), it would be expected to be faithfully replicated on the sister chromatid (Fig. 5d). We would expect the AB of pre-sister DNMs to be 2/3 in the triploidies/trisomies (Fig. 5d), whereas DNMs not replicated on the sister chromatid (post-sister) would have an AB of 1/3. A cut-off of 50% is symmetric between the pre-sister and post-sister ABs and can be used to differentiate them. High-AB (>50%) DNMs within sister segments are a combination of pre-sister mutations and misclassified ones (false-positive DNMs or AB measurement errors), whereas high-AB mutations within homologous segments probably consist of only misclassified mutations as the expected AB is always 1/3. Thus, the difference in the prevalence of high-AB mutations between homologous and sister segments serves as an estimate of the fraction of pre-sister mutations (probably an underestimate; Supplementary Note 4). For maternally transmitted triploidies and trisomies, we found that the prevalence of high-AB DNMs was significantly higher within sister segments (9.3%; 95% CI = 8.0–10.7%, block jackknife) than homologous segments (2.7%; 95% CI = 2.2–3.3%, block jackknife; Fig. 5e and Extended Data Fig. 8). This indicates that 6.6% (95% CI = 5.1–8.0%, block jackknife) of the maternal mutations are pre-sister mutations.
Notably, we did not observe the sister/homologous state difference for high-AB DNMs in the paternally transmitted triploidies (0.1%; P = 0.77). On the paternal side, most DNMs are expected to accumulate in mitosis of spermatogonia before the formation of sister chromatids. We would therefore expect to see a high AB for half of the DNMs in the paternally transmitted triploidies if the extra set of chromosomes was derived from the same spermatogonium (Extended Data Fig. 9). However, we saw little or no high-AB DNMs within sister segments on the father’s side, indicating that the spermatozoa are derived from distinct spermatogonia (Fig. 5d), consistent with the equal fraction of homologous/sister states observed at centromeres of paternally transmitted triploidies (Fig. 3e).
We contrasted the mutational spectrum of DNMs from chromosomes with extra copies (trisomy–mother, triploid–father and triploid–mother) to DNMs from euploid fetuses and pairs of chromosomes not affected by trisomies/monosomies (euploid–chromosome pair; Fig. 5f; 3 × 3 × 8 = 72 tests). C>G mutations were found at a greater frequency among maternal trisomies compared with in the euploid–chromosome pair set (P = 0.002; euploid–chromosome pair). No other comparisons passed multiple-testing correction (Holm). Duplication of chromosome 16 is frequent among the aneuploidies (Fig. 3a), and we have shown previously6 that C>G mutations in the maternal germline accumulate at high rates on chromosomes 8 and 16. Furthermore, these C>G mutations at these C>G-enriched regions are more likely to occur in strand-coordinated mutational clusters near non-crossover gene conversions6,8,30, which are the products of double-stranded breaks (DSBs). As DSBs can cause translocations, we searched the translocation cases and found strand-coordinated clusters at chromosomes 15 and 16 for the chromosome 15–chromosome 16 case (Fig. 3b), mostly consisting of C>A, C>G and C>T mutations (87%) of maternal origin (22 out of 22 phased DNMs). This indicates that chromosome 16 is susceptible to both point mutations and aneuploidies in the ageing oocyte due to DSBs.
Pathogenic genotypes
We searched for pathogenic SSVs in the DNM set (366 pregnancy loss cases) and found 26 SSV genotypes in as many loss cases (45 fetal samples) that classified as pathogenic or likely pathogenic based on criteria from the American College of Medical Genetics and Genomics38. Of these, 23 were DNMs and 3 were biallelic predicted loss of function (pLoF) variants (Methods and Supplementary Table 4). For 11 of the lost fetuses carrying pathogenic DNMs, at least 1 proper fetal sample had been sequenced; for the remaining 12, only villus samples were available. One of the DNMs, a splice donor indel in FBLN1, was a post-zygotic mutation confined to a villus sample. Nonetheless, the mean AB of all pathogenic DNMs (0.495; non-triploid loss cases) was comparable to that of DNMs in proper fetal samples (0.489; non-triploid loss cases; P = 0.95), and the majority of phased variants was of paternal origin (6 out of 8, 75%), consistent with attributes of germline mutations5.
We compared the frequency of pathogenic SSVs in the cases of pregnancy loss to that in adult controls (7,760 trios; Methods) and found an enrichment of pathogenic SSVs in pregnancy loss compared with in the controls (OR = 2.98, P = 5.7 × 10−6, Fisher’s exact test). This enrichment was also significant after correcting for parental age (P = 4 × 10−3, χ2 test, logistic regression) and was not observed for synonymous DNMs (P = 0.36, χ2 test, logistic regression). The enrichments were 3.63 (P = 3.0 × 10−4) and 2.59 (P = 2.2 × 10−3) when restricting to euploid and aneuploid/triploid pregnancy loss cases, respectively. Of the 26 pathogenic SSVs identified, 12 were in euploid fetuses (12 out of 141; Supplementary Table 5 and Supplementary Note 5).
Considering the possibility that not all genes that are essential to early human development have been linked to postnatal phenotypes, we also searched the cases of euploid pregnancy loss for pLoF DNMs in genes highly constrained for LoF variants (Methods) and found an increase over the adult controls (OR, 4.0; P = 0.006, Fisher’s exact test). Three of the pLoF variants were in genes without an established link to disease: DDX5, ZMYM4 and HECTD1 (Supplementary Table 6). To better understand the role of these variants, as well as that of the pathogenic SSVs, in pregnancy loss, we annotated the genes with regards to previous evidence of lethality, relevance of the associated phenotypes to developmental defects, fetal expression of the genes39 and evidence of lethality from mouse knockouts (Supplementary Tables 7–9). We found that, on average, the genes mutated in euploid fetuses had higher expression in fetal tissue than the ones in aneuploid/triploid fetuses (P = 6.9 × 10−196, t-test; Extended Data Fig. 10).
One of the euploid fetuses was homozygous for a pLoF variant in DHCR7, a gene linked to autosomal recessive Smith–Lemli–Opitz syndrome40. The variant is a splice acceptor variant (NM_001360.2, c.964-1G>C) that is described to result in miscarriage or neonatal death in homozygous state40. DHCR7 c.964-1G>C exhibits a deficit of homozygosity among adults, that is, a lower number of observed homozygous individuals than expected based on frequency12. In our study of 1.52 million genotyped European adults12, DHCR7 c.964-1G>C had the most prominent homozygous deficit, with 92 homozygotes expected but none observed. Here we observed the c.964-1G>C variant in a homozygous state in 1 out of 366 trios, the majority of whom have European ancestry, with an expectation of 0.022 homozygotes (95% CI = 0–0.053; supplementary data 4 from ref. 12). This finding is consistent with our previously estimated fivefold increased risk of miscarriage among couples who are both heterozygous carriers of DHCR7 c.964-1G>C12.
We detected one other biallelic pLoF genotype among the euploid lost fetuses, a compound heterozygous genotype in CPLANE1 (NM_023073.3, c.6354dup (p.Ile2119TyrfsTer2) and c.493del (p.Ile165TyrfsTer17)). CPLANE1 pathogenic genotypes have been detected in fetuses from early pregnancy loss41, and another pLoF variant in CPLANE1 had a suggestive deficit of homozygosity in our previous study (supplementary table 18 from ref. 12). We did not find any other homozygous deficit variants (supplementary table 21 from ref. 12) presenting as biallelic genotypes in our cohort. The biallelic genotypes in DHCR7 and CPLANE1 infer a high recurrence risk (at least 25%) and, in both cases, the couple had a history of recurrent pregnancy loss (three consecutive losses; Supplementary Note 6). DNMs are generally expected to have a lower risk of recurrence, unless there is evidence of parental mosaicism42,43, which we did not observe in this set.
Discussion
Here we performed WGS analysis of 1,439 samples from 664 cases of early pregnancy loss and characterized the sequence diversity in 467 of the loss cases for which samples were available for the full trio. We found probable genetic causes in 254 out of 467 loss cases (55%), the majority of which were aneuploidies (44.1%), followed by triploidies (6.4%), pathogenic SSVs (3.3%) and de novo CNVs (1.3%) (Supplementary Table 5).
Most of the genetic causes originated on the maternal chromosome, mainly in the form of triploidies and trisomies, consistent with the well-described meiotic errors accompanying oogenesis. We observed an indication of interplay between point mutations and large structural variation. The oocytes are in dictyate arrest for decades1 and accumulate mutation clusters in specific regions of the genome6,44, for example, the p-arm of chromosome 8 and termini of chromosome 16. We found a translocation between chromosomes 15 and 16 (Fig. 3b) containing clusters of strand-coordinated DNMs, a likely byproduct of faulty DSB repair.
The extra chromosomes in the trisomy and triploidy cases gave us a unique view into recombination and mutational processes in the germline. We found that triploid lost fetuses carried 31.8 more DNMs on average than euploid lost fetuses, as expected given the extra set of chromosomes. However, these mutations stem from both homologous chromosomes of the parents in contrast to the DNMs observed in adult offspring, providing a more comprehensive assessment of the diploid germ cells. The presence of sister chromatids enabled us to estimate that 6.6% of maternal DNMs occurred before the formation of sister chromatids. Furthermore, we mapped crossover locations for the trisomies and triploidies, providing a complementary view of the recombination map of the human genome.
We found a threefold enrichment of pathogenic SSVs in the trios affected by pregnancy loss in comparison to adult trios, indicating that two-thirds of the pathogenic SSVs identified in the euploid lost fetuses are truly causative, explaining around 6% of those losses (8 out of 141). Assuming that approximately 1 in 17 (1/0.06) euploid loss cases is explained by SSVs and 1 in 8 pregnancies is lost with a euploid fetus, our findings indicate that at least 1 in every 136 (17 × 8) pregnancies will be lost due to a pathogenic SSV. There is an excess of DNMs in the trios affected by pregnancy loss compared with adult trios, partly due to false-positive calls and partly attributed to post-zygotic mutations specific to villi. Nevertheless, most of our pathogenic SSVs were consistent with germline origin and were found in greater frequency in trios affected by pregnancy loss than in adult trios. This shows that the contribution of DNMs to pregnancy loss is determined by whether they occur in essential genomic sequences rather than their overall count. Larger cohorts of sequenced euploid fetuses are needed to identify variants with incomplete penetrance and/or polygenic effects. We realize that high-depth sequencing is cost prohibitive at scale, but low-pass sequencing (around 1×) coupled with imputation is comparable to chip genotyping in terms of cost and quality45. Furthermore, long-read sequencing could aid detection of structural variants in euploid lost fetuses without a pathogenic SSV.
Studies of the contribution of pathogenic SSVs to pregnancy outcomes17,18 have mainly focused on pregnancies with structural abnormalities of the fetus, typically discovered after the first trimester. It is hard to compare such phenotypes with the ones found in cases of early pregnancy loss, including gestational age, fetal anatomical structure, and placental morphology. Notably, we found that one of the pathogenic DNMs was a post-zygotic mutation confined to villi, which are a precursor of the fetal part of the placenta. The mutation occurred in FBLN1 (also known as fibulin-1), which encodes a structural component of blood vessel walls46 and has particularly high expression in placental trophoblast giant cells (Supplementary Table 7), suggesting that perhaps this pregnancy loss was triggered by abnormal placental function.
We detected homozygous and biallelic genotypes in genes with a strong deficit of biallelic loss of function in adults, and a high risk of recurrence. We also considered the possibility that many genes that are important for early development might not yet be identified as such. We found pLoF DNMs in three euploid lost fetuses in constrained genes that are highly expressed during the fetal stage39. Two of these genes, DDX5 and HECTD1, encode proteins that are involved in cellular processes that are essential for early development, such as DNA replication47 and ubiquitylation48, but they were not associated with postnatal phenotypes at the time of submission. During revision of this Article, variants in HECTD1 were described to cause neurodevelopmental defects and even infant death49, further illustrating the potential of sequencing fetal material from pregnancy loss cases to identify gene–disease connections.
There are considerable differences between aneuploidies detected in blastocysts and in fetuses from pregnancy loss cases3. We are therefore probably still missing substantial sequence diversity that is lost in the period from implantation to a clinically recognized pregnancy. Here we have nonetheless shown that sequencing early products of conception that succumb to negative selection can identify essential genes and characterize their function in early development.
Pregnancy loss has been considered to be the natural fate of fetuses with severe genetic defects, making therapeutic interventions unwarranted. However, we are unable to explain 45% of sporadic unselected clinical pregnancy loss cases using WGS, pointing to other factors in the aetiology of pregnancy loss. Given the high incidence of euploid pregnancy loss cases without a clear monogenic cause in the fetus, there is an urgent need to understand their aetiologies and develop therapeutic interventions that can ensure healthy live births. This provides value not only for the couples wanting a child, but also for the many societies that are challenged by decreasing fertility rates and rapidly ageing populations50. Continued sampling of trios and biomarkers throughout pregnancy is needed to disentangle the complex interplay between the fetal and maternal genomes and the environment, and the cascade of events sparked by a single fertilized egg.
Methods
Cohort
The COPL study is a prospective cohort study, with the first patients recruited in November 2020 (described in detail previously20). Women with a confirmed or suspected pregnancy loss were referred by a physician to a gynaecological department at one of three hospitals in Denmark: Copenhagen University Hospital Hvidovre, Herlev or North Zealand.
Women were included in the study if older than 18 years, if the pregnancy loss was before gestational age 22 weeks + 0 days (154 days) and if an intrauterine pregnancy had been confirmed by ultrasound, including anembryonic pregnancy as per definition. The participants provided both oral and written informed consent. Excluded from the study were pregnancies of unknown location, extrauterine pregnancies and molar pregnancies. The study was approved by the Health Research Ethics Committees for the Capital Region of Denmark (H-18024745) and followed the General Data Protection Regulation governance.
Sampling
The woman and her male partner (if possible) had venous blood drawn in 4 ml EDTA tubes that were kept at 5 °C until transport in tubes with cushioning material. All of the blood samples from the woman were drawn before initiating treatment for the pregnancy loss while fetal material was in utero, or within 24 h of passage of the fetal material. In cases of spontaneous or medically induced (mifepristone plus misoprostol) emptying of the uterus (53%), the woman was instructed (both orally and in writing) to collect all pregnancy loss tissue at home and place it in the fridge at 5 °C before bringing it to the hospital within 1 week. If she had a surgical removal (47%), COPL staff collected the material at the hospital and placed it at 5 °C. In cases of second-trimester loss, induction and delivery took place at the hospital, and clinical staff collected biopsies from the fetal foot. COPL staff packed and shipped the tissue in padded envelopes to the University of Copenhagen (maximum transportation time 2 h), where it was stored at 4 °C.
We used a previously described method to extract DNA from fetal material20. In brief, fetal tissue samples were washed several times in sterile PBS to remove maternal blood before being examined under a stereomicroscope to identify fetal tissue and/or chorionic villi. In some cases, the tissue could not be identified as either and was designated unknown. Some of these unknown samples were probably blood clots and maternal decidua. The fetal tissue or villi were dissected into pieces of 0.5 cm2 and then subjected to three consecutive PBS washes before being placed into a 2 ml cryotube (Nunc, Thermo Fisher Scientific, 10577391), submerged in liquid nitrogen for snap-freezing and then stored at −80 °C. For DNA extraction, we used the DNeasy Blood and Tissue 96 with TissueLyser LT (Qiagen, 69504) according to the instructions provided by the supplier. In brief, the samples were put on dry ice and the frozen tissue was moved to a 2 ml Safe-Lock Eppendorf tube (Eppendorf, 0030 123.344), as recommended by the supplier. A single bead (Stainless Steel Beads, Qiagen, 69989) and appropriate buffers from the DNeasy kit were added to the tube containing the frozen tissue together with appropriate buffers, which was bead milled for 1 min at 30 Hz using a 5 mm steel bead on a TissueLyser LT (Qiagen, 85600). The remaining steps were performed according to the recommendations of the supplier. Parental blood samples and extracted fetal DNA were shipped to deCODE, Iceland, on dry ice.
All information on the participants and samples was stored in a secure cloud (REDCap) in the Capital Region of Denmark. Blood tubes were labelled with individual bar codes, a pseudonymized code, and whether it was a maternal or paternal sample. Information about the last menstruation (gestational age–last menstruation; GA–LM), pregnancy loss type (missed miscarriage or spontaneous ongoing miscarriage) and gestational age at the time of loss was gathered by interview and patient records on inclusion.
WGS analysis
Sequencing libraries were prepared using the NEBNext Ultra II PCR-free kit (New England Biolabs). In brief, 0.5–1 µg of genomic DNA was fragmented in 96-well TPX-AFA plates (Covaris) to a mean target size of 450–550 bp using the Covaris LE220plus instrument. End repair and A-tailing was performed in a single step followed by ligation of unique dual-indexed sequencing adaptors (IDT for Illumina) and two rounds of SPRI-bead purification (0.6×) using the Hamilton STAR NGS liquid handler. The quality (concentration and insert size) of the sequencing libraries was determined using the LabChip GX (96-samples) instrument (Perkin Elmer). Sequencing libraries were pooled appropriately using Hamilton STARlet liquid handlers and sequenced using the Illumina’s NovaSeq 6000 instrument. We performed paired-end sequencing on the S4 flowcell (v1.0 chemistry), resulting in read lengths of 2 × 150 cycles of incorporation and imaging, in addition to 2*8 index cycles. Real-time analysis involved conversion of image data to base calling in real time. All of the steps in the workflow were monitored using an in-house laboratory information management system with barcode tracking of all of the samples/plates and reagents.
Alignment and sequence variant calling
The WGS data were aligned against the human reference genome (hg38) as previously described51 and managed with an in-house laboratory information management system. In brief, reads were aligned with BWA mem52 (v.0.7.10) and marked for duplicates with Picard tools (v.1.117). The aligned reads in BAM format for fetal and parental samples were used as input to GraphTyper53 (v.2.7.5), the average coverage of the BAM files is shown in Supplementary Fig. 2.
Paternal fraction estimation
VCF files from GraphTyper were parsed to identify genotypes where the mother was homozygous, and where the father carried an allele absent from the mother. Sites where the genotype of the parents passed the following quality thresholds were considered for downstream analysis; a genotype quality greater than 20 and depth greater than 20. Furthermore, we required the AB to be within [30%, 95%] for heterozygous calls; and within a range of [0, 5%] or [95%, 100%] for homozygous reference or alternative calls, respectively.
For sites that passed these quality criteria, we counted the number of reads in the fetus supporting the paternal allele (Y), conditioned on the genotype of the father and the DP genotype field from the fetus (D). We then regressed the number of reads supporting the paternal allele in the fetus on allele counts of the paternal allele in the father (A). For an uncontaminated euploid fetus, the slope and intercept of this regression (Y = D × A) would be expected to be 1 and 0, respectively. The slope is expected to decrease with lower paternal contribution, the same as for maternal contamination or maternal triploidies.
Coverage of the sex chromosomes (allosomes) can be informative about the maternal contamination in cases of pregnancy loss with male fetuses (Supplementary Fig. 10). Consistent with expectation, we found that fetal samples with 20% of the allosome coverage on the Y chromosome were predominantly of low contamination (90.0% paternal fraction) compared with those without Y chromosome coverage (55.9% paternal fraction; P = 2.7 × 10−61).
Estimation of genetic ancestry
Genetic ancestry of parental samples was estimated by running ADMIXTURE54 v.1.3.0 in supervised mode using 1000 Genomes55 CEU (Utah Caucasian, European), CHB (Beijing Han, East Asian), ITU (Indian Telugu, South Asian), PEL (Peruvian, Native American) and YRI (Nigerian Yoruba, African) as training populations. Markers were pruned for pairs in strong linkage disequilibrium by excluding long-range high-linkage-disequilibrium regions56 and running PLINK57 v.1.90b6.15 --indep-pairwise 200 25 0.4. ADMIXTURE was run on batches of ten test individuals at a time.
Coverage-based CNV calling
The CNV caller used for the pregnancy loss set is based on sequence coverage. Its aim is to find CNVs that are long enough for coverage to give enough evidence for calling them. To guard against too high variance in determining the copy-number state at a locus, the sequencing read coverage is binned into 1 kb regions, which is further normalized to overall genomic coverage (using the autosomes).
Sequence coverage shows dependency on G/C content and AT repeat content per region and depends on the fragment length distribution in sequence libraries. Most of the dependency can be captured by the number of G/C base pairs and the number of repeated AT motifs in each read pair fragment58. To correct for G/C content, we considered 400 bp non-overlapping windows across the genome, resulting in a table of number of reads starting at each 400 bp window, classified according to the number of G/C base pairs and the number of AT repeats. After coverage normalization, this gives a correction factor for each 400 bp window. Each 1 kb bin consists of a sequence of overlapping 400 bp fragments, each with a corresponding correction factor from the before mentioned table. The average of those correction factors is used as the correction factor for the 1 kb bin. This correction factor is applied to correct each bin, giving a G/C–AT-corrected normalized binned coverage in 1 kb bins, which are then used in the downstream CNV analysis.
The input data to the CNV caller consist of a series of G/C–AT-corrected normalized 1 kb coverage values for each sample on each chromosome. Supplementary Fig. 11 shows four such series at a locus. Each datapoint has an underlying count of sequencing reads that are assumed to be approximately Poisson distributed. Normalization and G/C–AT correction apply a scaling factor resulting in a scaled Poisson variable. The mean of the Poisson variable depends on the copy-number state of the sample in the bin, giving a mixture distribution. A few parameters specify the mean of each component in the mixture:
where y is the normalized coverage per 1 kb bin, μk is a multiplier per copy-number state, α is the inverse scaling factor, β is a bias factor (usually giving the mean of the copy-number 0 state since μ0 is usually 0), λ controls the distance between copy number states and πk is the prior probability of copy-number state k. For Illumina sequencing data, we use 7 states, with μk = 0.5 × k.
Each bin gets their own α, β, λ and πk values, which are set to maximum-likelihood estimates for the data for all samples within the bin. A further shift parameter is sometimes used that specifies a shifting of the data on the Poisson scale (when going to the Poisson scale from the y scale, the shift is applied). The main purpose of that parameter is to fit a higher variance in the copy-number 0 cluster than a Poisson variable with mean close to 0 can explain. Supplementary Fig. 12 shows histograms for the CNV in Supplementary Fig. 11. The copy-number clustering for copy-number states 0 and 1 (as well as 2 of course) are very clear.
The actual CNV calling is then run when the model above has been fitted. Each sample is run with a hidden Markov model (HMM) with states corresponding to the copy-number states59. Within each state, the model above is used for the emission probability given the copy-number state. The transition probabilities have a parameter specifying the probability of transitioning to a different state. The Viterbi HMM algorithm is used to find the most probable path through the HMM for each sample and, where the Viterbi path diverges from the normal copy number (2 for autosomal chromosomes), a CNV is called. The algorithm is run twice, first with a stringent transition probability (1 × 10−6) to find longer variants without splitting them into a series of shorter ones, and then with a more relaxed transition probability (0.001) to allow finding shorter variants. CNVs with similar boundaries are grouped together and the HMM is used to estimate which CNVs correspond to the same event and what is the most likely boundary for each such group. The probability model can be used to calculate the posterior likelihood for each copy-number state for each CNV, which can be used in downstream phasing and imputation.
Note that informative structural variants not captured by our copy-number analysis may have been missed because additional rearrangement analysis was not performed.
Sensitivity of the coverage-based CNV detection
To assess our sensitivity for detecting CNVs in the presence of contamination, we simulated duplications by switching between a pair of uncontaminated fetal sample with trisomy 16 and maternal sample along the chromosome (Supplementary Fig. 13). To simulate deletions, we switched between a male fetal sample and maternal sample along chromosome X. Contamination was simulated by taking the weighted average of the maternal and the fetal corrected coverage value for simulated CNV segments. We considered the following combinations of CNV sizes (5 kb, 10 kb, 20 kb, 50 kb 100 kb, 200 kb and 500 kb) and contamination values (0, 10%, 20%, 30%, 40%, 50%, 60% and 70%). We determined that we found a simulated CNV if 50% or more of the windows within the CNV were called in deletion state for deletions and duplication state for duplications in the output from the HMM program. We found 89.4% (95% CI = 79.4–95.6%) of the simulated duplications and 96.3% (95% CI = 94.5–97.6%) of the simulated deletions of size 10 kb at 30% contamination; other combinations are shown in Supplementary Fig. 13.
Combining segments and phasing
The calls from the HMM algorithm in the fetal samples were in the form of a series of small segments. Similar to the Battenberg algorithm60, we joined these disjoint segments through phase-informative sequence variants. For the joining, we assessed the allele-specific depth in the fetal sample limited to variants where parents were homozygous for different alleles.
Letting CMM be the measured maternal coverage, CM the actual maternal coverage and CCM the coverage that is due to maternal contamination, then
Letting CP be the paternal coverage, then:
CT represents the total coverage of the phase-informative variants (limiting to homozygous variants in either parent) in the fetal sample.
Assuming a diploid fetal sample, that is,
Then, we can get an estimate of the global maternal contamination coefficient CGLOB
For each segment, the maternal contribution was then corrected for CGLOB. If the maternal and paternal contributions deviated from the expected 50:50 ratio, within the predefined segments, those segments were assigned either a duplication or deletion status and assigned to the corresponding parent (phased). All calculations for the refinement of aneuploidies and CNVs were performed in R v.4.4.3, refined segments were combined into CNVs using Python v.3.9.21.
The pathogenicity of identified CNVs was estimated based on criteria from the ACMG61.
CNV breakend annotation with GraphTyper2
We discovered structural variants with Manta (v.1.6.0), merged the variant sites over all individuals with svimmer (git hash 2af9ccf1ac056b9516c25ec4a0121657a9f9ab8e), and finally genotyped them jointly across the entire set with GraphTyper2 (v.2.7.7). We considered all types of structural variants within 5 kb of CNV breakends. We searched for structural variant DNM candidates using the same criteria as described in the ‘DNM detection’ section with two exemptions: a more permissive allelic balance cut-off (10%) and a lower minimum number of reads supporting the DNM allele (6). If we found a DNM candidate among the structural variants of the CNV carrier within this region, then we set that as the position of the CNV breakpoint Supplementary Table 2. To search for balanced translocations present in the parents, we manually curated the translocation calls by GraphTyper with an allele count less than 10. We found exact breakpoints of the two balanced translocations that are present in parents (Supplementary Figs. 6 and 7).
Kinship and oversharing
We estimated kinship between fetal samples and parental samples using KING62 (v.2.1.5) on SNVs in subsampled 50 kb regions of the genome (1%). We considered only biallelic SNVs, with allele counts greater than four and an AAScore from GraphTyper of greater than 0.5.
We searched for indication of triploidy based on oversharing between the fetal sample and either of the parental samples, that is, a higher kinship coefficient than expected for parent–offspring relationship (k = 0.25). If kmat represents the kinship coefficient between the fetal sample and the maternal sample and kpat represents the kinship coefficient between the fetal sample and the paternal sample, we considered the following scenarios:
as potential triploidies. We then used the change in variance of allelic balance, as described in the next section, across the genome to further refine these potential triploidies.
Detecting crossovers between chromosomal states
We defined phase-informative variants as variants present in heterozygous state in one parent and homozygous in the other. In the following method, we restricted to phase-informative heterozygous variants in the parent transmitting the extra chromosome, where the other parent had a homozygous genotype.
If two sister chromatids from the transmitting parent were present at a given locus, we expected the AB of HS alleles at that locus to be either 0 or 2/3, depending on whether or not the sisters carried the HS allele (Fig. 2b). If both homologous chromosomes were present at a locus, we expected the AB to always be 1/3, considering the presence of an HS-allele in the transmitting parent. The state of homologous chromosomes has much lower variance than the state of sister chromatids, as there is no variability in sampling the homologous chromosomes. The sampling variance of the homologous chromosome state should be due to only sequencing variability, that is, reads supporting the alternative allele in the fetal sample.
We let Y be the observed coverage of the phase-informative variant in the fetal sample. T is a Bernoulli variable denoting the transmission of the phase-informative variant from the parent to the fetus. Then, by the law of total variance, we can partition the variance based on transmission T:
If we assume the read coverage in the fetus given transmission is determined with a Poisson variable:
Then:
The other part of the sum will be:
Taken together:
If both homologous chromosomes are present (Pi = 1) at 30× coverage in the fetus (μ = 10) then we expect:
By contrast, in the sister state (Pr = 0.5) the variance is much higher:
We used this change in variance of the allelic balance along the genome to find crossovers between the homologous chromosome states and sister chromatid states. We binned the number of phase-informative variants in 1 kb windows, and calculated the average allelic balance. We excluded 1 kb windows that overlapped larger 100 kb windows with sequence variants with low average quality metrics (AAScore < 0.8 or Carrier_regression_beta < 0.7; see the ‘DNM detection’ section for details of AAScore and Carrier_regression_beta). We then used a changepoint technique on the 1 kb windows that passed the exclusion criteria to detect changes in variance in the 1 kb windows (cpt.var from the changepoint package in R). We used the following criteria:
-
A minimum segment length of 1000 1 kb windows (1 Mb)
-
A maximum of eight changepoints per chromosome
-
The PELT method for detecting multiple changepoints
-
Our own penalty function (penalty = “manual”) with 10 × log(n) (pen.value = “10⋅log(n)”) where n is the number of windows per chromosome.
DNM detection
We searched sequence variants and extracted DNM candidates in a similar manner as before6,8 for 9,651 Icelandic trios by comparing the genotypes of the parents and offspring. The DNM candidates included both SNVs and indels (Supplementary Fig. 14). In brief, we defined a DNM candidate with permissive cut-offs for the genotype of the proband requiring an allele balance greater than 0.25 and depth of 12 reads at the position (supporting either the reference or alternative allele). For the genotypes of the parents, we required at least 12 reads, a maximum of one read supporting the alternative allele and the allelic balance to be less than 5%. Likely (NLIK) and possible (NPOSS) carriers of the DNM allele outside the descendants of the parent pair were defined as previously6. We restricted to DNM candidates with less than 50 likely carriers and either less than 10 possible carriers or a NLIK/NPOSS ratio greater than 80%. We tuned the DNM candidate filtering by using segregation of DNM candidates in three generation families (2,042 probands) and the following quality covariates in a generalized additive model with a logistic link:
-
AAScore: prediction probability from GraphTyper that the variant is a true positive.
-
Carrier_regression_beta: slope from the alternative allele depth regression for the sequence variant. To estimate the quality of the sequence variants, we regressed the alternative allele counts (AD) on the depth (DP) conditioned on the genotypes (GT). For a well-behaving sequence variant, the mean alternative allele count for a homozygous reference genotype should be 0; for a heterozygous genotype, it should be DP/2; and, for a homozygous alternative genotype, it should be DP. Under the assumption of no sequencing or genotyping error, the expected value of AD should be DP conditioned on the genotype; in other words, an identity line (slope 1 and intercept 0). Deviations from the identity line indicate that the sequence variant is spurious or somatic.
-
Carrier_regression_alpha: intercept from the alternative allele depth regression for the sequence variant.
-
Proband_het_AB: the allelic balance of the proband.
-
MaxAAS: the maximum read support for the sequence variant across all individuals.
-
Alignment_Alt_Reads: the number of reads supporting the alternative allele. This covariate and the following covariates were derived by identifying the reads in the BAM files supporting DNM allele.
-
Alignment_Alt_Unique_Positions: the unique number of starting positions for the reads supporting the alternative allele.
-
Alignment_Alt_Soft_clipped: the number of soft clipped bases (S in CIGAR string).
-
Alignment_Alt_Matched_bases: the number of matched bases (M in CIGAR string).
-
Alignment_Alt_Score_diff: the difference of the alignment score of the best and the second best hit as reported by BWA mem.
-
Alignment_Alt_Pair_sw_nm: the pairwise mismatches between reads supporting the alternative allele using Smith Waterman implementation in SeqAn63.
-
Alignment_Alt_Pair_align: the number of bases in the pairwise alignments.
With the following formula for the gam function from the mgcv R package64:
threegen_Consistent_hs~ I(cut(alignment_Alt_Unique_Positions,c(-1,2,4,8,10,Inf)))+ s(I(AAScore))+ s(Carrier_regression_beta)+ s(Carrier_regression_alpha)+ I(ifelse(alignment_Alt_Reads>0, (alignment_Alt_Score_diff/alignment_Alt_Reads)>10, FALSE))+ I(ifelse(alignment_Alt_Pair_align>0, (alignment_Alt_Pair_sw_nm/alignment_Alt_Pair_align)>0.05, TRUE))+ I(ifelse(alignment_Alt_Matched_bases>0, alignment_Alt_Soft_clipped/alignment_Alt_Matched_bases>0.5, TRUE))+ s(Proband_het_AB)+ s(ifelse(MaxAAS>15, 16, MaxAAS))+ I(NPOSS == 0)
We restricted to instances of the DNM candidates in cases in which we saw both of the proband’s haplotypes at a locus transmitted to the offspring of the proband (see figure 1c in ref. 6). In brief, if the DNM is a true germline variant, then the allele of the DNM candidate should be present in one of the proband’s offspring. On the other hand, if it is absent from the children, then this assay suggests that the DNM candidate is a false-positive DNM call (a more detailed description was reported previously6). Like before, we fitted the generalized additive model using the mgcv R package64.
We used the cut-off of 0.5 to call high-quality DNM candidates for the predicted probability of correct segregation in three generation families. To validate the false-positive detection rate of the DNMs, we also used the genotype consistency between pairs of monozygotic twins. We found that 3.8% of DNMs were unobserved in the monozygotic twin of the proband. Note that this approach overestimates the false-positive rate as there are instances of high frequency post-zygotic mutations that differ between pairs of monozygotic twins65.
Identifying DNMs in COPL trios
The AB of DNM candidates is a crucial quality-control metric for discerning between false positive DNMs (that is, somatic and genotyping artifacts) and DNMs that transmit to the next generation6. However, in the case of the triploidies, trisomies and the maternally contaminated samples, the AB is expected to be lower than 50% for genuine DNMs. This is not compatible with the generalized additive model applied on the deCODE trios that uses the AB; we therefore decided on a simpler threshold strategy to define high-quality DNMs in the COPL data. We defined a high-quality DNM in the COPL set as one that satisfies AB of proband > 0.25; NPOSS < 10 and AAScore > 0.5. To compare the subsets in COPL, we calculated the detection power to detect DNMs per fetal sample using the ratio of the phase-informative markers and a binomial distribution (Supplementary Fig. 9). We considered only samples with paternal fraction > 25% and excluded a single sample with more than 800 DNMs after quality filtering.
Phasing of mutations in Icelandic trios
Like before, we used two complementary approaches to phase DNMs6,8: one by tracking of the transmission of the DNMs to the offspring of the proband (142,069 DNMs; three-generation phasing) and another one using read tracing of the DNM allele to phase-informative alleles (246,877 DNMs; read phasing). We combined the phase of the DNMs from the different approaches, resulting in 333,365 phased DNMs; if the phase of DNMs differed between the approaches, they were set to unphased DNMs in the downstream analysis.
Phasing of mutations in COPL trios
We read-traced the DNM allele as with deCODE data, except for one crucial difference—we considered only reads linking the DNM allele to nearby phase-informative sequence variants, if the reads were linked to an allele private to one of the parents. This was done to avoid phase ambiguity due maternal contamination or extra chromosomes.
Annotation of pathogenic variants
Sequence variants were annotated on the basis of release 100 of the Variant Effect Predictor (VEP)66 using RefSeq gene annotations67. We included SNVs and small indels (<50 bp) at coding and splicing regions in the autosome. We required that the variants were supported by at least six sequence reads, with at least three reads supporting the alternative allele. For indels, we required an AB of at least 0.4. For heterozygous variants, we considered pLoF (stop-gained, frameshift indels, splice essential variants) and missense DNMs in known autosomal-dominant disease genes (human phenotype ontology code HP:0000006 for autosomal-dominant disease genes). For missense DNMs, we also required either that the gene was highly constrained for missense variants (95th percentile over all genes, corresponding to a gnomAD68 v.4.1.0 missense z score of over 3) or that the variant was nearby (within <10 bp) a previously reported pathogenic/likely pathogenic missense variant on ClinVar (with at least one ClinVar submission supporting the variant as pathogenic/likely pathogenic and no benign/likely benign submissions).
We also included homozygous and compound heterozygous pLoF variants in known autosomal recessive disease genes (based on the code HP:0000007). For homozygous variants, we required the position to be covered by at least eight sequence reads, and for the alternative allele to be supported by at least eight reads. For compound heterozygous variants, we required the positions to be covered by at least four reads, and each variant to be supported by at least two sequence reads. To detect compound heterozygous genotypes, we performed phasing of the variants to paternal and maternal alleles. We also included genotypes where we could not phase both variants, allowing for the possibility of a DNM on one allele. We used minor allele frequencies from 56,969 Icelandic genomes69 and 730,947 exomes and 76,215 genomes from gnomAD11 to restrict to variants with frequencies <2%, and considered only genotypes present in fewer than three genomes. All pathogenic genotypes detected in the trios affected by pregnancy loss were confirmed by manual inspection of BAM files.
The same approach was used for defining pathogenic genotypes in a set of adult control trios. For the purpose of pathogenic SSV analysis, individuals with neurodevelopmental disorders, including epilepsy, autism and intellectual disability, were removed from the set of 9,651 Icelandic trios. Those patients, who were whole-genome sequenced as part of ongoing projects at deCODE genetics and have been described previously70, were ascertained through the State Diagnostic Counselling Center and the Department of Child and Adolescent Psychiatry and diagnosed based on ICD-10 criteria. Individuals with a clinical diagnosis of a rare genetic disease, as described previously69, were also removed from downstream analyses. Removal of these patients from the set of Icelandic trios resulted in a set of 7,760 adult control trios.
Constrained genes
To investigate DNMs in genes that have not been linked to any postnatal phenotypes but that might be essential to early developmental processes, we looked at genes that are highly constrained for pLoF variants. We used constraint data from gnomAD68 v.4.1.0, ranking genes (n = 17,481 protein-coding genes) based on their pLI scores. We defined highly constrained genes as the genes in the 90th percentile of all ranked genes.
Age regression
To compare the accumulation of mutations with advancing parental age at conception between pregnancy loss and the Icelandic trios, we regressed the number of DNMs against the parental ages at conception with the following formula:
Nr_of_DNMs~ fathers_age+ mothers_age+ tissue_villi
Note that, we are using a shared intercept model. This is due the fact DNMs detected in the fetal samples are only partially phased through the read-tracing, which results in unidentifiable sex-specific intercepts. We restricted to the subset of the pregnancy loss cohort that is more comparable to the Icelandic set, in terms of quality, that is, we restricted to non-triploid samples with 95% power of detecting DNMs.
We ran separate regressions for the different mutational classes (C>A, C>G, C>T, CpG>TpG, Indel, T>A, T>C and T>G) along with the total number of DNMs per fetal sample/proband. The estimates of the age effects, villus contribution and intercepts are provided in Extended Data Fig. 7 and Supplementary Table 3. The CIs and P values are derived from a block jackknife procedure.
Mutational signatures
To estimate the contribution of COSMIC mutational signatures to the 96-class mutation spectra of the pregnancy loss samples, we used the Fit() function provided by the signature.tools.lib (v.2.4.4) package71 in R (https://github.com/Nik-Zainal-Group/signature.tools.lib). Note that the mutation burden of each pregnancy loss sample is modest, which limits power for signature detection. The purpose of this exercise was not to discover mutational signatures de novo but, rather, to specifically test the hypothesis that SBS18 was elevated in some villus samples as reported previously34. The mutation spectrum of spermatogonia is dominated by SBS1 and SBS5/40 (ref. 72) and this is mirrored in the mutational spectrum of germline variants (see figure 5 of ref. 73). With this in mind, we fitted only SBS1, SBS5 and SBS18 onto the mutational catalogues. Signatures were fitted using 100 bootstraps.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The DNMs identified in the Icelandic cohort are available at Zenodo74 (https://doi.org/10.5281/zenodo.14025565) along with recombination maps, proband information, gene conversions and observed non-crossovers. Crossover recombination data are available online (https://www.science.org/doi/suppl/10.1126/science.adh2531/suppl_file/science.adh2531_data_s1.zip and https://www.science.org/doi/suppl/10.1126/science.adh2531/suppl_file/science.adh2531_data_s2.zip). The raw sequencing data from the Icelandic cohort cannot be shared publicly due to Icelandic legislation and the regulations set by the Icelandic Data Protection Authority, which restrict the dissemination of individual-level or personally identifiable information. Access to raw data can be granted at the facilities of deCODE genetics in Iceland, for scientific purposes only, and subject to Icelandic legislation regarding use of the data. To gain access to the Icelandic data contact H.J. or K.S. Requests for access are generally considered monthly. The DNMs identified in the COPL cohort are available at Zenodo75 (https://doi.org/10.5281/zenodo.15183295). All clinical summary data for the COPL cohort can be found in a previous publication20. Raw data are available under Danish legal provisions and appropriate material transfer agreements pending The Health Research Ethics Committees for the Capital Region of Denmark (H-18024745) and General Data Protection Regulation governance. To gain access to the Danish data contact E.H. or H.S.N. Requests for access are generally considered monthly. Data from the following publicly available datasets were also used in the study: VEP (https://www.ensembl.org/info/docs/tools/vep/index.html), 1000 Genomes reference genome assembly (https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/GRCh38_reference_genome/); gnomAD (https://gnomad.broadinstitute.org/), OMIM (https://omim.org/) and HPO (https://hpo.jax.org/). All other data presented in the study are provided in the Article and its Supplementary Information. Source data are provided with this paper.
Code availability
The processing of the sequencing data is based on the methods described above, as well as the following open-source software: BWA (v.0.7.10; https://github.com/lh3/bwa), Picard tools (v.1.117; https://github.com/broadinstitute/picard), GraphTyper (v.2.7.5; https://github.com/DecodeGenetics/graphtyper), Manta (v.1.6.0; https://github.com/Illumina/manta), svimmer (https://github.com/DecodeGenetics/svimmer), ADMIXTURE (v.1.3.0; https://dalexander.github.io/admixture/), PLINK (v.1.90b6.15; https://www.cog-genomics.org/plink/1.9/), KING (v.2.1.5; www.kingrelatedness.com), R (v.4.4.3; https://www.r-project.org/) and Python (v.3.9.21; https://www.python.org/).
References
Gruhn, J. R. & Hoffmann, E. R. Errors of the egg: the establishment and progression of human aneuploidy research in the maternal germline. Annu. Rev. Genet. 56, 369–390 (2022).
Sahoo, T. et al. Comprehensive genetic analysis of pregnancy loss by chromosomal microarrays: outcomes, benefits, and challenges. Genet. Med. 19, 83–89 (2017).
McCoy, R. C. et al. Meiotic and mitotic aneuploidies drive arrest of in vitro fertilized human preimplantation embryos. Genome Med. 15, 77 (2023).
Finley, J. et al. The genomic basis of sporadic and recurrent pregnancy loss: a comprehensive in-depth analysis of 24,900 miscarriages. Reprod. BioMed. Online 45, 125–134 (2022).
Waldvogel, S. M., Posey, J. E. & Goodell, M. A. Human embryonic genetic mosaicism and its effects on development and disease. Nat. Rev. Genet. https://doi.org/10.1038/s41576-024-00715-z (2024).
Jónsson, H. et al. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature 549, 519–522 (2017).
Kong, A. et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475 (2012).
Halldorsson, B. V. et al. Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science 363, eaau1043 (2019).
Hinch, R., Donelly, P. & Hinch, A. G. Meiotic DNA breaks drive multifaceted mutagenesis in the human germ line. Science 382, eadh2531 (2023).
Halldorsson, B. V. et al. The sequences of 150,119 genomes in the UK Biobank. Nature 607, 732–740 (2022).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Oddsson, A. et al. Deficit of homozygosity among 1.52 million individuals and genetic causes of recessive lethality. Nat. Commun. 14, 3453 (2023).
Nielsen, R., Hellmann, I., Hubisz, M., Bustamante, C. & Clark, A. G. Recent and ongoing selection in the human genome. Nat. Rev. Genet. 8, 857–868 (2007).
Nagaoka, S. I., Hassold, T. J. & Hunt, P. A. Human aneuploidy: mechanisms and new insights into an age-old problem. Nat. Rev. Genet. 13, 493–504 (2012).
Quenby, S. et al. Miscarriage matters: the epidemiological, physical, psychological, and economic costs of early pregnancy loss. Lancet 397, 1658–1667 (2021).
Steinthorsdottir, V. et al. Variant in the synaptonemal complex protein SYCE2 associates with pregnancy loss through effect on recombination. Nat. Struct. Mol. Biol. https://doi.org/10.1038/s41594-023-01209-y (2024).
Byrne, A. B. et al. Genomic autopsy to identify underlying causes of pregnancy loss and perinatal death. Nat. Med. 29, 180–189 (2023).
Lord, J. et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet 393, 747–757 (2019).
Levy, B. et al. Genomic imbalance in products of conception: single-nucleotide polymorphism chromosomal microarray analysis. Obstetr. Gynecol. 124, 202–209 (2014).
Schlaikjær Hartwig, T. et al. Cell-free fetal DNA for genetic evaluation in Copenhagen Pregnancy Loss Study (COPL): a prospective cohort study. Lancet 401, 762–771 (2023).
Wang, Y. et al. Clinical application of SNP array analysis in first-trimester pregnancy loss: a prospective study. Clin. Genet. 91, 849–858 (2017).
Demko, Z. P., Simon, A. L., McCoy, R. C., Petrov, D. A. & Rabinowitz, M. Effects of maternal age on euploidy rates in a large cohort of embryos analyzed with 24-chromosome single-nucleotide polymorphism-based preimplantation genetic screening. Fertil. Steril. 105, 1307–1313 (2016).
Wang, B. T. et al. Abnormalities in spontaneous abortions detected by G-banding and chromosomal microarray analysis (CMA) at a national reference laboratory. Mol. Cytogenet. 7, 33 (2014).
Graw, S. L., Sample, T., Bleskan, J., Sujansky, E. & Patterson, D. Cloning, sequencing, and analysis of inv8 chromosome breakpoints associated with recombinant 8 syndrome. Am. J. Hum. Genet. 66, 1138–1144 (2000).
Eggertsson, H. P. et al. GraphTyper2 enables population-scale genotyping of structural variation using pangenome graphs. Nat. Commun. 10, 5402 (2019).
Chen, X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222 (2016).
Verdoni, A. et al. Reproductive outcomes in individuals with chromosomal reciprocal translocations. Genet. Med. 23, 1753–1760 (2021).
Watanabe, Y. Geometry and force behind kinetochore orientation: lessons from meiosis. Nat. Rev. Mol. Cell Biol. 13, 370–382 (2012).
Zaragoza, M. V. et al. Parental origin and phenotype of triploidy in spontaneous abortions: predominance of diandry and association with the partial hydatidiform mole. Am. J. Hum. Genet. 66, 1807–1820 (2000).
Palsson, G. et al. Complete human recombination maps. Nature https://doi.org/10.1038/s41586-024-08450-5 (2025).
Hassold, T. et al. Failure to recombine is a common feature of human oogenesis. Am. J. Hum. Genet. 108, 16–24 (2021).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Sondka, Z. et al. COSMIC: a curated database of somatic variants and clinical data for cancer. Nucleic Acids Res. 52, D1210–D1217 (2024).
Coorens, T. H. H. et al. Inherent mosaicism and extensive mutation of human placentas. Nature 592, 80–85 (2021).
Gao, Z. et al. Overlooked roles of DNA damage and maternal age in generating human germline mutations. Proc. Natl Acad. Sci. USA 116, 9491–9500 (2019).
Kaplanis, J. et al. Genetic and chemotherapeutic influences on germline hypermutation. Nature 605, 503–508 (2022).
Goldmann, J. M. et al. Germline de novo mutation clusters arise during oocyte aging in genomic regions with high double-strand-break incidence. Nat. Genet. 50, 487–492 (2018).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Cao, J. et al. A human cell atlas of fetal gene expression. Science 370, eaba7721 (2020).
Lazarin, G. A., Haque, I. S., Evans, E. A. & Goldberg, J. D. Smith–Lemli–Opitz syndrome carrier frequency and estimates of in utero mortality rates. Prenat. Diagn. 37, 350–355 (2017).
Bozhinovski, G., Terzikj, M., Kubelka-Sabit, K. & Plaseska-Karanfilska, D. High incidence of CPLANE1-related joubert syndrome in the products of conceptions from early pregnancy losses. Balkan Med. J. 41, 97–104 (2024).
Jónsson, H. et al. Multiple transmissions of de novo mutations in families. Nat. Genet. 50, 1674–1680 (2018).
Campbell, I. M. et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am. J. Hum. Genet. 95, 173–182 (2014).
Seplyarskiy, V. B. & Sunyaev, S. The origin of human mutation in light of genomic data. Nat. Rev. Genet. 22, 672–686 (2021).
Rubinacci, S., Hofmeister, R. J., Sousa da Mota, B. & Delaneau, O. Imputation of low-coverage sequencing data from 150,119 UK Biobank genomes. Nat. Genet. 55, 1088–1090 (2023).
Singh, U. et al. Expression and functional analysis of fibulin-1 (Fbln1) during normal and abnormal placental development of the mouse. Placenta 27, 1014–1021 (2006).
Mazurek, A. et al. DDX5 regulates DNA replication and is required for cell proliferation in a subset of breast cancer cells. Cancer Discov. 2, 812–825 (2012).
Vaughan, N., Scholz, N., Lindon, C. & Licchesi, J. D. F. The E3 ubiquitin ligase HECTD1 contributes to cell proliferation through an effect on mitosis. Sci. Rep. 12, 13160 (2022).
Zerafati-Jahromi, G. et al. Sequence variants in HECTD1 result in a variable neurodevelopmental disorder. A. J. Hum. Genet. https://doi.org/10.1016/j.ajhg.2025.01.001 (2025).
Organisation for Economic Co-operation and Development. Declining fertility rates put prosperity of future generations at risk. OECD www.oecd.org/en/about/news/press-releases/2024/06/declining-fertility-rates-put-prosperity-of-future-generations-at-risk.html (2024).
Jónsson, H. et al. Whole genome characterization of sequence diversity of 15,220 Icelanders. Sci. Data 4, 170115 (2017).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Eggertsson, H. P. et al. Graphtyper enables population-scale genotyping using pangenome graphs. Nat. Genet. 49, 1654–1660 (2017).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Consortium, R. E. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Price, A. L. et al. Long-range LD can confound genome scans in admixed populations. Am. J. Hum. Genet. 83, 132–135 (2008).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015).
Benjamini, Y. & Speed, T. P. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 40, e72 (2012).
Rabiner, L. R. A tutorial on hidden Markov models and selected applications in speech recognition. Proc. IEEE 77, 257–286 (1989).
Nik-Zainal, S. et al. The life history of 21 breast cancers. Cell 149, 994–1007 (2012).
Riggs, E. R. et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med 22, 245–257 (2020).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Döring, A., Weese, D., Rausch, T. & Reinert, K. SeqAn an efficient, generic C++ library for sequence analysis. BMC Bioinform. 9, 11 (2008).
Wood, S. N. Fast stable restricted maximum likelihood and marginal likelihood estimation of semiparametric generalized linear models. J. R. Stat. Soc. B 73, 3–36 (2011).
Jónsson, H. et al. Differences between germline genomes of monozygotic twins. Nat. Genet. 53, 27–34 (2021).
McLaren, W. et al. The Ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Pruitt, K. D., Tatusova, T. & Maglott, D. R. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, D501–D504 (2005).
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625, 92–100 (2024).
Arnadottir, G. A. et al. Population-level deficit of homozygosity unveils CPSF3 as an intellectual disability syndrome gene. Nat. Commun. 13, 705 (2022).
Weiss, L. A. et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 358, 667–675 (2008).
Degasperi, A. et al. A practical framework and online tool for mutational signature analyses show inter-tissue variation and driver dependencies. Nat. Cancer 1, 249–263 (2020).
Moore, L. et al. The mutational landscape of human somatic and germline cells. Nature 597, 381–386 (2021).
Rahbari, R. et al. Timing, rates and spectra of human germline mutation. Nat. Genet. 48, 126–133 (2015).
Pálsson, G. DecodeGenetics/PalssonEtAl_Nature_2024: Initial release of data. Zenodo https://doi.org/10.5281/zenodo.14025565 (2024).
hakon-jon-deCODE. hakon-jon-deCODE/Arnadottir_et_al_2025_Nature: Initial release of data. Zenodo https://doi.org/10.5281/zenodo.15183295 (2025).
Acknowledgements
We thank everyone who agreed to participate in this study; all colleagues at the Gynaecological Acute Clinics who helped us inform patients about the project and arranged inclusions; S. B. Blom, I. M. Søndergaard, S. S. Dandanell, C. Brinkmann, M. M. B. S. Petersen, Y. I. E. Sammaa-Aru, N. Vinterberg and E. S. Juel for their effort in including participants and collecting and packing the whole-blood samples and pregnancy tissue; and M. Visser for preparing the samples in the laboratory. We acknowledge deCODE genetics/Amgen, Department of Cellular and Molecular Medicine at University of Copenhagen, and Department of Obstetrics and Gynaecology at Copenhagen University Hospital, Amager Hvidovre for financial support for this study; and Ole Kirks Foundation, Danish National Research Foundation (DNRF115), AP Moller Foundation, Independent Research Fund Denmark (0134-00299B and 2034-00263B), BioInnovation Institute Foundation (grant number: BII22SG1021020), and the Novo Nordisk Foundation (grant numbers: NNF15OC0016662, NNF22OC0077221, NNF23OC0087269, NNF22OC0074308 and NNF20SA0064340).
Author information
Authors and Affiliations
Consortia
Contributions
G.A.A., H.J., T.S.H., J.R.G., E.R.H., H.S.N. and K.S. designed the study. G.A.A., H.J., T.S.H., J.R.G., D.W., A.C.-H.C., L.l.R., G.B.W., H.S., J.F.P., G.L.N., J.S., O.T.M., S.B., K.B., E.R.H. and H.S.N. collected samples and data. G.A.A., H.J., T.S.H., J.R.G., A.C.-H.C., L.l.R., J.S., O.T.M., S.B. and H.S.N. performed experiments. G.A.A., H.J., T.S.H., J.R.G., A.G., V.S., K.H.S.M., S.O., A.H., O.T.M., B.V.H., S.B., D.F.G., M.N., P.S., U.T. and E.R.H. designed the methods. G.A.A., H.J., T.S.H., J.R.G., P.L.M., A.G., D.W., A.C.-H.C., A.O., L.S., K.H.S.M., S.O., P.I.O., G.H.H., S.A.G., G.P., B.O.J., R.F., P.D.R. and K.B. analysed the data. G.A.A., H.J., T.S.H., J.R.G., E.R.H., H.S.N. and K.S. wrote the manuscript. All of the authors contributed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors affiliated with deCODE genetics/Amgen declare competing financial interests as employees.
Peer review
Peer review information
Nature thanks Christopher Barnett, Ziyue Gao and the other, anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Schematic view of meiosis I error.
Missegregation of homologous chromosome during meiois I. See Fig. 2 for meiosis II error.
Extended Data Fig. 2 Crossover locations in paternal triploidies.
See Fig. 4b for the crossover locations in maternal triploidies.
Extended Data Fig. 3 Spectra of DNMs present in single/multiple fetal samples.
Here, we restricted to pregnancy losses with multiple fetal samples and aggregated the DNMs based on whether they were present in a single sample or in multiple samples per mutation class. For comparison, the bottom panel is the mutational spectrum for DNMs identified in Icelanders.
Extended Data Fig. 4 Number of DNMs as a function of allelic balance.
Aggregation was done on the fetal sample level and the entire DNM set was used.
Extended Data Fig. 5 Number of DNMs per fetal sample, tissue type and pathogenic carrier status against paternal age at conception.
The black line is the regression line from the Icelandic set, the coloured lines are regression lines for subsets of the COPL cohort and the shaded areas represent 95% CIs. a, all fetal samples regardless of karyotype. b, excluding triploidies. c, only triploidies. d, same as b but coloured according to whether the fetus is a carrier of a pathogenic DNM.
Extended Data Fig. 6 Paternal ages at conception.
a, the parental ages at conception for the pregnancy losses. b, the parental ages at conception for the Icelandic set. c-d, histogram of the parental ages at conception for the pregnancy losses. e-f, histogram of the parental ages at conception for the Icelandic set.
Extended Data Fig. 7 Age regression - Poisson shared intercept/slopes models.
The analysis restricted to non-triploid samples with 95% power of detecting DNMs. a, paternal age effect. b, maternal age effect. c, shared intercept at conception. d, shared intercept at onset of puberty (13 years). e, villi coefficient. f, deCODE vs COPL-villi difference against deCODE vs COPL-non-villi difference. In f, the age effects are shared across the COPL and the deCODE cohorts, the intercept is shared between the sexes and the regression is done separately per mutational class. In a-d and f the solid lines are identity lines. The centres represent means and error bars are 95%-CIs based on a Block-Jackknife procedure. The estimates are based on 293 pregnancy loss trios and 9,430 Icelandic trios.
Extended Data Fig. 8 Allele balance of DNMs conditioned on sister chromatid or homologous chromosome state.
Extended version of Fig. 5e with both sister chromatid and homologous chromosome state. The vertical line is an allelic balance of 50%.
Extended Data Fig. 9 Schematic overview of dispermy derived from a single spermatogonium or distinct spermatogonia.
Paternal triploidies caused by spermatozoa from the same spermatogonium fertilizing the oocyte are expected to have pre-sister mutations in 2/3 allelic balance. For distinct spermatogonia the allelic balance of pre-sister and post-sister mutations will be the same, i.e. 1/3.
Extended Data Fig. 10 Expression of genes mutated in euploid versus aneuploid/ triploid fetuses.
Based on fetal tissue expression data from Cao et al., showing expression for 15 fetal tissues for n = 16 genes mutated in euploid fetuses and n = 16 genes mutated in aneuploid/triploid fetuses. Genes mutated in euploid fetuses are on average more highly expressed in fetal tissues than genes mutated in aneuploid/triploid fetuses. The box plots show the median expression value (transcripts per million), limits correspond to the first (25%) and third (75%) quartiles, and whiskers extend to the smallest and lowest values observed, limiting to 1.5 times the interquartile range.
Supplementary information
Supplementary Information
Supplementary Notes, Supplementary Figures, Supplementary Tables 1 and 5, legends for Supplementary Tables 1–9 and Supplementary References.
Supplementary Tables
Supplementary Tables 2–4 and 7–9.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Arnadottir, G.A., Jonsson, H., Hartwig, T.S. et al. Sequence diversity lost in early pregnancy. Nature 642, 672–681 (2025). https://doi.org/10.1038/s41586-025-09031-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09031-w
This article is cited by
-
Anaysis of the association between chromosomal abnormalities in early missed abortion embryos and maternal age and AMH levels based on CNV-Seq
BMC Pregnancy and Childbirth (2025)
-
Fetal and parental genomes offer mechanistic insights into pregnancy loss
Nature (2025)
-
Genomics pioneer fired from firm he founded: ‘It was not easy to domesticate me’
Nature (2025)
