
Abstract
The surface landscapes of cells differ as a function of cell type and are frequently altered in disease contexts1,2,3. Exploiting such differences is key to many therapeutic strategies and is the basis for developing diagnostic and basic-science tools. State-of-the-art strategies typically target single surface antigens, but each individual receptor rarely defines the specific cell type4,5. The development of programmable molecular systems that integrate multiple cell-surface features to convert on-target inputs to user-defined outputs is therefore highly desirable. Here we describe an autonomous decision-making device driven by proximity-gated protein trans-splicing that allows local generation of an active protein from two otherwise inactive polypeptide fragments. We show that this protein-actuator platform can perform convergent protein ligation on designated cell surfaces, allowing highly selective generation of active proteins, which can either remain physically associated with the cell surface on which they were manufactured or be released into the surrounding milieu. Because of its intrinsic modularity and tunability, we demonstrate that the technology is compatible with different types of input, targeting modality and functional output, allowing for the localized interrogation or manipulation of cellular systems.
Similar content being viewed by others

Main
Cell-surface heterogeneity is exploited in nature as a central strategy for discrimination in multicellular systems with the purpose of modulating biological functions in time and space. Designing protein tools that emulate this paradigm with new and highly selective stimulus–response functions provides opportunities to study complex cell biochemistry and to create sophisticated synthetic biology outputs. Current synthetic systems use macromolecular targeting modalities, such as antibodies, to selectively deliver activities that affect downstream cellular function6,7. Despite the notable successes of this strategy, it is limited by the fact that single antigens rarely define cell or tissue types, and the wrongful commitment of these agents can therefore complicate their use in more complex settings4,5. Protein Boolean logic gates8,9 have been implemented to define targeting more finely10,11, whereby engineered antibody-based platforms or synthetic protein switches use the concurrent binding of two or more targeting vectors to recruit a third effector modality12,13,14. Although these are exciting developments, many effector modalities (such as cytokines and enzymes) are highly active in ways that are independent of any Boolean-based recruitment, and they will therefore exert activities unconstrained by target binding. It therefore remains an outstanding challenge to turn on protein function in situ from an inactive state in response to cell-surface cues. Developing a system that allows for the local generation of effector proteins is one way of achieving this goal. Fragmented proteins are often inactive but their function can be gained by proximity-induced complementation or ligation15,16,17. Thus, a control strategy could be devised that uses the identity of cell-surface antigens to recruit inactive precursor polypeptides, which in proximity would template the reconstitution of an effector molecule on a target cell by in situ protein ligation. In this scenario, activation of the desired protein function occurs only on the subset of target cells displaying the predefined antigen combination, in essence making the effector activity stringently cell selective.
Conditional protein splicing (CPS) is an attractive strategy to control protein structure and function because it uses the autocatalytic excision–ligation reaction of split inteins to synthesize functional proteins on demand from precursor polypeptides16 (Supplementary Fig. 2a,b). We previously engineered the conditional reactivity of the ultra-efficient split inteins Npu, gp41-1, gp41-8 and NrdJ-1 (ref. 18). This was done by caging each split intein fragment with a truncated segment of the matching partner (Supplementary Fig. 2c), thereby strongly inhibiting the initial fragment–fragment complexation required for splicing. However, because each caged split-intein fragment exists in an incomplete and therefore semi-frustrated structural state, the pair will readily undergo a spontaneous intermolecular domain-swapping event to produce the functional intein fold when colocalized19 (Supplementary Fig. 2c). Any input that induces such colocalization can in principle program this strategy, theoretically limited only by which targeting vectors can be appended onto the caged split-intein fragment pair. Importantly, CPS serves as a broadly applicable control strategy for the on-demand generation of a wide array of protein functions20, with the caged split intein thereby functioning as an actuator of a rewired stimulus–response relationship. We therefore proposed an approach that uses the unique features of a cell-surface landscape to conditionally template in situ protein trans-splicing, thereby achieving cell-selective protein function.
Here, we develop a protein actuator system called splicing-modulated actuation upon recognition of targets (SMART), which discriminates between various cell populations on the basis of their surface landscapes, such that cells with correct antigen combinations template protein trans-splicing on their surface (Fig. 1a). We show that the system is compatible with a wide variety of targeting modalities, from antibody fragments to small molecules that drive the cell-selective ligation of proteins. The localized activities of these proteins can be used in applications as varied as gated-proximity labelling and the induction of localized cellular signalling outputs.

a, Complementary caged split intein fragments (IntNcage and IntCcage) are fused to targeting vectors (anti-Ag1 and anti-Ag2) to different cell-surface antigens (Ag1 and Ag2). Colocalization on the cell surface leads to protein trans-splicing and generation of a functional protein nested between anti-Ag1 and anti-Ag2. The system works as an AND-gated input-to-output actuator. b, As a functional output, SMART-SpyCatcher was designed by splitting SpyCatcher003 at residues 73–74 and fusing the fragments to IntNcage and IntCcage. The reconstituted SpyCatcher003 binds covalently to SpyTag003 through an isopeptide bond. c, Live-cell imaging of individual K562 cell lines (wild type, HER2+, EGFR+ and HER2+/EGFR+) treated with anti-HER2–SpyN and SpyC–anti-EGFR (100 nM each, 2 h) followed by SpyTag003 labelled with Alexa Fluor 594 (SpyTag003–AF594, 100 nM, 20 min). Cells were stained with Hoechst; HER2 and EGFR expression were visualized by eGFP and iRFP, respectively. Scale bar, 20 μm. d, Schematic of SMART-SpyCatcher operating through [HER2 AND EGFR] logic on a mixed population of the four K562 cell lines. e, Flow cytometry analysis of the mixed K562 population (combined at equal amounts) either treated with SpyTag003–AF594 alone (top) or following treatment with SMART-SpyCatcher (bottom, [HER2 AND EGFR] logic). The four cell types were gated based on HER2–eGFP AND EGFR–iRFP expression and each cell type is plotted as number of cells versus AF594 relative fluorescence units (RFU); histograms are normalized to the mode. f, Dose–response experiment on the mixed K562 population with a dilution series of SMART-SpyCatcher (1 µM to 1 nM) and excess SpyTag003–AF594 (100 nM). Fold change in AF594 median fluorescence intensity (MFI) were calculated relative to untreated cells. Data shown in panels c,e,f are representative of three independent experiments. All other data are presented as mean ± s.e.m. (n = 3 independent biological replicates).
Engineering a protein actuator
Although CPS can in principle generate any desired protein output, we initially focused our efforts on designing a SMART protein that would convert two antigen inputs presented on the same cell into a unique protein recruitment dock. This would allow us to monitor and characterize the protein design using fluorescence-based methods and later deliver a broader arsenal of functionalities. To achieve this, we concentrated on developing a SMART version of SpyCatcher003 (ref. 21), a protein superglue that spontaneously forms an isopeptide bond with the 16-amino-acid peptide SpyTag003 (Fig. 1b).
We began by systematically screening multiple potential split sites in SpyCatcher003 (designated SpyN and SpyC) for compatibility with CPS (Extended Data Fig. 1a), initially using the caged Npu split intein (NpuNcage and NpuCcage) in conjunction with the FKBP/rapamycin/FRB three-hybrid system (Supplementary Fig. 3 and Supplementary Table 1). To simulate cell-surface colocalization, we induced in-solution dimerization of SpyN–NpuNcage–FKBP and FRB–NpuCcage–SpyC by adding rapamycin (Extended Data Fig. 1b). Our screening process revealed several pairs with the ability to fully recapitulate SpyCatcher003–SpyTag003 reactivity after splicing (Extended Data Fig. 1c and Supplementary Fig. 4). We chose the SpyN1–73–SpyC74–113 pair, because this combination had minimal background reactivity with the peptide probe before splicing, that is, spontaneous complementation was minimal (Extended Data Fig. 1c). The Npu split intein contains four cysteines in its structure and leaves a cysteine substitution in the spliced product. Anticipating that these features could be deleterious to applications in the oxidizing environment of the cell surface, we exchanged Npucage for a caged version of the NrdJ-1 split intein22, which leaves a serine at the splice site. The resulting constructs SpyN–NrdJ-1Ncage–FKBP and FRB–NrdJ-1Ccage–SpyC yielded almost complete splicing when rapamycin was added (Extended Data Fig. 1d), whereas no splicing or reactivity with SpyTag003 was observed when the caged split intein was inactivated (Extended Data Fig. 1e). To enable further engineering of this system, we generated a fusion of NrdJ-1 and solved its structure at 1.95 Å by X-ray crystallography (Protein Data Bank (PDB): 8UBS; Extended Data Fig. 1f, Supplementary Fig. 5 and Supplementary Table 2). Using this structural information, we were able to design a fully active mutant of NrdJ-1cage in which the non-catalytic Cys76 is replaced by a valine (Extended Data Fig. 1f–j and Supplementary Fig. 6). We expected that this mutant would be better suited to applications on the cell surface. We refer hereafter to the individual components SpyN–NrdJ-1N(C76V)cage and NrdJ-1Ccage(C76V)–SpyC as SpyN and SpyC, respectively, and their sum as SMART-SpyCatcher.
On-cell performance of SMART-SpyCatcher
Next, SMART-SpyCatcher was refitted for on-cell activation by removing both FKBP and FRB and installing DARPins23,24,25 targeting the two model antigens HER2 and EGFR, thereby generating anti-HER2–SpyN and SpyC–anti-EGFR (Supplementary Fig. 3 and Supplementary Table 1). The ability of SMART-SpyCatcher to perform [HER2 AND EGFR] logic was assessed on K562 leukaemia cells coexpressing HER2–eGFP and EGFR–iRFP12. Treatment of these cells with anti-HER2–SpyN and SpyC–anti-EGFR followed by a SpyTag003 Alexa Fluor 594 conjugate (SpyTag003–AF594) resulted in the expected fluorescence signal, albeit a weak one, on the cell surface (Extended Data Fig. 2a); using Npucage as the actuator component also led to the expected output (Supplementary Fig. 7). With the aim of improving the reaction, we tested an enhanced version of NrdJ-1(C76V)cage (referred to as eNrdJ-1cage) with re-engineered cages to improve the recruitment of SpyTag003–AF594 (Supplementary Fig. 6). This strategy afforded noticeably brighter AF594 fluorescence on the cell surface (Fig. 1c and Extended Data Fig. 2a). We therefore continued to use this actuator variant for all subsequent experiments unless otherwise stated. To exclude in-solution or at-solution-surface interphase activation, we also evaluated the activity of SMART-SpyCatcher on K562 cell lines naive (wild type) or single positive for either HER2–eGFP or EGFR–iRFP, all of which failed to elicit any response (Fig. 1c and Extended Data Fig. 2b,c). We further tested the on-target/off-target specificity of SMART-SpyCatcher when presented simultaneously with multiple decisions, and applied a mixed-population flow cytometry assay for quantification (Fig. 1d and Supplementary Figs. 8 and 9). When incubated concurrently with equal amounts of the four cell lines in a mixture, SMART-SpyCatcher retained its target specificity (Fig. 1e, Extended Data Fig. 2d and Supplementary Fig. 9), even over a broad concentration range (nanomolar to micromolar), producing a sigmoidal dose–response curve only for K562HER2+/EGFR+ (Fig. 1f). Further experimental evidence supports a three-step mechanism of action: first, anti-HER2–SpyN and SpyC–anti-EGFR bind to the target cell; second, SpyCatcher003 is spliced, owing to colocalization; and third, SpyCatcher003 reacts with SpyTag003. Blocking any of these steps leads to a complete loss of the AF594 signal (Extended Data Fig. 3a–c). Furthermore, although SMART-SpyCatcher worked under various conditions (Extended Data Fig. 3d) and was AND gated (Extended Data Fig. 3e), its decision-making ability was lost when the cages were omitted from NrdJ-1, instead inducing massive crosslinking between single- and double-positive K562 cells (Extended Data Fig. 3f).
We also generated SpyN and SpyC constructs that target epithelial cell adhesion molecule (EpCAM; Supplementary Fig. 3 and Supplementary Table 1). When tested on mixed K562 cell lines expressing either low endogenous or high ectopic levels of EpCAM in combination with different profiles of HER2 and EGFR (Supplementary Figs. 9–11), we found that SMART-SpyCatcher computed AND functions on cells that fulfilled the assigned logic gates (Extended Data Fig. 4a). This observation held true when SMART-SpyCatcher was evaluated on a variety of cancer cell lines with endogenous levels of HER2, EGFR and EpCAM (Extended Data Fig. 4b,c and Supplementary Tables 3 and 4). Furthermore, we found that the responsiveness of SMART-SpyCatcher, as measured by the magnitude of SpyTag003–AF594 recruitment, was correlated with the quantity of the less-expressed antigen used in each AND gate (Extended Data Fig. 4d). This implies that the level of SMART actuation is predominantly dictated by the quantities of each target antigen. Finally, with multiple antigen inputs available, we could also use SpyN–anti-Ag1/SpyN–anti-Ag2/SpyC–anti-Ag1/SpyC–anti-Ag2 in OR and SpyN–anti-Ag1/SpyC–anti-Ag2/SpyC–anti-Ag3 in AND/OR targeting strategies (Extended Data Fig. 4e–h).
Tuning SMART response dynamics
Encouraged by the ability of our protein device to make correct decisions, we created a suite of SMART-SpyCatcher pairs with a dynamic response range. Guided by the crystallographic information on NrdJ-1 (Fig. 2a), we manipulated the cage of eNrdJ-1Ncage while maintaining unaltered eNrdJ-1Ccage as a constant. This afforded multiple pairs with increased or decreased activities (Fig. 2b, Extended Data Fig. 5, Supplementary Fig. 12 and Supplementary Tables 5 and 6), without any compromise in target specificity. For instance, introducing K114A and K116A mutations in the cage sequence increased SpyTag003–AF594 recruitment by 150%, whereas A119K led to a 25% drop. Alternatively, elongating or shortening the cage length of eNrdJ-1Ncage resulted in variants with activities ranging between 13% and 150% of that of the standard 35-amino acid toehold.

a, Summary of the diverse interactions between NrdJ-1N and NrdJ-1C forming the hedgehog–intein fold (PDB: 8UBS). Electrostatic surface potential mapping (PyMOL, adaptive Poisson–Boltzmann solver; kB = Boltzmann constant; T = temperature (K) and ec = elementary charge (C)) reveals an electrostatic interface between the basic N terminus of NrdJ-1C and an acidic groove of NrdJ-1N (i); further inserts detail electrostatic interactions (ii), residue hydrogen bonding (iii,iv), backbone hydrogen bonding (v) and hydrophobic packing (vi) stabilizing the complex. b, The eNrdJ-1Ncage variants were tested on K562HER2+/EGFR+ cells using anti-HER2–SpyN (variant indicated), SpyC–anti-EGFR (eNrdJ-1Ccage) and SpyTag003–AF594 (100 nM each). The AF594 MFI values were normalized to the response with standard eNrdJ-1Ncage. c, Anti-EGFR-Decoy was introduced as a NOT operator to obstruct AND logic output on HER2+/EGFR+/EpCAM+ cells by splicing with SpyC to generate defective SpyCatcher003. d, A SMART-SpyCatcher implementing [HER2 AND EpCAM NOT EGFR] logic was tested on mixed K562 cells (phenotypes indicated). Cells were treated with anti-EGFR–Decoy (concentrations indicated), anti-HER2–SpyN/SpyC–anti-EpCAM/SpyTag003–AF594 (100 nM each) and analysed by flow cytometry. The AF594 MFI is plotted relative to control cells lacking anti-EGFR–Decoy. e, Schematic of AND-gated cell depletion. Cells are decorated with SpyTag003-biotin via SMART-SpyCatcher enabling recruitment of a Streptavidin–Saporin disulfide conjugate (Strep–Saporin), leading to cell death upon internalization. f, Mixed K562 cells (phenotypes indicated) were treated with a two-dose regimen of SMART-SpyCatcher/SpyTag003-biotin ([HER2 AND EGFR] logic, 100 nM each) and Streptavidin–Saporin (20 nM) at a 24-h interval. Cell viability was assessed after 72 h by flow cytometry and normalized to untreated wild-type cells. g, A431 cells (HER2low, EGFRhi and EpCAMhi) were treated as in f, using the indicated SMART-SpyCatcher system (eNrdJ-1cage(K114A/K116A)). Cell viability was determined by XTT assay. Data are mean ± s.e.m. (n = 3 independent biological replicates). Statistical analysis: unpaired two-sided t-test (b). NS, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
The ability to tune the responsiveness of the SMART system by engineering the caging element expands the types of application possible using the approach. Notably, we were able to perform more logic operations, including a NOT gate, where we exploited a decoy protein featuring a weakened cage (Fig. 2c,d, Extended Data Fig. 6a and Supplementary Fig. 13), and for selectively targeting breast cancer cell lines based on their surface antigen presentations (Extended Data Fig. 6b–f and Supplementary Figs. 14–16).
Cell-selective killing
The ability to stringently deliver a payload to cells has many applications. Interestingly, when K562HER2+/EGFR+ cells were treated with anti-HER2–SpyN/SpyC–anti-EGFR/SpyTag003-biotin, it allowed for the cell-selective recruitment and endosomal internalization of a NeutrAvidin Rhodamine Red-X conjugate (Extended Data Fig. 7a–c). We reasoned that targeted cell tagging with a protein toxin (such as Saporin)26 could allow for selective depletion in a heterogeneous population (Fig. 2e). To test this, mixed K562 cells were first treated with SMART-SpyCatcher assigned for [HER2 AND EGFR] logic and SpyTag003-biotin, then cultured with a Streptavidin–Saporin disulfide conjugate, and subsequently sampled for variations in cell composition after 72 h (Extended Data Fig. 7d,e). A one-dose regimen led to a 53% reduction of K562HER2+/EGFR+ cells (Extended Data Fig. 7f and Supplementary Fig. 17), and a two-dose regimen gave a 78% depletion of the subpopulation (Fig. 2f and Supplementary Fig. 18). Importantly, the wild type, K562HER2+ and K562EGFR+ cell lines were not depleted to a similar extent in the mixed population. We also tested the depletion of A431 cells (HER2low, EGFRhi and EpCAMhi) and used SMART-SpyCatcher with eNrdJ-1cage(K114A/K119A) to enhance the recruitment of SpyTag003-biotin. Compared with an untreated control, A431 cells were depleted by 92% when using the two-dose regimen with SpyTag003-biotin, Streptavidin–Saporin and SMART-SpyCatcher operating by [EGFR AND EpCAM] logic (Fig. 2g). As expected, an identical workflow with SMART-SpyCatcher acting by [HER2 AND EGFR] or [HER2 AND EpCAM] logic failed to achieve a comparable reduction in the cell viability of this cell line, and no reduction was observed when the toxin conjugate was omitted. Thus, SMART-SpyCatcher enables logic-gated cell-depletion strategies27,28,29.
Diversifying vector modalities
Many modalities including antibody fragments and mimetics, peptides and small molecules can be used to target cell-surface receptors (Fig. 3a). The ability to use these unlocks new potential targets of SMART. For instance, while we earlier fused SpyN and SpyC to DARPins, we found that single-domain antibody (sdAb)30 and single-chain fragment variable (scFv)12 fusions could easily be produced by recombinant bacterial expression (Supplementary Fig. 3 and Supplementary Table 1). Furthermore, introducing a carboxy-terminal cysteine in eNrdJ-1Ccage (which lacks any other cysteines) provided facile chemical installation of synthetic ligands (Supplementary Figs. 19–21). For example, we conjugated SpyC to the peptidyl antagonist BKT140 (ref. 31) of the cytokine receptor CXCR4, and to the small-molecule antagonist SCH58261 (ref. 32) that targets the G-protein-coupled receptor ADORA2A, correspondingly making SpyC–anti-CXCR4 and SpyC–anti-A2A. To test the generated sdAb and scFv fusions, as well as the synthetic ligand conjugates, we used K562HER2+/EGFR+, wild-type OE19 cells (HER2hi, EGFRlow, EpCAMhi, CEACAM6hi, CXCR4low and ADAORA2Alow), and the two stable cell lines OE19CXCR4DOX and OE19A2ADOX made for the doxycycline-inducible expression of either CXCR4 or ADORA2A, respectively (Supplementary Fig. 22). Without altering our previous SMART-SpyCatcher protocol, we found these new vectors to be fully compatible with our previous DARPin variants, in each case affording the expected logic-gated outputs (Fig. 3b–d, Supplementary Fig. 23 and Supplementary Table 4). Thus, the SMART platform is highly modular with respect to targeting vector modalities.

a, SMART targeting modalities encompass antibody fragments (AbFs), antibody mimetics (AbMs) and synthetic ligands (peptides and small molecules). b–d, SMART-SpyCatcher was tested with an anti-CEACAM6 single-domain antibody, a CXCR4-targeting cyclic peptide BKT140, an ADORA2A-targeting small molecule SCH58261 and the DARPins anti-HER2, anti-EGFR and anti-EpCAM. OE19 (HER2hi, EGFRlow and EpCAMhi; b) or derivatives stably expressing CXCR4 (OE19CXCR4DOX; c) or ADORA2A (OE19A2ADOX; d) were treated with SMART-SpyCatcher and fluorescently labelled SpyTag003 (100 nM each), followed by flow cytometry. e, Schematic of AND-gated protein proximity labelling by the delivery of a SpyTag003 catalyst. f, K562HER2+/EGFR+, OE19, A431 and OE19CXCR4DOX were treated with SMART-SpyCatcher (100 nM), SpyTag003–APEX2 (300 nM) and then biotin-phenol (250 μM) and H2O2 (100 mM, 2 min) before western blot analysis. AND gates predicted to enable labelling are shown in red; asterisks indicate endogenously biotinylated proteins. g, Densitometry analysis was done using FIJI (National Institutes of Health). Signals were normalized to tubulin (loading control). a.u., arbitrary units. h, Schematic of cell-selective photocatalytic proximity labelling using SMART-SpyCatcher and a SpyTag003-Ir conjugate. i, Mixed K562 cells (phenotypes indicated) were treated with SMART-SpyCatcher (100 nM) and SpyTag003-Ir (200 nM). After washing, cells were incubated with biotin-diazirine (100 μM), irradiated with 450-nm light (5 min), stained with Streptavidin-Alexa Fluor 546 and analysed by flow cytometry; histograms are normalized to the mode. j, Quantification of the data in i; mean ± s.e.m. (n = 3 independent biological replicates). Statistical analysis: applied unpaired two-sided t-test (b–d); applied one-way analysis of variance (ANOVA) followed by Dunnett’s test (j) (Supplementary Tables 7 and 8).
Logic-gated proximity labelling
Many surface proteins localize into specialized networks, the biological functions of which are often difficult to discern. With our diverse set of SMART-SpyCatcher pairs in hand, we reasoned that logic operations could be coupled to protein proximity labelling strategies to elucidate such microenvironments (Fig. 3e). We initially established a workflow that involves the delivery of SpyTag003–APEX2 for the APEX2/H2O2-catalysed generation of long-lived phenoxyl radicals (a t½ of milliseconds)33. First, we treated K562HER2+EGFR+ cells with SMART-SpyCatcher for [HER2 AND EGFR] logic, followed thereafter with SpyTag003–APEX2 (Extended Data Fig. 8a, Supplementary Fig. 3 and Supplementary Table 1). After washing out excess SpyTag003–APEX2, a biotin-phenol probe was added and activated by H2O2, which led to robust cell biotinylation (Extended Data Fig. 8b). Importantly, no labelling was observed in the various controls (Extended Data Fig. 8b). We validated these results on various cell lines by performing several AND-gated logic combinations involving HER2, EGFR, EpCAM, CEACAM6, CXCR4 and ADORA2A for the delivery of SpyTag003–APEX2 before proximity labelling. Only when SMART-SpyCatcher was matched with the correct antigen profile of the individual cell line did we observe APEX2-dependent biotinylation (Fig. 3f,g and Extended Data Fig. 8c–j).
Our ability to conduct AND-gated proximity labelling was extended to the photocatalytic proximity labelling platform μMap34, which uses an iridium-centred photocatalyst to activate an aryl diazirine by blue-light irradiation for the generation of short-lived singlet carbene species (t½ = 2 ns). In this case, K562 cells expressing HER2 and EGFR were treated with anti-HER2–SpyN and SpyC–anti-EGFR followed by SpyTag003 conjugated to the iridium photocatalyst (Extended Data Fig. 9a–d and Supplementary Table 1). These initial studies confirmed that treatment of these cells with the diazirine-biotin probe followed by blue-light irradiation led to robust cellular labelling that was dependent on the SMART reaction (Extended Data Fig. 9e,f and Supplementary Fig. 24). Encouraged by this, we then performed a similar photocatalytic proximity labelling experiment using mixtures of K562 cell lines expressing different receptor combinations and used flow cytometry as the read-out (Fig. 3h). We found that labelling occurred only on cells for which the SMART-SpyCatcher was programmed to react with the assigned antigen profile (Fig. 3i,j and Supplementary Fig. 25). Collectively, these data demonstrate that the SMART system can be used to conduct photocatalytic proximity labelling experiments in a context-specific manner, potentially creating new opportunities for proteomics mapping of cell surfaces in complex settings such as tissues.
Developing SMART-IL-1β
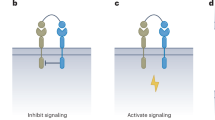
The SMART platform has the potential to respond to antigen inputs with different protein functional outputs through their templated trans-splicing. To examine this further, we next focused on generating a new SMART protein. Cytokines are a potent class of immune-system modulators, and their on-demand function has become a pursuit of protein design13. Interleukin-1β (IL-1β) is a key pro-inflammatory cytokine with a broad range of immune activities, including the stimulation of antigen-presenting cells, natural killer cells and CD4+/CD8+ T cells35 (Extended Data Fig. 10a). We explored whether SMART could be used as a platform to generate IL-1β in response to a combination of surface antigens with the spliced cytokine being released into the surrounding microenvironment (Fig. 4a). Because IL-1β represents a challenging target for SMART, owing to its complex β-barrel structure, we first systematically screened for potential split sites using the FKBP/rapamycin/FRB three-hybrid system (Extended Data Fig. 10b–f, Supplementary Fig. 3 and Supplementary Table 1). This revealed IL-1βN1–44 and IL-1βC45–153 as a candidate pair, with IL-1β activity on cultured HeLa cells only when spliced. Specifically, we found that NF-κB localized to the nucleus (Extended Data Fig. 10g,h), a clear sign of activated IL-1 receptor 1/IL-1 receptor accessory protein (IL-1R1/IL-1RAcP) signalling induced by biologically active IL-1β. We therefore refer to the individual components IL-1βN1–44-eNrdJ-1Ncage and eNrdJ-1Ccage-IL-1βC45–153 as IL-1βN and IL-1βC, respectively, and their sum as SMART-IL-1β.

a, Colocalization of SMART-IL-1β (IL-1βN–anti-Ag1/anti-Ag2–IL-1βC) on a target-cell template ligation and release of the cytokine IL-1β, which activates IL-1R1/IL-1RAcP on neighbouring cells. b, OE19 cells (HER2hi, EGFRlow and EpCAMhi) were treated with SMART-IL-1β (eNrdJ-1cage, 20 nM) and the conditioned media supplemented to HeLa cells to stimulate IL-1R1 receptor signalling (top). Control experiments included: adding anti-HER2/anti-EpCAM DARPins (500 nM); use of splicing-deficient SMART-IL-1β (eNrdJ-1N(C1A)cage, 20 nM); and IL-1RA (IL-1R1 antagonist) pretreatment of HeLa cells. NF-κB localization was assessed by immunofluorescence imaging. c–f, HEK-Blue IL-1β cells, reporting IL-1β stimulation by SEAP, were exposed to conditioned media. SMART-IL-1β was supplied to OE19 (**P = 0.0072) (c), K562HER2+/EGFR+ (d) and individual cultures of the specified K562 cell lines (e) at the indicated concentrations. The conditioned media were then transferred to HEK-Blue IL-1β cells for 24 h before SEAP quantification. Controls are as in b. K562HER2+/EGFR+ cells were used in dose–response experiments with SMART-IL-1β using eNrdJ-1cage, eNrdJ-1cage_1–38 (strengthened cage) or eNrdJ-1cage_1–27 (weakened cage) (f). g, OE19 cells were co-cultured with HeLaeGFP+ cells, stably expressing eGFP, and treated with SMART-IL-1β (eNrdJ-1cage, 20 nM). IL-1R1 pathway signalling was determined as in b. h, HEK-Blue IL-1β cells co-cultured with K562HER2+/EGFR+ cells were treated with SMART-IL-1β (eNrdJ-1cage, 20 nM) and SEAP activity determined after 24 h (***P = 0.0003). Controls are as in b. Data are mean ± s.e.m. (n = 3 independent biological replicates). Statistical analysis: unpaired two-sided t-test. Data in b and g are representative of two independent experiments. Scale bars: b, 40 μm; g, 20 μm.
SMART-IL-1β actuation on cells
Our original SMART mechanism retains the spliced product on the cell surface by having targeting vectors fused onto the N and C termini of the ExtN and ExtC fragments, respectively (anti-Ag1–ExtN-IntNcage and IntCcage-ExtN–anti-Ag2). For SMART-IL-1β, however, we wanted to release IL-1β after its antigen-templated ligation to mimic the natural release of a cytokine in a real biological context (Fig. 4a). We therefore rearranged the domains of the protein chimeras by fusing the targeting vectors onto the caged split intein fragments, replacing FKBP and FRB, thus generating the general designs ExtN-IntNcage–anti-Ag1 and anti-Ag2–IntCcage-ExtN. Based on this, we proceeded by making IL-1βN–anti-HER2 and anti-EpCAM–IL-1βC. We then incubated these with an OE19 (HER2hi, EGFRlow or EpCAMhi) culture and tested the produced medium for IL-1β activity. When HeLa cells were supplemented with this medium, we found that IL-1R signalling was activated, indicated by the nuclear translocation of NF-κB (Fig. 4b and Extended Data Fig. 10i); by contrast, no activation was seen when SMART-IL-1β was blocked from binding to the OE19 cells, when the splicing-deficient mutant eNrdJ-1N(C1A)cage was used, or when the HeLa cells were pretreated with IL-1 receptor 1 antagonist (IL-1RA), which blocks IL-1β binding. SMART-IL-1β therefore actuates the ligation and release of functional IL-1β in response to defined antigen inputs. We quantified this further using a cell reporter line (HEK-Blue IL-1β), which produces secreted embryonic alkaline phosphatase (SEAP) when stimulated with IL-1β and IL-1R1/IL-1RAcP activation. Robust SEAP activity was detected when the reporter cell line was supplemented with medium produced from a combination of OE19/IL-1βN–anti-HER2/anti-EpCAM–IL-1βC (Fig. 4c) or K562HER2+/EGFR+/IL-1βN–anti-HER2/anti-EGFR–IL-1βC (Fig. 4d). Importantly, no-to-low signal was measured in control experiments. SEAP activity was furthermore detectable over a broad concentration range of SMART-IL-1β (Fig. 4e), with little activity related to medium from control K562 cell lines. By tuning the cage of eNrdJ-1cage, we could adjust the effective concentration of SMART-IL-1β needed on K562HER2+/EGFR+ cells to yield a 50% response on HEK-Blue IL-1β cells (Fig. 4f).
Finally, we also tested SMART-IL–1β in co-culture experiments. First, we co-cultured HeLaeGFP+ with OE19 cells (which importantly do not respond to IL-1β; Extended Data Fig. 10j) and then treated these with IL-1βN–anti-HER2/anti-EpCAM–IL-1βC. This led to the nuclear localization of NF-κB in the HeLaeGFP+ cells (Fig. 4g). When we co-cultured HEK-Blue IL-1β with K562HER2+/EGFR+ and treated these with IL-1βN–anti-HER2/anti-EGFR–IL-1βC, we measured robust SEAP activity, which was absent in control experiments (Fig. 4h). We believe that SMART-IL-1β constitutes the first example of an on-demand system capable of post-translationally generating an active cytokine (IL-1β) in response to local stimuli and mediating its release into the cell microenvironment.
Discussion
In this study, we present a synthetic biology platform we call SMART that, at its core, functions as a protein actuator based on conditional protein trans-splicing with tunable responsiveness to cellular inputs. SMART can sense multiple cell surface features and converts these to a user-defined functional output based on Boolean logic operations. Specifically, we show that programmable protein ligation is achievable on cell surfaces in response to the presence of two receptors. A key attribute of our SMART system is its modularity in terms of possible inputs and outputs. We have demonstrated that the platform is compatible with a variety of protein, peptide and small-molecule targeting agents, enabling SMART activation following the co-engagement of a range of surface proteins, such as growth-factor receptors, cell-adhesion molecules and G-protein-coupled receptors.
In principle, SMART can generate many types of output, leveraging the broad sequence tolerance of the NrdJ-1 split intein that functions as the protein ligase in the system. In the present work, we have illustrated this through the generation of a functional SpyCatcher003 protein and the cytokine IL-1β. SpyCatcher003 serves as a protein dock that allowed us to recruit a variety of activities to designated cells. These two examples also illustrate another feature of the SMART system, namely the ability to control whether the reaction product remains associated with the manufacturing cells (as in the SpyCatcher example) or is released into the extracellular environment (as shown for IL-1 β) through a ligation-and-retain and ligation-and-release mechanism. The latter represents a unique feature of SMART that distinguishes the approach from other protein logic devices that rely on the recruitment of pre-existing factors to a location of interest. By coupling protein trans-splicing to the localized release of an active protein, we can achieve exquisite control over protein activation. We imagine that the scope of SMART will be expanded by implementing efficient screening approaches to identify split sites in target proteins36, in combination with the use of orthogonal inteins from expanded libraries37,38. Finally, although we so far focused on cell-surface applications of SMART, we expect that the platform will find applications in other contexts, including logic-gated imaging, proximity proteomics and possibly even genomics in permeabilized cells.
Methods
General materials
Common reagents and chemicals were purchased from MilliporeSigma unless stated otherwise. The 2,4-dinitrochlorobenzene (CAS number 97-00-7), tert-butyl bromoacetate (CAS 5292-43-3), N-hydroxysuccinimide (CAS 6066-82-6), N,N′-dicyclohexylcarbodiimide (CAS 538-75-0), Celite 545 (CAS 68855-54-9), ethyl 2-bromoacetate (CAS 105-36-2) and 1-(2-aminoethyl)maleimide hydrochloride (CAS 134272-64-3) were purchased from MilliporeSigma. The 2-[2-(2-aminoethoxy)ethoxy]ethanol (CAS 86770-74-3), biotin-PEG3-amine (CAS 359860-27-8), 2-(furan-2-yl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine (CAS. 288-13-1) and 4-[3-(trifluoromethyl)-3H-diazirine-3-yl]benzylamine hydrochloride (CAS 1258874-29-1) were purchased from Ambeed. Triethylamine (CAS 121-44-8) was purchased from Thermo Fischer Scientific. Dithiothreitol (DTT, CAS 3483-12-3), isopropyl-β-thiogalactopyranoside (IPTG, CAS 367-93-1) and bovine serum albumin (BSA) were obtained from Gold Biotechnology. Alexa Fluor 594 C5 maleimide, biotin-PEG3-NHS ester (CAS 1253286-56-4), hydroxy-PEG10-tert-butyl ester (CAS 778596-26-2) and chloroacetamido-PEG4-NHS ester (CAS 1353011-95-6) were purchased from BroadPharm. Biotin maleimide (CAS 116919-18-7) was purchased from MilliporeSigma. Alexa Fluor 568 DBCO was purchased from Lumiprobe.
Oligonucleotide primers were purchased from MilliporeSigma. The gBlock gene fragments were purchased from Integrated DNA Technologies. PrimeSTAR HS DNA polymerase was purchased from Takara Bio. Gibson Assembly Master Mix and the restriction enzymes NheI, NotI and T7 DNA Ligase were purchased from New England Biolabs. DNA purification kits were purchased from Qiagen. PCR purification and gel extract columns were purchased from Thomas Scientific. All the plasmid sequencing was done by GENEWIZ or Plasmidsaurus. DH5α competent cells and One Shot Stbl3 chemically competent Escherichia coli cells were purchased from Thermo Fisher Scientific. SHuffle T7 competent E. coli cells were purchased from New England Biolabs. Plasmids for lentiviral preparations were obtained from Addgene.
Nickel nitrilotriacetic acid (Ni-NTA) resin was obtained from Thermo Fisher Scientific. MOPS-SDS running buffer was obtained from Boston Bioproducts. Criterion cassettes, acrylamide, ammonium persulfate, tetramethylethylenediamine and Econo-Pac 10DG columns were obtained from Bio-Rad. Nitrocellulose membrane (0.45 μm) for western blotting was purchased from Thermo Fisher Scientific. Empore solid-phase extraction stage tips were purchased from Thermo Fisher Scientific.
Primary antibodies were purchased from Santa Cruz Biotechnology, Abcam, Cell Signaling Technology, Thermo Fisher Scientific and MilliporeSigma. Secondary antibodies were purchased from LI-COR Biotechnology.
NeutrAvidin Rhodamine Red-X and Streptavidin-Alexa Fluor 546 conjugates were purchased from Thermo Fischer Scientific. Streptavidin–Saporin (Streptavidin-ZAP) was purchased from Advanced Targeting Systems.
Dulbecco’s Modified Eagle medium (Gibco), RPMI-1640 medium (Gibco), McCoy’s 5a medium modified (Gibco), F-12K medium (Gibco), Dulbecco’s phosphate-buffered saline (DPBS, Gibco), Penicillin-Streptomycin (5,000 U ml−1), trypsin-EDTA (0.25%), trypsin-EDTA (0.05%), Lipofectamine 3000 transfection reagent, l-glutamine (200 mM), puromycin dichloride (10 mg ml−1) and Falcon standard tissue culture dishes were purchased from Thermo Fisher Scientific. A mammary epithelial cell growth medium kit was purchased from MilliporeSigma. Fetal bovine serum (heat inactivated) was purchased from Bio-Techne. Doxycycline (hyclate) was purchased from StemCell Technologies. An XTT cell viability kit was purchased from Cell Signal Technologies. Hoechst 33342 solution was obtained from Invitrogen. Glass-bottom plates were purchased from Cellvis. QUANTI-Blue solution was purchased from InvivoGen.
Human cell lines K562EpCAMlow (wild type, CCL-243), K562HER2+, K562EGFR+, K562HER2+/EGFR+, K562HER2+/EpCAMhi and K562HER2+/EGFR+/EpCAMhi were gifts from D. Baker. Human cell lines MCF-10a (CRL-10317), MCF-7 (HTB-22), LoVo (CCL-229), A594 (CCL-185) were gifts from Y. Kang. Human cell line Sk-br-3 (HTB-30) was a gift from S. Lipkowitz. Human cell line OE19 (JROECL19) was purchased from MilliporeSigma. Human cell lines HCT-116 (CCL-247) and A431 (CRL-1555) were purchased from ATCC. Human cell line HEK-Blue IL-1β was purchased from Invivogen. The PiggyBac transposase plasmid was a gift from C. Kadoch.
Coomassie-stained SDS–PAGE gels and western blots were imaged on an Odyssey system (LI-COR). See Supplementary Fig. 1 for uncropped source data. Densitometry measurements were performed using FIJI (National Institutes of Health)39. Molecular graphics and analyses were performed with PyMOL v.2.5, developed by Schrödinger. Graph plots and statistical analysis were made in GraphPad Prism v.9.2.0 (121).
High-performance liquid chromatography
Analytical-scale reverse-phase high-performance liquid chromatography (RP-HPLC) was done on an Agilent 1100 series or an Agilent 1260 Infinity system equipped with a C18 Vydac column (5 mM, 4.6 × 150 mm) at a flow rate of 1 ml min−1. Semi-preparative RP-HPLC was done on an Agilent 1260 Infinity system equipped with a Waters XBridge BEH C18 column (5 mM, 10 × 250 mm) at a flow rate of 4 ml min−1. Preparative-scale RP-HPLC was done on a Waters prep LC system consisting of a Waters 2545 binary gradient module and a Waters 2489 ultraviolet (UV)-visible detector equipped with a C18 Vydac column (10 mM, 22 × 250 mm). The HPLC solvents were H2O with 0.1% TFA (solvent A) and 90% acetonitrile in water with 0.1% trifluoroacetic acid (TFA) (solvent B). Applied solvent gradients are specified in detail in the relevant sections.
Mass spectrometry
Proteins and peptides were characterized by electrospray ionization mass spectrometry (ESI-MS) on a Bruker Daltonics MicroTOF-Q II mass spectrometer by direct injection after isolation by RP-HPLC.
Peptide synthesis
The CXCR4 antagonist peptide ligand BTK140 was synthesized manually using a standard Fmoc solid-phase peptide synthesis method on a 0.2 mmol scale using H-Rink Amide resin (ChemMatrix). The solid-phase synthesis cycle included Fmoc deprotection at room temperature for 20 min with 20% v/v piperidine in dimethylformamide (DMF) containing 0.1 M 1-hydroxybenzotriazole hydrate and coupling at room temperature for 30 min using 5 equivalents of Fmoc protected amino acid in DMF with 5 equivalents of hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU) and 10 equivalents of N,N-diisopropylethylamine (DIPEA). All couplings were performed twice. For non-proteogenic amino acids and for coupling of the Arg–Arg junction, the coupling step was performed with 3 equivalents of the specific Fmoc protected amino acid in DMF with 3 equivalents of (7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyAOP) and 10 equivalents of DIPEA for 3 h (if the coupling step was done once) or 2 × 2 h (if double couplings were done). D-Lys(Dde) deprotection was done on resin at room temperature for 30 min by adding 2% v/v hydrazine in DMF under N2 agitation. Two successive coupling steps were done at room temperature for 1 h with 5 equivalents of azido-PEG3-acid, or alternatively azido-PEG10-acid, in DMF with 5 equivalents of HATU and 10 equivalents of DIPEA. Deprotection of Cys(Acm) was done on resin using either thallium triflate or in solution phase with I2. Final peptide cleavage and side-chain deprotection was done at room temperature for 3 h using a cleavage cocktail of 95% v/v TFA, 2.5% v/v triisopropyl silane, 2.5% v/v H2O. The peptide was pelleted by adding ice-cold diethyl ether and subsequent centrifugation at 4,000g for 10 min at 4 °C. The crude peptide was then dissolved in aqueous acetonitrile 0.1% v/v TFA and purified by preparative RP-HPLC using aqueous 0.1% v/v TFA as solvent A and 90% v/v acetonitrile, 0.1% v/v TFA as solvent B, with a gradient of 0–73% over 40 min. Pure fractions were identified by analytical RP-HPLC and ESI-TOF MS, and pooled and lyophilized.
Peptide conjugation
To BTK140-PEG3-azide (370 µg, 123 nmol) dissolved in 1 ml MeCN we added a slight excess of AF568-DBCO (124 nmol) dissolved in DMSO to 1 mM, and the reaction was left overnight at room temperature away from light. The crude mixture was analysed by RP-HPLC and diluted with 9 ml of solvent A (aqueous 0.1% v/v TFA) on full conversion to BTK-AF568, which was further purified by preparatory-scale RP-HPLC (0–73% solvent B for 40 min). The collected product fractions were combined and lyophilized away from light. The peptide was redissolved in 100 mM NaH2PO4 (pH 7.2), 150 mM NaCl, 1 mM EDTA and 10% v/v glycerol to a final concentration of 100 µM when used for cell-phenotyping experiments.
Chemical synthesis
Synthetic protocols of SCH58261 and its maleimide functionalization, the azido-PEG10-acid linker, the iridium photocatalyst and its chloroalkane functionalization, and the biotin-phenol and biotin-diazirine probes are provided in the Supplementary Methods.
DNA cloning
All bacterial expression vectors were based on a pET backbone vector with a gene for kanamycin resistance. DNA fragments amplified by PCR were inserted into a gene cassette using Gibson Assembly. The gene cassette included an upstream T7 promoter under regulation of a lac operator, an ATG start codon in frame with an N-terminal His6-SUMO fusion tag when specified and ended with a TAA stop codon and a T7-terminator sequence.
Site-directed mutagenesis (including missense substitutions, insertions and deletions), as well as amplifications of gene inserts and plasmid backbones, was done using PCR. To a solution of 10 µl of 1 ng µl−1 plasmid, 1 µM forward primer and 1 µM reverse primer we added 10 µl 2× PrimeSTAR DNA polymerase master mix. The mixture was used in a PCR reaction. The completed PCR reaction was restriction digested with 20 units of DpnI for 1 h at 37 °C before the PCR amplicon was isolated by a general PCR clean-up protocol. The purified PCR amplicon was then used in a Gibson assembly reaction (see below). Alternatively, full plasmid amplicons from site-directed mutagenesis were used directly in heat-shock transformations of chemically competent E. coli DH5α cells, which were plated on LB-agar plates supplemented with 50 µg ml−1 kanamycin. Single colonies were picked, grown and the plasmid isolated before being verified by Sanger sequencing.
DNA components prepared by PCR using primers designed to have 15–20 base pair overlaps were used in Gibson Assembly reactions. Gibson Assembly reactions were set up by mixing 2 µl of DNA fragments (100 ng plasmid backbone amplicon, 3–5 molar excess gene insert amplicon) with 2 µl 2× NEBuilder HiFi DNA Assembly master mix. The reaction was incubated for 15 min at 50 °C, cooled and thereafter used in a heat-shock transformation of chemically competent E. coli DH5α, which was plated on LB-agar plates supplemented with 50 µg ml−1 kanamycin. Single colonies were picked, grown and the plasmid isolated before being verified by Sanger sequencing.
The PiggyBac transposon plasmids used in the generation of stable cell lines were generated by standard restriction cloning. Gene fragments containing the desired transgene sequence were synthesized to contain NheI and NotI restriction sites. Separate restriction digest reactions with NheI and NotI were prepared for the plasmid backbone and the two gene inserts encoding SPHA-Flag-CXCR4-GFP and SPHA-Flag-ADORA2A-mCherry. Reactions were done for 1 h at 37 °C and the digest products isolated. The linearized plasmid backbone was mixed with either of the digested gene inserts and T7 DNA ligase and the reactions incubated for 1 h at 37 °C. The ligation reactions where then used to transform One Shot Stbl3 chemically competent E. coli cells, which were plated on LB-agar plates supplemented with ampicillin (100 µg ml−1). Single colonies were picked, grown and the plasmid isolated before being verified by either Sanger sequencing or full plasmid sequencing.
Recombinant protein expression and purification
Chemically competent E. coli BL21(DE3) cells were heat-shock transformed with a pET vector carrying the gene cassette for the protein of interest. Cells were grown overnight at 37 °C in 8 ml LB medium supplemented with 50 µg ml−1 kanamycin. This overnight culture was used to inoculate an expression culture of 1 l LB medium supplemented with 50 µg ml−1 kanamycin. In general, the expression culture was incubated at 37 °C until it reached an optical density at 600 nm (OD600) of 0.4, after which it was cooled for 20 min at 18 °C. Protein expression was then induced by the addition of 0.1 ml 1 M IPTG and the culture was left overnight at 18 °C. For expression of full-length NrdJ-1, used in the crystallography study and for any SpyTag003 and stand-alone DARPin constructs, the expression culture was incubated at 37 °C until it reached an OD600 of 0.6. The expression was then induced by the addition of 0.1 ml 1 M IPTG and the culture was left for 4 h at 37 °C. In all cases, cells were collected by centrifugation at 3,500g at 18 °C for 20 min and suspended in lysis buffer containing 20 ml 50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, 20 mM imidazole, supplemented with 1 mM DTT and 1 mM phenylmethylsulfonyl fluoride (PMSF). Cell suspensions were then either stored at −20 °C until further use or used directly. Soluble protein was extracted by subjecting the cell suspension to sonication using a duty cycle of 20 s on, 30 s off at 30% amplitude while cooled on an ice bath, after which a cleared lysate was produced by centrifugation at 35,000g at 4 °C for 20 min. The cleared lysate was passed through a pre-equilibrated Ni2+-nitrilotriacetic acid (NTA) column (2 ml resin slurry per litre of culture) and the flow-through discarded. The column was then washed with 50 ml lysis buffer, before the protein was eluted using 6 ml lysis buffer supplemented with 250 mM imidazole. His6–SUMO tagged proteins were treated overnight with His6–Ulp1 protease while being dialysed against lysis buffer supplemented with 1 mM DTT. The dialysed sample was then passed through a pre-equilibrated Ni2+-NTA column to remove any cleaved His6–SUMO tag and His6–Ulp1 protease. The flow-through and an additional 6 ml lysis buffer passed through the column was collected, combined and concentrated to 0.5 ml. The concentrated sample was filtered through a 0.22 μm spin filter. Size-exclusion chromatography was done at 4 °C with a flow rate of 0.5 ml min−1 on an ÄKTA Fast Performance liquid chromatography (GE Healthcare) system using a Superdex 200 10/300 GL (Cytiva Life Sciences) column with 100 mM NaH2PO4 (pH 7.2), 150 mM NaCl, 1 mM EDTA and 1 mM DTT as the eluent. Pure fractions were identified by SDS–PAGE, validated by analytical RP-HPLC and the protein mass was confirmed by ESI-TOF MS. Pure fractions were supplemented with 10% v/v glycerol, aliquoted and flash-frozen in liquid nitrogen before being stored at −80 °C until further use. Analytical data for all proteins is shown in Supplementary Fig. 3, Extended Data Fig. 9 and Supplementary Table 1. The fully annotated amino acid sequences of overexpressed constructs used in this study are given in Supplementary Table 9.
SHuffle T7 competent E. coli cells were used for the cytosolic expression of His6–SUMO–anti-HER2scFv–SpyN. Chemically competent cells were heat-shock transformed with a pET vector carrying the gene cassette for the protein of interest. Cells were grown overnight at 37 °C in 8 ml LB medium supplemented with 50 µg ml−1 kanamycin. This overnight culture was used to inoculate an expression culture of 1 l LB medium supplemented with 50 µg ml−1 kanamycin. The culture was incubated at 37 °C until it reached an OD600 of 0.4, after which it was cooled for 30 min at 16 °C. Protein expression was then induced by the addition of 0.3 ml 1 M IPTG and the culture was expressed overnight at 16 °C. Cells were collected and the protein purified from the soluble fraction as mentioned above.
Western blotting
Samples were run on an SDS–PAGE gel. For western blotting, the gel was used for a transfer reaction onto a nitrocellulose membrane, which was subsequently blocked with TBS-T (25 mM Tris, 150 mM NaCl, 0.1% v/v Tween-20, pH 7.7) supplemented with 4% w/v skimmed-milk powder for 1 h at room temperature. The membrane was washed 3 times for 5 min with TBS-T and incubated with primary antibodies at specified dilution (Supplementary Table 10) on an orbital shaker for 1 h at room temperature or alternatively overnight at 4 °C. After washing 3 times for 5 min with TBS-T, the IRDye secondary antibody or alternatively IRDye Streptavidin was applied for 30 min to 1 h at room temperature, before imaging on a Li-Cor Odyssey imager (Li-Cor).
X-ray crystallography
Crystallography studies used a fused version of NrdJ-1 containing C1A and N145A inactivating mutations and SGG and SEI as N- and C-terminal extein sequences, respectively. The protein was dialysed against a buffer containing 25 mM HEPES (pH 7.5), 150 mM NaCl, then concentrated to 40 mg ml−1, and finally flash-frozen as aliquots in liquid nitrogen before being stored at −80 °C for further use. Initial crystallization conditions were established using a SaltRx HT screen (Hampton Research) with a Phoenix crystallization robot (Art Robbins). Crystals were grown at 4 °C by the sitting-drop vapour-diffusion method. A focused screen was centred on conditions with increasing concentrations of sodium formate at various pH values. Optimal crystals were obtained after one week using a solution of 4.5 M sodium formate (pH 7.0). Diffraction data were obtained at the National Synchrotron Light Source II (Brookhaven National Laboratory), beamline 17-ID-1. The data were processed using the XDS package40. The phase information was determined by molecular replacement using PHASER in the CCP4 suite41 and using an in silico AlphaFold2 (refs. 42,43) model of full-length NrdJ-1 as input. Iterative rounds of model building in Coot44 and refinements in PHENIX Refine (v.1.17_3644)45 were performed to obtain the final structure. Data collection and refinement statistics are displayed in Supplementary Table 2.
Protein conjugation reactions
Purified protein containing a C-terminal cysteine (SpyTag003-Cys, SpyTag003D117A-Cys, anti-HER2-Cys DARPin, anti-EGFR-Cys DARPin, anti-EpCAM-Cys DARPin, anti-CEACAM6-Cys single-domain antibody and SpyC-Cys) or catalytic cysteine (anti-HER2-SpyN-eNrdJ-1Ncage) was reduced with 1 mM DTT for 20 min on ice to prepare it for conjugation chemistry. The sample was washed with buffer (100 mM NaH2PO4 (pH 7.2), 150 mM NaCl, 1 mM EDTA) using spin-filtration to remove small-molecule thiols. The reduced protein was either flash-frozen in liquid nitrogen and stored at −80 °C for later use or used directly in conjugation reactions as described in the following sections.
The reduced protein was mixed with a molar excess of the required alkylating agent (iodoacetamide, Alexa Fluor 594 maleimide, Alexa Fluor 568 maleimide, Alexa Fluor 488 maleimide, biotin-maleimide, DBCO-maleimide or SCH58261-maleimide) and incubated for 20 min at room temperature in the dark. Reactions were monitored by RP-HPLC and ESI-TOF MS and quenched with 1 mM DTT when completed. The reaction mixture was then passed over an Econo-Pac 10DG column using 100 mM NaH2PO4 (pH 7.2), 150 mM NaCl, 1 mM EDTA, 10% v/v glycerol as the eluent. The product was characterized by RP-HPLC and ESI-TOF MS, flash-frozen and stored at −80 °C. Analytical data for protein conjugations are provided in Supplementary Fig. 3 and Supplementary Table 1, and for the SpyC-anti-A2A conjugate in Supplementary Fig. 21 and Supplementary Table 1.
SpyC-DBCO was generated by reacting SpyC-Cys with DBCO-maleimide according to the protocol described above and used subsequently in a strain-promoted alkyne-azide cyclo-addition reaction with the synthetically prepared CXCR4 antagonist peptide BTK140 functionalized with an azide. SpyC-DBCO (1.7 mg, 94 nmol) was reacted with BTK-PEG10-azide (4.4 mg, 133 nmol) in 100 µl of 100 mM NaH2PO4 (pH 7.2), 150 mM NaCl, 1 mM EDTA, 10% v/v glycerol. The reaction was incubated at 4 °C and monitored by RP-HPLC and ESI-TOF MS. When full conversion was observed, the reaction was washed with buffer (100 mM NaH2PO4 (pH 7.2), 150 mM NaCl, 1 mM EDTA, 10% v/v glycerol) by spin filtration to remove excess peptide and then finally concentrated to 1 ml. Size-exclusion chromatography was done at 4 °C at a flow rate of 0.5 ml min−1 on an ÄKTA Fast Performance liquid chromatography (GE Healthcare) system using a Superdex 200 10/300 GL (Cytiva Life Sciences) column with 100 mM NaH2PO4 (pH 7.2), 150 mM NaCl, 1 mM EDTA, 10% v/v glycerol as the eluent. Pure fractions were identified by SDS–PAGE, validated by analytical RP-HPLC and the protein mass was confirmed by ESI-TOF MS. Pure fractions were flash-frozen in liquid nitrogen before being stored at −80 °C until further use. Analytical data for the final SpyC–anti-CXCR4 conjugate are provided in Supplementary Fig. 21 and Supplementary Table 1.
Anti-CEACAM6–AF594 was prepared by reacting anti-CEACAM6–SpyCatcher003 (0.33 mg, 12.3 nmol) with SpyTag003–AF594 (0.2 mg, 12.5 nmol) in 100 µl of 100 mM NaH2PO4 (pH 7.2), 150 mM NaCl, 1 mM EDTA, 10% v/v glycerol. The reaction was incubated away from light at room temperature and monitored by RP-HPLC and ESI-TOF MS. When full conversion was observed, the reaction was washed with buffer (100 mM NaH2PO4 (pH 7.2), 150 mM NaCl, 1 mM EDTA, 10% v/v glycerol) by spin filtration to remove excess peptide and then finally concentrated to reach 100 µM of the conjugate. The product was validated by RP-HPLC and ESI-TOF MS, flash-frozen and stored at −80 °C.
SpyTag003-HaloTag13 was used in reactions with the iridium catalyst functionalized with a chloroalkane linker (Ir-PEG4-C6H12Cl). Specifically, one equivalents of SpyTag003-HaloTag13 (585 µg, 15 nmol) was reacted with three equivalents of Ir-PEG4-C6H12Cl (62 µg, 50 nmol) in 100 mM NaH2PO4 (pH 7.2), 150 mM NaCl, 1 mM EDTA, 1 mM DTT for 1 h at room temperature. The reaction was monitored by RP-HPLC. When all the protein had been consumed to generate the SpyTag003–HaloTag13-Ir conjugate (referred to as SpyTag003-Ir), the reaction was washed in 100 mM NaH2PO4 (pH 7.2), 150 mM NaCl, 1 mM EDTA, 1 mM DTT by spin filtration to remove excess Ir-PEG4-C6H12Cl. The conjugate was then aliquoted, flash-frozen and stored at −80 °C.
Rapamycin-induced protein trans-splicing reactions
Screening for the optimal split site within SpyCatcher003 was done in splicing buffer (100 mM NaH2PO4 (pH 7.2), 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 10% v/v glycerol) by combining complementary protein chimeras Flag-SpyN1−x-NpuNcage-FKBP and FRB-NpuCcage-SpyCy-113–Myc (1 µM of each), with x and y denoting the last and first residue of the two fragments (the split site). When indicated, samples were supplemented with SpyTag003 or conjugates thereof (2 µM) and either rapamycin (a final concentration of 10 µM from a 50 µM DMSO stock) or TEV protease (10 units). Reaction mixtures were incubated for 24 h at 37 °C before being analysed by SDS–PAGE or western blotting.
Screening for the optimal split site in IL-1β was done in splicing buffer (100 mM NaH2PO4 (pH 7.2), 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 10% v/v glycerol) by combining complementary protein chimeras Flag-IL-1βN1−x-eNrdJ-1Ncage-FKBP and FRB-eNrdJ-1Ccage-IL-1βCy−153–Myc (1 µM of each), with x and y denoting the last and first residue of the two fragments (the split site). When indicated, samples were supplemented with rapamycin (final concentration of 10 µM from a 50 µM DMSO stock). Reaction mixtures were incubated at 37 °C before being analysed by SDS–PAGE or western blotting.
Generation of stable cell lines
To generate stable cell lines expressing CXCR4- and ADORA2A-related constructs, low-passage OE19 cells were transfected with a PiggyBac transposon plasmid encoding for the desired transgene under the control of a doxycycline-inducible promoter and a PiggyBac transposase plasmid (2:1 ratio). Lipofectamine 3000 transfection reagent was used for the transfection according to the manufacturer’s instructions. The transfected cells were allowed to recover in RPMI-1640 supplemented with 2 mM l-glutamine, 10% v/v FBS, 100 U ml−1 penicillin/streptomycin for 30 h after transfection. The cells were then cultured for 5 days in RPMI-1640 supplemented with 2 mM l-glutamine, 10% v/v FBS, 100 U ml−1 penicillin/streptomycin and 1 µg ml−1 puromycin. Transfected OE19 cells that survived puromycin selection were further expanded and maintained in RPMI-1640 supplemented with 2 mM l-glutamine, 10% v/v FBS, 100 U ml−1 penicillin/streptomycin and 1 µg ml−1 puromycin, with cultures being discarded after 30 passage cycles. Transgene expression was achieved by supplementing the culture with 400 ng ml−1 doxycycline for 24 h. Protein expression was confirmed by western blot, immunofluorescence microscopy and flow cytometry. The primary structures of the CXCR4- and ADORA2A-related constructs are given in Supplementary Table 11.
Lenti-X 293 T cells (Takara Bio, 632180) were cultured in DMEM supplemented with 10% (v/v) FBS and 100 U ml−1 penicillin/streptomycin in 6-well plates to approximately 70% confluency. The cells were then transfected in culture medium without penicillin/streptomycin using Lipofectamine 2000 with: a transfer plasmid encoding the genes for HER2Δ-eGFP and blasticidin resistance (4 µg); the envelope plasmid pMD2.G (1.2 µg); and the packaging plasmid psPAX2 (3.6 µg). After 24 h, the culture was aspirated and DMEM supplemented with 10% (v/v) heat FBS and 100 U ml−1 penicillin/streptomycin added. Lentivirus was collected the following day by isolating and filtering (0.45 µm) the culture supernatant. The lentivirus preparation was stored at −80 °C until further use. Wild-type HeLa cells were cultured in DMEM supplemented with 10% (v/v) FBS and 100 U ml−1 penicillin/streptomycin to a confluency of 50% in a 6-well plate. Then, 1 ml of the produced lentivirus preparation was added along with 1 ml of fresh medium and further supplemented with 4 µg ml−1 polybrene. After 24 h, an extra 1 ml of virus was added to the HeLa cells. The following day, the medium was changed and the cells were allowed to recover for another 24 h. The day after that, selection was initiated by adding 1 µg ml−1 blasticidin. After several passages, the cells were sorted by fluorescence-activated cell sorting on a BD FACSymphony A3 Cell Analyzer using the eGFP signal for gating to isolate the HeLaeGFP+ population. The primary structure of HER2Δ-eGFP is given in Supplementary Table 11.
Mammalian cell culture
Mammalian cell lines were cultured in media as detailed in the Supplementary Table 12 in an incubator at 37 °C and 5% CO2. All cell lines were regularly tested free of mycoplasma.
Confocal microscopy
Suspension cell lines
The specific cell line or mixture was treated as specified in the relevant section. After treatment, cells were suspended in DBPS, 1% w/v BSA, 2 mM CaCl2 supplemented with Hoechst 33342 (10 µg ml−1) and incubated for 30 min at room temperature before being washed twice in buffer. The cells were then transferred to a glass-bottom plate and allowed to settle before being imaged using 40× magnification on a Nikon A1/HD25 microscope (Nikon Instruments). Processing of fluorescence microscopy images was done using FIJI (National Institutes of Health)39.
Adherent cell lines
The specific cell line was plated on a glass-bottom plate, cultured and then treated as specified in the relevant section. After treatment, DBPS, 1% w/v BSA, 2 mM CaCl2 supplemented with Hoechst 33342 (10 µg ml−1) was added to the plate and the adherent cells incubated for 30 min at room temperature before being washed twice in buffer. The cells were then imaged as described above.
Flow cytometry
Cell lines were suspended as 1,000,000 cells per ml in cold DBPS, 1% w/v BSA, 2 mM CaCl2 (supplemented with 50 mM EDTA for adherent cell cultures), filtered through a cell strainer and kept on ice. Flow cytometry data acquisition was obtained on a BD LSR II flow cytometer and the results were analysed using FlowJo 10.8.1. Single-colour controls were included for compensation adjustments. Examples for the gating strategy used for flow cytometry analysis of mixed K562 populations are given in Supplementary Figs. 8–11. Examples for the gating strategy used for flow cytometry analysis of mixed mammary populations are given in Supplementary Figs. 14–16.
Cell surface labelling using SMART-SpyCatcher
Suspension cell lines
Individually cultured K562 cell lines were spun down and counted to estimate the cell concentration. Then approximately 200,000 cells of an individual K562 cell culture were isolated, washed twice with a buffer containing DPBS, 1% w/v BSA and 2 mM CaCl2 and then suspended in 0.4 ml of the same buffer or in an appropriate medium. To this sample was added the specified SMART-SpyCatcher system at the stated concentration by adding each individual component (SpyN and SpyC) from stock solutions dissolved in 100 mM NaH2PO4 (pH 7.2), 150 mM NaCl, 1 mM EDTA and 1 mM DTT. The sample was then incubated for 2 h at 37 °C in 5% CO2 to allow for cell-surface receptor binding and protein trans-splicing. The specified SpyTag003 conjugate was then added (100 nM final concentration) and the SpyTag003–SpyCatcher003 reaction was allowed to proceed in the dark at room temperature for 20 min. The cells were then washed twice with cold DPBS, 1% w/v BSA, 2 mM CaCl2 and incubated on ice until further analysis by confocal microscopy or flow cytometry. Samples subjected to SDS–PAGE or western blotting were washed twice with DPBS alone to remove excess BSA. Control experiments examining the mechanism of action in Extended Data Fig. 3a–c included the following steps before the addition of SpyTag003–AF594: first, addition of competing DARPins (500 nM) blocking SMART-SpyCatcher binding; second, addition of the Cys1-alkylated eNrdJ-1Ncage component, unable to perform protein trans-splicing; or third, pre-addition of unlabelled SpyTag003 (500 nM) to block the reaction with SpyTag003–AF594. In reactions with SpyTag003D117A–AF594, the mutation D117A disables the formation of an isopeptide bond with SpyCatcher003.
Adherent cell lines
Individually cultured adherent cell lines were lifted using trypsin, washed with complete medium and counted to estimate the cell concentration. Then 200,000 cells were seeded in 24-well plates and allowed to attach and recover for 24 h in complete media. The assay protocol for SMART-SpyCatcher actuation and SpyTag003 recruitment followed that outlined above for the suspension cell lines. After treatment, the adherent cells were washed twice with DPBS, 1% w/v BSA, 2 mM CaCl2 and then lifted with a non-enzymatic solution of DPBS, 1% w/v BSA, 50 mM EDTA and incubated on ice before analysis by flow cytometry. For SDS–PAGE or western blotting, cells were lifted as described and then washed with DPBS alone to remove excess BSA.
Mixed-population experiments using SMART-SpyCatcher
Suspension cell lines
Two distinct mixed populations of K562 cell lines were generated (K562 expresses low endogenous levels of EpCAM):
-
Mixed K562 population 1
-
K562 (wild-type cell line)
-
K562EGFR+
-
K562HER2+
-
K562HER2+/EGFR+
-
-
Mixed K562 population 2
-
K562 (wild-type cell line)
-
K562EGFR+
-
K562HER2+/EpCAMhi
-
K562HER2+/EGFR+/EpCAMhi
-
The mixed populations were generated by combining the specified four cell lines (50,000 cells each) in a reaction tube. The SMART-SpyCatcher system (SpyN and SpyC) was then added at the specified concentration and the sample was incubated for 2 h at 37 °C and 5% CO2 to allow for cell-surface receptor binding and protein trans-splicing. In experiments involving NOT gating, the decoy was added at 100 nM just before the addition of SMART-SpyCatcher. The required SpyTag003 conjugate was then added (100 nM final concentration) and the SpyTag003–SpyCatcher003 reaction was allowed to proceed in the dark at room temperature for 20 min. The cells were then washed twice with cold DPBS, 1% w/v BSA, 2 mM CaCl2 and incubated on ice until further analysis by confocal microscopy or flow cytometry. When appropriate for data representation, the individual data sets from the two distinct mixed populations were combined into one common bar graph.
Adherent cell lines
The three adherent cell lines MCF-10a, MCF7 and Sk-br-3 were cultured individually. Each cell line was lifted with a non-enzymatic solution of DPBS, 1% w/v BSA, 50 mM EDTA and counted to estimate the cell concentrations. MCF-10a was then mixed in equal numbers with either MCF-7 or Sk-br-3, and the two mixed mammary populations washed with either complete DMEM medium (MCF-10a + MCF-7) or complete McCoy’s 5a Medium Modified medium (MCF-10a + Sk-br-3) before being aliquoted at 200,000 cells per well in a 24-well plate. The cells were incubated for 6 h at 37 °C and 5% CO2. The assay protocol for SMART-SpyCatcher actuation and SpyTag003 recruitment follows that outlined above for the suspension cell lines. After treatment, the adherent cells were washed twice with DPBS, 1% w/v BSA, 2 mM CaCl2 and then lifted with a non-enzymatic solution of DPBS, 1% w/v BSA, 50 mM EDTA and incubated on ice before analysis by flow cytometry.
Cell antigen phenotyping
Suspension cell lines
Individually cultured K562 cell lines were spun down and counted to estimate the cell concentration. Then approximately 200,000 cells of an individual K562 cell culture were isolated, washed with DBPS, 1% w/v BSA, 2 mM CaCl2 and incubated for 30 min at room temperature in the same buffer supplemented with 100 nM Alexa Fluor 594 DARPin conjugate targeting the antigen EpCAM. The cells were washed twice with DBPS, 1% w/v BSA, 2 mM CaCl2 and incubated on ice until further analysis by flow cytometry.
Adherent cell lines
Plated cells were lifted with trypsin, washed with complete medium and counted to estimate the cell concentration. Then 200,000 cells were seeded in a 24-well plate format and allowed to recover and attach for 24 h in complete media. The cells were then washed with DBPS, 1% w/v BSA, 2 mM CaCl2 and incubated for 30 min at room temperature in the same buffer supplemented with 100 nM Alexa Fluor 594 DARPin conjugate targeting one of the three antigens HER2, EGFR or EpCAM, or alternatively 100 nM Alexa Fluor 594 single-domain antibody conjugate targeting CEACAM6. The doxycycline-inducible cell lines OE19CXCR4DOX and OE19A2ADOX were incubated with 100 nM of the synthetic conjugates BTK140-PEG3-AF568 or SCH58261-AF488 respectively. The cells were washed twice with DBPS, 1% w/v BSA, 2 mM CaCl2 before being lifted with a non-enzymatic solution of DPBS, 1% w/v BSA, 50 mM EDTA and incubated on ice until further analysis by flow cytometry.
NeutrAvidin Rhodamine Red-X recruitment assay
The cell sample was prepared following the assay protocol detailed for single population or mixed populations of suspension cells described above. After incubation with SMART-SpyCatcher (SpyN and SpyC each at 100 nM final concentration) for 2 h, SpyTag003 labelled with biotin (SpyTag003-Biotin, 100 nM) was added to the cells and the SpyTag003–SpyCatcher003 reaction allowed to proceed for 20 min at room temperature in the dark. The cells were then washed twice with cold DPBS, 1% w/v BSA, 2 mM CaCl2 and then incubated with NeutrAvidin Rhodamine Red-X (1:500) for 30 min. The cells were then washed with DBPS, 1% w/v BSA, 2 mM CaCl2 before being imaged by confocal microscopy or analysed by flow cytometry. For the experiment studying the intake of NeutrAvidin Rhodamine Red-X, the sample was imaged immediately and after an extra incubation period of 4 h at the specified temperature.
Cell-depletion assay
Mixed K562 population depletion
The sample was prepared following the assay protocol detailed for mixed populations of suspension cells described above. After incubation with SMART-SpyCatcher (SpyN and SpyC each at 100 nM final concentration) for 2 h, SpyTag003 labelled with biotin (SpyTag003-biotin, 100 nM) was added to the cells and the SpyTag003–SpyCatcher003 reaction allowed to proceed for 30 min at room temperature. The cells were washed with complete RPMI-1640 medium. Cells were then aliquoted into a 96-well plate (12,500 cells per well) and an extra 100 μl of complete RPMI-1640 media containing Streptavidin–Saporin (20 nM final concentration) was added. The cells were then cultured for 72 h for the single-dose regimen. For the two-dose regimen, the cells were cultured for 24 h before being subjected to the treatment described above a second time and then cultured for an extra 72 h. Samples were then analysed by flow cytometry.
Subpopulation percentages obtained from the flow cytometry analysis were normalized using the following formula: percentage viability = X1/(X0 × (WT1/WT0)) × 100, where X0 is the percentage of the specific subpopulation in the negative untreated sample, X1 is the percentage of the specific subpopulation, the relevant sample, WT0 is the percentage of the wild-type subpopulation in the negative untreated sample, and WT1 is the percentage of the wild-type subpopulation in the relevant sample. The derived percentage is taken to be the viability of the specified subpopulation.
Single A431 population depletion
For the A431 cell-depletion assay, the cultured cells were lifted with trypsin, counted and then seeded at 5,000 cells per well in a 96-well plate before further culturing for 24 h at 37 °C and 5% CO2. The cells were then washed with DPBS, 1% w/v BSA, 2 mM CaCl2 and incubated with the indicated SMART-SpyCatcher (SpyN and SpyC, each at 100 nM final concentration) for 2 h at 37 °C and 5% CO2. SpyTag003-biotin (100 nM) was added and the sample was incubated for 30 min at room temperature. The cells were gently washed with complete DMEM medium before being suspended in 200 μl DMEM medium containing 20 nM Streptavidin–Saporin conjugate as indicated. The cells were cultured for 24 h at 37 °C and 5% CO2 before the complete treatment described above was repeated to give a two-dose regimen. Finally, the cells were cultured for an extra 72 h. Each well was then washed twice with complete DMEM medium and 200 μl of complete DMEM medium added. An XTT cell viability assay was used to quantify the metabolic activity of each sample according to the protocol prescribed by the manufacturer. The conversion of XTT to formazan was measured on a SpectraMax iD5 Multi-Mode Microplate Reader (Molecular Devices) using well-scan mode at 465 nm at room temperature. The obtained absorbance values were normalized to the negative untreated control.
Protein proximity labelling
APEX2 proximity labelling
The cell sample, prepared following the assay protocol detailed for single populations, was treated with the SMART-SpyCatcher reactants (SpyN and SpyC each at 100 nM final concentration) for 2 h. The cells were then incubated with SpyTag003-APEX2 (300 nM) for 30 min at room temperature before being washed twice with DPBS. To initiate APEX proximity labelling, the cells were treated with 1 ml DPBS containing biotin-phenol (250 µM final concentration) followed by the addition of 10 µl of freshly prepared DPBS containing 100 mM H2O2, and the reaction was allowed to proceed at room temperature under gentle swirling for the indicated time. The reaction was quenched by the addition of 200 µl DPBS, 10 mM sodium ascorbate and 5 mM Trolox, and the cells washed twice with 1 ml of the same buffer. The cells were lifted (when necessary) by the addition of 1 ml DPBS, 50 mM EDTA, pelleted and suspended in 200–500 µl 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% v/v NP-40, 0.5% w/v sodium deoxycholate, 0.1% w/v sodium dodecyl sulfate supplemented with 1× Halt Protease Inhibitor Cocktail and 1 mM PMSF, then sonicated briefly and centrifuged for 20 min at 15,000g at 4 °C. The supernatant was isolated and analysed by SDS–PAGE/western blotting.
AND-gated μMap photocatalytic proximity labelling
For the mixed-cell experiment, the mixed K562 population 1 was prepared as described above in DPBS, 1% w/v BSA, 2 mM CaCl2. The cells were incubated with the SMART-SpyCatcher (100 nM, eNrdJ-1cage variant) in 1 ml DPBS, 1% w/v BSA, 2 mM CaCl2 for 2 h at 37 °C, 5% CO2. SpyTag003-Ir was added to a final concentration of 200 nM in DPBS and the cells were incubated for 30 min at room temperature. The cells were washed twice with DPBS before incubation in DPBS supplemented with 250 µM biotin-diazirine for 5 min at room temperature. The cell sample was then irradiated using a Kessil PR160L lamp (LED 440 nm) for 5 or 10 min. The cells were washed again twice with DPBS and then treated with Streptavidin–Alexa Fluor 546 (1:2,000 v/v ratio) and incubated for 30 min. Finally, the cells were washed twice with DPBS and analysed by flow cytometry.
Cell experiments using SMART-IL-1β
On-cell splicing and release
OE19 cells were cultured to full confluency on a 12-well plate and washed twice with DBPS supplemented with 1% w/v BSA and 2 mM CaCl2. Alternatively, one million cultured K562 cells were collected and washed twice with DBPS, 1% w/v BSA, 2 mM CaCl2. In both cases, the cells were incubated with each SMART-IL-1β fragment (at the specified concentration) in 0.5 ml with DBPS, 1% w/v BSA and 2 mM CaCl2 for 2 h at 37 °C and 5% CO2. The supernatant was then withdrawn, cleared of any cell debris by centrifugation and used further.
IL-1β stimulation of HeLa cultures
HeLa cells were cultured to about 30–50% confluency in a 24-well glass-bottom plate. The supernatant from OE19 or K562 cultures, prepared as described above, was applied to the cell culture with an additional 500 µl fresh DMEM supplemented with 10% v/v FBS, 100 U ml−1 penicillin/streptomycin for 30 min at 37 °C at 5% CO2. The cells were then washed with DBPS before being fixed by incubation in 0.5 ml 5% v/v formaldehyde for 10 min at room temperature. Excess formaldehyde was quenched by the addition of 0.5 ml 150 mM glycine followed by washes with DPBS. The cells were subsequently permeabilized by incubation with 0.5 ml DPBS and 0.5% v/v Triton X-100. The fixed and permeabilized cell sample was then washed and blocked for 1 h at room temperature by the addition of DPBS, 4% w/v BSA and 0.1% v/v Tween-20, and then washed with DPBS, 4% w/v BSA and 0.1% v/v Tween-20. The cell sample was thereafter treated first with rabbit anti-NF-κB (p65) antibody in DPBS and 1% w/v BSA for 1 h at room temperature, washed and then incubated with goat anti-rabbit dye conjugate and Hoechst (1:2,000 dilution) in DPBS and 1% w/v BSA for 30 min in the buffer. Finally, the cell sample was subjected to immunofluorescence microscopy.
HEK-Blue IL-1β cell assay
Cultured HEK-Blue IL-1β cells were gently lifted by trypsinization, counted and plated as 100,000 cells per well in a 24-well plate, after which they were allowed to recover overnight. After aspiration, 500 µl supernatant from OE19 or K562 cultures, prepared as described above, was added with an additional 500 µl of fresh medium without Normocin/Zeocin. The cell cultures were then incubated for 24 h at 37 °C and 5% CO2. From each of the treated HEK-Blue IL-1β cell cultures, 50 µl supernatant was transferred to a 96-well plate. To each well we added 150 µL QUANTI-Blue solution, after which the plate was incubated for 30 min at 37 °C. SEAP levels were then determined using a plate reader measuring the absorbance at 630 nm.
Mixed cells K562 and HEK-Blue
One million K562 cells and 100,000 HEK-Blue IL-1β cells were collected, mixed and washed and suspended in 0.5 ml DPBS, 1% w/v BSA and 2 mM CaCl2. The cells were then co-incubated in a 12-well plate. SMART-IL-1β was added to the cell sample, which was further incubated for 2 h at 37 °C and 5% CO2. Then 500 µl RPMI medium supplemented with 5% FBS, 100 U ml−1 penicillin-streptomycin and 25 mM HEPES buffer was added and the cells were co-cultured for 24 h at 37 °C and 5% CO2. The SEAP assay was then done as described above.
Mixed cells OE19 and HeLaeGFP+
OE19 and HeLaeGFP+ cells were seeded in a 24-well glass-bottom plate in a 5:1 ratio and cultured in a mixture of 50% DMEM, 50% RPMI media supplemented with 2 mM l-glutamine, 10% w/v FBS and 100 U ml−1 penicillin-streptomycin. When nearly confluent, the cells were washed with DPBS, 1% w/v BSA and 2 mM CaCl2 and incubated with SMART-IL-1β for 2 h at 37 °C and 5% CO2. The cells were then washed, fixed, permeabilized and analysed by fluorescence microscopy as described above.
Statistics and reproducibility
All statistical analyses were done in GraphPad Prism v.9.2.0. P-values were determined by either two-sided t-test or one-way ANOVA followed by Dunnett’s test, as listed in the figure legends. The statistical significances of differences (NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001) are specified throughout the figures and legends. Heatmaps were generated using GraphPad Prism v.9.2.0. All experiments analysed by SDS–PAGE/western blotting and/or flow cytometry were repeated at least 2–3 times (independent biological replicates). All experiments analysed by microscopy were repeated at least twice (independent biological replicates). All results were reproducible.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the data supporting the findings of this study are available in the paper and the Supplementary Information. Coordinates and structure files have been deposited to the Protein Data Bank (PDB: 8UBS).
References
Bausch-Fluck, D. et al. The in silico human surfaceome. Proc. Natl Acad. Sci. USA 115, E10988–E10997 (2018).
Dannenfelser, R. et al. Discriminatory power of combinatorial antigen recognition in cancer T cell therapies. Cell Syst. 11, 215–228 (2020).
Hu, Z. et al. The Cancer Surfaceome Atlas integrates genomic, functional and drug response data to identify actionable targets. Nat. Cancer 2, 1406–1422 (2021).
Kichloo, A. et al. Systemic adverse effects and toxicities associated with immunotherapy: a review. World J. Clin. Oncol. 12, 150–163 (2021).
Tarantino, P., Ricciuti, B., Pradhan, S. M. & Tolaney, S. M. Optimizing the safety of antibody–drug conjugates for patients with solid tumours. Nat. Rev. Clin. Oncol. 20, 558–576 (2023).
Porta, C., Paglino, C. & Mutti, L. Ranpirnase and its potential for the treatment of unresectable malignant mesothelioma. Biologics 2, 601–609 (2008).
Tsuchikama, K., Anami, Y., Ha, S. Y. Y. & Yamazaki, C. M. Exploring the next generation of antibody–drug conjugates. Nat. Rev. Clin. Oncol. 21, 203–223 (2024).
Chen, Z. et al. De novo design of protein logic gates. Science 368, 78–84 (2020).
Vishweshwaraiah, Y. L., Chen, J., Chirasani, V. R., Tabdanov, E. D. & Dokholyan, N. V. Two-input protein logic gate for computation in living cells. Nat. Commun. 12, 6615 (2021).
MacKay, M. et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat. Biotechnol. 38, 233–244 (2020).
Savanur, M. A., Weinstein-Marom, H. & Gross, G. Implementing logic gates for safer immunotherapy of cancer. Front. Immunol. 12, 780399 (2021).
Lajoie, M. J. et al. Designed protein logic to target cells with precise combinations of surface antigens. Science 369, 1637–1643 (2020).
Quijano-Rubio, A. et al. A split, conditionally active mimetic of IL-2 reduces the toxicity of systemic cytokine therapy. Nat. Biotechnol. 41, 532–540 (2023).
Oostindie, S. C. et al. Logic-gated antibody pairs that selectively act on cells co-expressing two antigens. Nat. Biotechnol. 40, 1509–1519 (2022).
Shekhawat, S. S. & Ghosh, I. Split-protein systems: beyond binary protein-protein interactions. Curr. Opin. Chem. Biol. 15, 789–797 (2011).
Shah, N. H. & Muir, T. W. Inteins: nature’s gift to protein chemists. Chem. Sci. 5, 446–461 (2014).
Dagliyan, O. et al. Computational design of chemogenetic and optogenetic split proteins. Nat. Commun. 9, 4042 (2018).
Gramespacher, J. A., Stevens, A. J., Nguyen, D. P., Chin, J. W. & Muir, T. W. Intein zymogens: conditional assembly and splicing of split inteins via targeted proteolysis. J. Am. Chem. Soc. 139, 8074–8077 (2017).
Gramespacher, J. A., Burton, A. J., Guerra, L. F. & Muir, T. W. Proximity induced splicing utilizing caged split inteins. J. Am. Chem. Soc. 141, 13708–13712 (2019).
Lee, G. & Muir, T. W. Distinct phases of cellular signaling revealed by time-resolved protein synthesis. Nat. Chem. Biol. 20, 1353–1360 (2024).
Keeble, A. H. et al. Approaching infinite affinity through engineering of peptide-protein interaction. Proc. Natl Acad. Sci. USA 116, 26523–26533 (2019).
Carvajal-Vallejos, P., Pallissé, R., Mootz, H. D. & Schmidt, S. R. Unprecedented rates and efficiencies revealed for new natural split inteins from metagenomic sources. J. Biol. Chem. 287, 28686–28696 (2012).
Stefan, N. et al. DARPins recognizing the tumor-associated antigen EpCAM selected by phage and ribosome display and engineered for multivalency. J. Mol. Biol. 413, 826–843 (2011).
Steiner, D., Forrer, P. & Plückthun, A. Efficient selection of DARPins with sub-nanomolar affinities using SRP phage display. J. Mol. Biol. 382, 1211–1227 (2008).
Zahnd, C. et al. A designed ankyrin repeat protein evolved to picomolar affinity to Her2. J. Mol. Biol. 369, 1015–1028 (2007).
Polito, L., Bortolotti, M., Mercatelli, D., Battelli, M. G. & Bolognesi, A. Saporin-S6: a useful tool in cancer therapy. Toxins 5, 1698–1722 (2013).
Bolshakov, A. P., Stepanichev, M. Y., Dobryakova, Y. V., Spivak, Y. S. & Markevich, V. A. Saporin from Saponaria officinalis as a tool for experimental research, modeling, and therapy in neuroscience. Toxins 12, 546 (2020).
Dittel, B. N. Depletion of specific cell populations by complement depletion. J. Vis. Exp. https://doi.org/10.3791/1487 (2010).
Lee, D. S. W., Rojas, O. L. & Gommerman, J. L. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat. Rev. Drug Discov. 20, 179–199 (2021).
Baral, T. N., Murad, Y., Nguyen, T.-D., Iqbal, U. & Zhang, J. Isolation of functional single domain antibody by whole cell immunization: implications for cancer treatment. J. Immunol. Methods 371, 70–80 (2011).
Tamamura, H. et al. Enhancement of the T140-based pharmacophores leads to the development of more potent and bio-stable CXCR4 antagonists. Org. Biomol. Chem. 1, 3663–3669 (2003).
Zocchi, C. et al. The non-xanthine heterocyclic compound SCH 58261 is a new potent and selective A2a adenosine receptor antagonist. J. Pharmacol. Exp. Ther. 276, 398–404 (1996).
Lam, S. S. et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54 (2015).
Geri, J. B. et al. Microenvironment mapping via Dexter energy transfer on immune cells. Science 367, 1091–1097 (2020).
Garlanda, C., Dinarello, C. A. & Mantovani, A. The interleukin-1 family: back to the future. Immunity 39, 1003–1018 (2013).
Ho, T. Y. H. et al. A systematic approach to inserting split inteins for Boolean logic gate engineering and basal activity reduction. Nat. Commun. 12, 2200 (2021).
Pinto, F., Thornton, E. L. & Wang, B. An expanded library of orthogonal split inteins enables modular multi-peptide assemblies. Nat. Commun. 11, 1529 (2020).
Palei, S., Becher, K. S., Nienberg, C., Jose, J. & Mootz, H. D. Bacterial cell-surface display of semisynthetic cyclic peptides. ChemBioChem. 20, 72–77 (2019).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D 67, 235–242 (2011).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019).
Kirsten, D. & Kritzer, J. A. HaloTag forms an intramolecular disulfide. Bioconjug. Chem. 32, 964–970 (2021).
Acknowledgements
This work was funded by NIH-GMS grant R01 GM086868 and by funds from the Ludwig Institute for Cancer Research. C.K. was supported by EMBO (ALTF 1189-2020). N.E.S.T. is supported by an NIH postdoctoral fellowship (GM149123). X.Y. was supported by a graduate fellowship from the China Scholarship Council (CSC). We thank members of the Muir laboratory for discussions and support; P. Jeffrey for technical assistance with setting up crystal trays, looping, data collection and data processing; C. DeCoste and K. Rittenbach for technical assistance with flow cytometry and cell sorting, and instrument use (Princeton University Flow Cytometry Resource Facility, Department of Microbiology, supported, in part, with funding from NCI-CCSG P30CA072720-5921 and 1S10OD028592-01A1); S. Wang and G. Laevsky for technical assistance with confocal microscopy and instrument use; D. Baker for sharing K562 cell lines; Y. Kang for sharing MCF-10a, MCF-7, LoVo and A594 cell lines; S. Lipkowitz for sharing the Sk-br-3 cell line; and C. Kadoch for sharing the PiggyBac transposon system.
Author information
Authors and Affiliations
Contributions
C.K. and T.W.M. conceived the work. C.K., N.E.S.T., G.E., X.Y., Y.L. and T.W.M. designed and executed the experiments. C.K. and T.W.M. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
T.W.M. is a scientific founder, scientific advisory board member and shareholder of SpliceBio and is a consultant for Merck. T.W.M., C.K., N.E.S.T., X.Y. and G.E. have filed a provisional US patent based on this work. Y.L. declares no competing interests.
Peer review
Peer review information
Nature thanks Baojun Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Systematic screening to identify SpyCatcher003 split site and optimization of SMART-SpyCatcher.
a, SpyCatcher003 was split at various sites and the cognate pairs used to generate FLAG-SpyN1-x-NpuNcage-FKBP and FRB-NpuCcage-SpyCy-113-Myc, with x and y denoting the last and first residue of the two fragments (i.e., the split site). The isopeptide bond (formed between D117 of SpyTag003 and K31 of SpyCatcher003) is shown in sticks in the structure of SpyTag-SpyCatcher (PDB: 4MLI). The primary structure of SpyCatcher003 is shown with split sites indicated. b, Schematic of the cell-free in vitro screen used to identify the optimal split site. Conditional protein splicing (CPS) was induced either by proteolytic decaging using TEV protease or through chemically induced FKBP/rapamycin/FRB hybridization, which simulates ideal colocalization on a target cell surface. c, The result of screening FLAG-SpyN1-73-NpuNcage-FKBP and FRB-NpuCcage-SpyC74-113-Myc generated from splitting SpyCatcher003 at position 73-74. Reactions were performed with FLAG-SpyN1-73-NpuNcage-FKBP (1 μM), FRB-NpuCcage-SpyC74-113-Myc, (1 μM) and His6-SpyTag003 (2 μM). CPS was induced by the addition of either TEV protease (10 units) or rapamycin (10 μM). The reactions were analyzed by Western blot after 24 hr incubation at 37 °C. FLAG-SpyCatcher003-Myc was used as a size standard for the spliced product; FLAG-SpyCatcher003-Myc (1 μM) reacted with His6-SpyTag003 (2 μM) was used as a size standard for the covalent complex between the two. The legend and experimental conditions apply for subsequent panels with alterations when noted. d, Npucage was swapped with NrdJ-1cage in SMART-SpyCatcher thereby giving FLAG-SpyN1-73-NrdJ-1Ncage-FKBP and FRB-NrdJ-1Ccage-SpyC74-113-Myc. A SpyTag003 Alexa Fluor 594 conjugate (SpyTag003-AF594) was used as the activity probe. The reactions were analyzed by Western blot. e, SpyN1-73-NrdJ-1Ncage-FKBP with the wildtype (WT) or a catalytically dead C1A mutant of NrdJ-1Ncage was mixed with FRB-NrdJ-1Ccage-SpyC74-113-Myc and SpyTag003-AF594. The reactions were analyzed by Western blot. f, The crystal structure of fusion NrdJ-1 (PDB: 8UBS), with sequences corresponding to NrdJ-1N and NrdJ-1C colored in cyan and orange respectively. A schematic representation of the fusion protein and the caged split intein is shown as well to indicate relevant domains in addition to their N- and C-termini and the position of the non-catalytic Cys76. g-h, A structural analysis was performed to predict the impact of introducing a C76V mutation in fusion NrdJ-1. g, The backbone of wildtype NrdJ-1 (crystal structure) and that of the NrdJ-1C76V mutant (in silico AlphaFold2 model) are superimposable with a root mean square deviation (RMSD) of 0.56 Å. h, Residue sidechains of Cys76 and Leu2 are shown as sticks and cavities in solid grey. The experimentally determined local environment of Cys76 is shown on the left, whereas the in silico predicted local environment of C76V is shown on the right and changes between the two are indicated with arrows. i, Combinations of FLAG-SpyN1-73-NrdJ-1Ncage-FKBP or FLAG-SpyN1-73-NrdJ-1NC76Vcage-FKBP with FRB-NrdJ-1Ccage-SpyC74-113-Myc or FRB-NrdJ-1Ccage(C76V)-SpyC74-113-Myc were tested as indicated using the reaction conditions described above. The reactions were analyzed by Western blot. j, Electrospray ionization time-of-flight mass spectrometry (ESI-TOF MS) characterization of the SpyCatcher003 spliced product produced by rapamycin triggered CPS between FLAG-SpyN1-73-NrdJ-1NC76Vcage-FKBP and FRB-NrdJ-1Ccage(C76V)-SpyC74-113-Myc. Reaction conditions followed those described above. Data shown in panels c-e, and i are representative of two independent experiments.
Extended Data Fig. 2 Confocal microscopy of K562 cell lines treated with SMART-SpyCatcher for [HER2 AND EGFR] logic.
a, K562HER2+/EGFR+ cells were treated with αHER2-SpyN (100 nM) and SpyC-αEGFR (100 nM) for 2 hr, followed by SpyTag003 labeled with Alexa Fluor 594 (SpyTag003-AF594, 100 nM) for 20 min. SMART-SpyCatcher employed either NrdJ-1N(C76V)cage/NrdJ-1Ccage(C76V) or NrdJ-1N(C76V)cage(K109EK119A)/NrdJ-1Ccage(C76VD66K) (referred to as eNrdJ-1cage). Following washing, the live cells were analyzed by confocal microscopy. Cell nuclei were stained with Hoechst, while HER2 and EGFR were tagged with eGFP and iRFP, respectively. Scale bar equals 20 μm. All subsequent panels apply similar reaction and analysis conditions as in panel a using the SMART-SpyCatcher (eNrdJ-1cage) [HER2 AND EGFR] system. b, K562HER2+, K562EGFR+, and K562HER2+/EGFR+ cells were treated and analyzed individually. Scale bars equal 10 µm. c, To determine the colocalization of HER2, EGFR and SpyTag003-AF594 the signal intensities of eGFP, iRFP, and AF594 derived from the cells in panel b were plotted (yellow dotted lines). d, A mixed population consisting of equal amounts of K562 (wildtype), K562HER2+, K562EGFR+, and K562HER2+/EGFR+ cells were treated and imaged. Representative single cells of each cell line from the treated mixture and their associated fluorescence signals are shown on the right. Scale bar equals 20 μm. Data are representative of two independent experiments.
Extended Data Fig. 3 The mechanism of action of SMART-SpyCatcher.
a, K562HER2+/EGFR+ cells were treated with αHER2-SpyN (100 nM) and SpyC-αEGFR (100 nM) for 2 hr, followed by SpyTag003 labeled with Alexa Fluor 594 (SpyTag003-AF594, 100 nM) for 20 min. SMART-SpyCatcher employed eNrdJ-1cage. Following washing, the live cells were analyzed by confocal microscopy. Cell nuclei were stained with Hoechst, while HER2 and EGFR were tagged with eGFP and iRFP, respectively. The cells in row 1 were treated as described above, whereas reaction conditions in the subsequent experiments were supplemented with DARPins targeting HER2 and EGFR (500 nM each, row 2), used an inactivated version of NrdJ-1Ncage (Cys1-alkylated, row 3), or were supplemented with unlabeled SpyTag003 (500 nM, row 4). Scale bar equals 20 μm. All subsequent panels apply similar reaction and analysis conditions as in panel a (row 1) using the SMART-SpyCatcher (eNrdJ-1cage) system with any alterations as noted. b, Western blot analysis of K562HER2+/EGFR+ cells treated with αHER2-SpyN and SpyC-αEGFR (identified by FLAG and Myc respectively), and SpyTag003 labeled with biotin (SpyTag003-biotin; identified by Strep-800). The inactivated version of NrdJ-1Ncage (Cys1-alkylated) was used as a negative control. c, Experiments were performed on K562HER2+/EGFR+ cells with the SMART-SpyCatcher (eNrdJ-1cage) [HER2 AND EGFR] system, and either SpyTag003-AF594 or inactive SpyTag003D117A labeled with Alexa Fluor 594 (SpyTag003D117A-AF594). Scale bar equals 20 μm. d, K562HER2+/EGFR+ cells were treated with SMART-SpyCatcher (100 nM, eNrdJ-1cage) in buffer (DPBS, 1% w/v BSA, 2 mM CaCl2), RPMI 1640 medium (cystine-free), or in medium supplemented with 5% v/v fetal bovine serum (5% v/v FBS) for 2 hr, followed by SpyTag003-AF594 for 20 min. Further image analysis was performed as described above. Scale bar equals 20 μm. e, Flow cytometry analysis of a mixed population consisting of equal amounts of K562 (wildtype), K562HER2+, K562EGFR+, and K562HER2+/EGFR+ cells following treatment with SMART-SpyCatcher (eNrdJ-1cage) operating through the AND logic indicated. The added SpyN and SpyC pairs (i.e., the targeting DARPins employed in the constructs) are indicated at the bottom. Data are presented as the mean of the AF594 median fluorescence intensities (MFI) from flow cytometry analysis with error bars signifying the standard error mean (n = 3 independent biological replicates; see Supplementary Table 13 for statistical one-way ANOVA followed by Dunnett’s test). f, The mixed K562 population described above were treated with αHER2-SpyN (100 nM) and SpyC-αEGFR (100 nM) employing eNrdJ-1cage or the uncaged split intein NrdJ-1, for 2 hr, followed by SpyTag003-AF594 (100 nM) for 20 min. Further image analysis was performed as described above. Scale bars are equal to 20 μm. Data shown in panels a-d, and f are representative of two independent experiments.
Extended Data Fig. 4 Extended SMART logic and three-input systems.
a, SMART-SpyCatcher (i.e., SpyN and SpyC) was assigned to operate through AND logic involving combinations of EpCAM with HER2 and EGFR. Two combinations of K562 cell lines were used to test for the actuation of SMART-SpyCatcher and thereby recruitment of SpyTag003-AF594. Anticipated target cells are highlighted in red. Mixed-population 1 consisted of equal amounts of K562 (wildtype), K562EGFR+, K562HER2+, and K562HER2+/EGFR+, whereas mixed-population 2 consisted of equal amounts of K562 (wildtype), K562EGFR+, K562HER2+/EpCAMhigh, and K562HER2+/EGFR+/EpCAMhigh. The antigen profile of each cell line is indicated below each bar plot (L and H designates low endogenous and high ectopic levels respectively for EpCAM), whereas the antigens targeted by the added SpyN and SpyC pairs (i.e. the targeting DARPins employed in the constructs) are indicated at the bottom. Experiments were performed with SMART-SpyCatcher (100 nM, eNrdJ-1cage), 100 nM SpyTag003-AF594. Data are presented as the mean of the AF594 median fluorescence intensities (MFI) from flow cytometry analysis with error bars signifying the standard error mean (n = 3 independent biological replicates; see Supplementary Table 13 for statistical one-way ANOVA followed by Dunnett’s test). b, Target cells were profiled for their relative surface levels of HER2, EGFR, and EpCAM. Cells were treated with DARPins labeled with AF594 followed by flow cytometry analysis. In parallel, combinations of SpyN and SpyC linked to targeting DARPins were used to solve the AND logic matrix for a given cell line. c, Top: Single cells lines were profiled for their relative surface levels of HER2, EGFR, and EpCAM using the three DARPin Alexa Fluor 594 conjugates αHER2-AF594, αEGFR-AF594, and αEpCAM-AF594. Cells were treated with the indicated DARPin-AF594 conjugate and analyzed by flow cytometry analysis. Bottom: Single cell lines were also treated with variations of SMART-SpyCatcher003 (100 nM, eNrdJ-1cage) operating through different combinations of AND logic targeting HER2, EGFR, and EpCAM. The heatmaps represents the mean MFI of the AF549 signal from three independent replicates (see Supplementary Tables 3 and 4 for individual values). The cell lines are categorized as displaying low (MFI < 1000) or high (MFI ≥ 1000) levels of the three antigens. d, The quantity of the lesser-expressed antigen used in each AND gate and the resulting recruitment of SpyTag003-AF594 is plotted for the data from panel c. Errors = standard error mean (n = 3 independent biological replicates). e, Schematic illustrating SMART-SpyCatcher operating through [(HER2 OR EGFR) AND (HER2 OR EGFR)] logic on a mixed K562 population; sets of SpyN and SpyC are used to achieve [HER2 AND HER2], [EGFR AND EGFR], [HER2 AND EGFR], and [EGFR AND HER2] gating, essentially resulting in OR logic. f, The SMART-SpyCatcher [HER2 OR EGFR] logic operation was tested on mixed K562 cell population 1. Cells were treated with SMART-SpyCatcher (i.e. αHER2-SpyN/αEGFR-SpyN/SpyC-αHER2/SpyC-αEGFR, each at 100 nM, employing eNrdJ-1cage), and SpyTag003-AF594 (100 nM) and analyzed by flow cytometry. Error bars signify the standard error mean (n = 3 independent biological replicates; see Supplementary Table 14 for statistical one-way ANOVA followed by Dunnett’s test). g, Schematic illustrating a 3-input logic operation, where SMART-SpyCatcher acts through the use of αAg1-SpyN/SpyC-αAg2/SpyC-αAg2 to achieve [Ag1 AND either Ag2 OR Ag3] cell targeting strategy. h, SMART-SpyCatcher (i.e. αAg1-SpyN/SpyC-αAg2/SpyC-αAg2) was used in mixed K562 cell experiments to evaluate its AND/OR logic function involving combinations of HER2, EGFR and EpCAM. The two mixed populations of K562 cell lines described above were used to test for the actuation of SMART-SpyCatcher and thereby recruitment of SpyTag003-AF594. Experiments were performed with SMART-SpyCatcher (100 nM, employing eNrdJ-1cage), and SpyTag003-AF594 (100 nM). Data are presented as AF594 MFI from flow cytometry analysis with error bars signifying the standard error mean (n = 3 independent biological replicates; see Supplementary Table 15 for statistical one-way ANOVA followed by Dunnett’s test).
Extended Data Fig. 5 Tuning SMART for an adjustable stimulus-response.
Several eNrdJ-1Ncage variants were made either by mutating ionizable sidechain residues (a-c) or adjusting the cage length (d-f); these were tested in standard mixed K562 cell experiments followed by flow cytometry analysis. Data are presented as the mean of the AF594 median fluorescence intensities (MFI) from flow cytometry analysis with error bars signifying the standard error mean (n = 3 independent biological replicates). Panels b and e: A mixed population consisting of equal amounts of K562 (wildtype), K562HER2+, K562EGFR+, and K562HER2+/EGFR+ cells was treated with αHER2-SpyN (using the indicated eNrdJ-1Ncage variant at 100 nM), SpyC-αEGFR (using the standard eNrdJ-1Ccage variant at 100 nM), and SpyTag003-AF594 (100 nM). The experimental median fluorescence intensity (MFI) values were normalized to those obtained employing eNrdJ-1Ncage for comparison (n = 3 independent biological replicates). The normalized AF594 MFI associated with the individual four cell lines across the different experiments are summarized in the form of two heatmaps (see Supplementary Table 13 for statistical one-way ANOVA followed by Dunnett’s test and Supplementary Tables 5 and 6 for individual values). Panels c and f: The most active variants from the two ways of cage tuning were tested in dose-response experiments.
Extended Data Fig. 6 Employing tuned SMART-SpyCatchers.
a, SpyCatcher003 utilizes its residue K31 to form an isopeptide bond with D117 of SpyTag003 upon binding. A mutant of SpyN carrying K31E was evaluated for its abilty to recruit SpyTag003-AF594 upon SMART actuation of SpyCatcher003. The data represent the flow cytometry analysis of K562HER2+/EGFR+ cells left untreated or treated with either SMART-SpyCatcher or SMART-SpyCatcherK31E (both at 100 nM, employing eNrdJ-1cage and operating through [HER2 AND EGFR] gating). RFU denotes relative fluorescence units. The relative mean AF594 MFI is given in the bar graph on the right with error bars signifying the standard error mean (n = 3 independent biological replicates) and statistical significance evaluated using an unpaired two-sided t-test (ns = not significant; ****P < 0.0001). b, The total cellular levels of HER2 and EpCAM for MCF-10a, MCF-7, and Sk-br-3 cell lines were determined by Western blotting (left) and quantified (right). Intensity values were corrected against the GAPDH signal for each cell line and further normalized to the overall highest level (Sk-br-3 for HER2, MCF-7 for EpCAM). c, The endogenous surface levels of HER2 and EpCAM on mammary cell lines MCF-10a, MCF-7, and Sk-br-3 were furthermore profiled and categorized as low (MFI < 1000) or high (MFI ≥ 1000). Errors = standard error mean (n = 3 independent biological replicates). d, MCF-10a cells either unstained or stained with the cell-permeable dye 5-Chloromethylfluorescein diacetate (CMFDA) were also phenotyped to determine any differences in the levels of surface HER2 and EpCAM. Errors = standard error mean (n = 3 independent biological replicates). Statistical significance was evaluated using an unpaired two-sided t-test (ns = not significant). e, SMART-SpyCatcher assigned for [HER2 AND EpCAM] logic was tested in flow cytometry experiments using mixed mammary cell population 1 (equal amounts of MCF-10aHER2low/EpCAMlow and MCF-7HER2low/EpCAMhigh) and mixed mammary population 2 (equal amounts of MCF-10aHER2low/EpCAMlow and Sk-br-3HER2high/EpCAMhigh). MCF-10a cells were pre-stained with CMFDA, which labels intracellular proteins. Each population was incubated with 100 nM SpyTag003-AF594 in the absence or presence of 100 nM αHER2-SpyN and SpyC-αEpCAM (employing eNrdJ-1cage with K114AK116A). Shown are representative flow cytometry plots of the recruitment of SpyTag-AF594 by the subpopulations under the different conditions. f, SMART-SpyCatcher003 using different versions of eNrdJ-1cage were tested on the cell lines (color coded as in panel e) and the quantified data presents the mean of the individual median fluorescence intensities (MFI) of the AF594 signal with error bars signifying the standard error mean (n = 3 independent biological replicates). Statistical analysis using paired two-sided t-test.
Extended Data Fig. 7 Targeted cell depletion using Boolean logic.
a, K562HER2+/EGFR+ cells were treated with SMART-SpyCatcher (100 nM, employing eNrdJ-1cage) for [HER2 AND EGFR] logic and SpyTag003 labeled with biotin (SpyTag003-biotin, 100 nM). Following washing, the cells were treated with a NeutrAvidin Rhodamine Red-X conjugate (NA-RRx, magenta) and then either visualized by confocal microscopy immediately or after further incubation for 4 hr at 37 °C or 4 °C. Cell nuclei were stained with Hoechst, while lysosomal compartments were stained with LysoTracker. Scale bar equals 20 μm. b, Schematic illustrating the proposed recruitment and internalization of NA-RRx enabled by the SMART-SpyCatcher system. c, A mixed population consisting of equal amounts of K562 (wildtype), K562EGFR+, K562HER2+, and K562HER2+/EGFR cells was used to test for the selective recruitment of SpyTag003-biotin and subsequent recruitment of NA-RRx. The cell mixture was treated as in panel b. The NA-RRx signal associated with the individual subpopulations was quantified by flow cytometry with the data presented as the mean of the NA-RRx median fluorescence intensities (MFI) with error bars signifying the standard error mean (P(WT vs HER2+) = 0.9950; P(WT vs EGFR+) = 0.9950; P(WT vs HER2+/EGFR+) < 0.0001; see Supplementary Table 13 for further statistical one-way ANOVA followed by Dunnett’s test). d, Schematic illustrating the selective cell depletion of a HER2/EGFR positive K562 cell line in a complex cell mixture using SMART-SpyCatcher, SpyTag003-biotin and a Streptavidin-Saporin disulfide conjugate. e, Summary of how the one-dose and two-dose regimens were performed. f, The mixed K562 cell population employed in panel c was treated with SMART-SpyCatcher (100 nM, employing eNrdJ-1cage and [HER2 AND EGFR] logic), SpyTag003-biotin (100 nM), and Streptavidin-Saporin (20 nM). Cells were then analyzed by flow cytometry and the data presented as percentage viability relative to untreated wildtype cells (ns = not significant; ****P < 0.0001). All flow cytometry data are presented as the mean with error bars signifying standard error mean (n = 3 independent biological replicates). Statistical significance in panel f was evaluated using an unpaired two-sided t-test. Data shown in panel a is representative of two independent experiments.
Extended Data Fig. 8 AND-gated proximity labeling.
a, Western blot analysis of K562HER2+/EGFR+ treated with αHER2-SpyN (100 nM, eNrdJ-1cage) and SpyC-αEGFR (100 nM) for 2 hr at 37 °C, followed by HA-tagged SpyTag003-APEX2 (300 μM) for 20 min. b, K562 (wildtype), K562HER2+, K562EGFR+, and K562HER2+/EGFR+ cells were treated individually as described in panel a. Following washing, APEX2 proximity labeling was induced by the addition of biotin-phenol (250 μM) and H2O2 (1 mM). The reactions were quenched with sodium ascorbate (10 mM) and Trolox (5 mM) at room temperature after two minutes. Samples were then analyzed by Western blot using Strep-800 to detect biotinylation. c-e, Time-course study of APEX2 labeling. The cells were treated as in panel b with labeling quenched at the indicated time points. αHER2-SpyN/SpyC-αEGFR was used with the K562HER2+/EGFR+ cells; αHER2-SpyN/SpyC-αEpCAM was used with the OE19 cells; αEGFR-SpyN/SpyC-αEpCAM was used with the A431 cells. SMART-SpyCatcher employed eNrdJ-1cage in all cases. f, Displayed are the employed AND gates used in the following experiments (note, lettering differs from that in Fig. 3). Each AND gate constitute a pair of SpyN and SpyC protein fusions and/or synthetic ligand conjugates; for instance, AND gate D is generated by αCEACAM6-SpyN and SpyC-αHER2. g-j, AND-gated proximity labeling using the described AND gates, SpyTag003-APEX2 and the workflow described under panels a-e. In each case, the lane expected to give proximity labeling is indicated in red. When applicable, cells were treated for 12 hr beforehand with doxycycline (DOX, 200 ng/mL) to induce the expression of FLAG-CXCR4-eGFP (OE19CXCR4DOX) or FLAG-ADORA2A-mCherry (OE19A2ADOX). SpyC-Cys indicates the unconjugated variant. Asterisks indicate endogenously biotinylated proteins. Data shown in panels a-e, and g-j are representative of two and three independent experiments, respectively.
Extended Data Fig. 9 Optimization of AND-gated μMap photocatalytic proximity labeling.
a, Illustration of the principle of AND-gated photocatalytic proximity labeling achieved using SMART-SpyCatcher targeting two cell surface antigens and the subsequent delivery of SpyTag003 conjugated to an iridium photocatalyst. Iridium enables the blue light activation of a biotin diazirine probe, which reacts with nearby proteins. b, Schematic of the preparation of SpyTag003-Ir from the precursors SpyTag003-HaloTag13 and the iridium photocatalyst Ir-PEG4-C6H12Cl. HaloTag13 is based on HaloTag9 with the additional mutations C61S and C262S to improve recombinant expression and solubility46. c, d, The protein preparation of SpyTag003-HaloTag13 and its conjugate with Ir-PEG4-C6H12Cl (i.e. SpyTag003-Ir) were analyzed by SDS-PAGE (left), RP-HPLC (0-70% B gradient over 30 min, middle), and ESI-TOF MS (right). e, The K562HER2+/EGFR+ cell line was treated with SMART-SpyCatcher (100 nM, eNrdJ-1cage) operating by [HER2 AND EGFR] logic and subsequently supplemented with SpyTag003-Ir (200 nM). Following a wash, the culture was incubated with biotin-diazirine (250 µM) and irradiated with blue light (LED 440 nm) for 5 min or 10 min. The cells were thereafter analyzed by flow cytometry using Streptavidin conjugated to Alexa Fluor 546 (AF546) to detect biotinylation. f, Quantification of the flow cytometry data from panel e for the samples irradiated for 5 min (****P < 0.0001). Data are presented as the mean of the AF546 median fluorescence intensities (MFI) with error bars signifying the standard error mean (n = 3 independent biological replicates). See Supplementary Table 16 for individual values. Statistical significance was evaluated using an unpaired two-sided t-test.
Extended Data Fig. 10 Systematic screen to make SMART-IL-1β.
a, The X-ray crystal structure (PDB: 4DEP) of interleukin 1β (IL-1β) bound to interleukin 1 receptor, type 1 (IL-1R1). b, IL-1β was split at various sites and the cognate pairs used to generate FLAG-IL-1βN1-x-eNrdJ-1Ncage-FKBP and FRB-eNrdJ-1Ccage-IL-1βCy-153–Myc, with x and y respectively denoting the last and first residue of the two fragments (i.e., the split site). Shown is the X-ray crystal structure (PDB: 9ILB) and primary structure of IL-1β with tested split sites indicated. c, Schematic of the in vitro screen used to identify the optimal split site that would render a conditional version of IL-1β: (i) The screen relied on chemically induced FKBP-rapamycin-FRB heterodimerization to trigger conditional protein splicing (CPS) with analysis by Western blotting to identify spliced IL-1β; (ii) spliced IL-1β variants were subsequently tested on cells to determine biological activity using immunofluorescence imaging to determine NF-κB localization. d, Western blot of reactions performed with FLAG-IL-1βN1-x-eNrdJ-1Ncage-FKBP (0.5 μM), FRB-eNrdJ-1Ccage-IL-1βCy-153–Myc (0.5 μM) and rapamycin (10 μM) for 2 hr at 37 °C. The expected mass of spliced IL-1β is 20.5 kDa. e-f, Additional experiments for the two pairs FLAG-IL-1βN1-44-eNrdJ-1Ncage-FKBP/FRB-eNrdJ-1Ccage-IL-1βC45-153-Myc and FLAG-IL-1βN1-69-eNrdJ-1Ncage-FKBP/FRB-eNrdJ-1Ccage-IL-1βC70-153-Myc to validate splicing of IL-1β being rapamycin- and CPS-dependent (the latter experiment used the splicing-deficient eNrdJ-1N(C1A)cage mutant). g, Cultured HeLa cells were incubated with recombinantly expressed IL-1β for 20 min at 37 °C to induce IL-1R1 signaling and NF-κB nuclear localization. Cells were then fixed (5% formaldehyde), permeabilized (0.5% Triton X100), and analyzed by confocal imaging using a primary rabbit anti-NF-κB (p65) antibody and a secondary goat anti-rabbit antibody Alexa Fluor 488 conjugate. Scale bar equals 100 μm. Subsequent experiments followed a similar analytical procedure. h, Cultured HeLa cells were treated with reaction mixtures of the pair FLAG-IL-1βN1-44-eNrdJ-1Ncage-FKBP/FRB-eNrdJ-1Ccage-IL-1βC45-153-Myc and analyzed as described above to determine IL-1R1 signaling activation. Scale bar equals 50 μm. i, HeLa cells were left untreated or incubated with recombinant IL-1β (0.5 nM). In experiments with SMART-IL-1β (i.e. IL-1βN/IL-1βC), HeLa cells were treated with a medium produced by incubating OE19 cells (HER2high, EGFRlow, EpCAMhigh) with IL-1βN-αHER2 (20 nM) and αEpCAM-IL-1βC (20 nM) for 2 hr at 37 °C. In all cases, HeLa cells were incubated for 30 min at 37 °C, before being collected and analyzed by Western blotting to determine the phosphorylation of NF-κB (pNF-κB) and IκB (pIκB). j, The localization of NF-κB was examined for OE19 cells untreated or incubated with recombinant IL-1β. Scale bar equals 40 μm. Data shown are representative of two independent experiments.
Supplementary information
Supplementary Fig. 1
Uncropped western blots.
Supplementary Table 2
Data collection and refinement statistics (molecular replacement).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kofoed, C., Erkalo, G., Tay, N.E.S. et al. Programmable protein ligation on cell surfaces. Nature 645, 793–800 (2025). https://doi.org/10.1038/s41586-025-09287-2
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41586-025-09287-2
This article is cited by
-
Temporal photoproximity labeling of ligand-activated EGFR neighborhoods using MultiMap
Nature Chemical Biology (2026)
-
Transforming cell-surface signatures into customizable protein functions
Nature Reviews Molecular Cell Biology (2026)
-
Improving CAR-T cell function through a targeted cytokine delivery system utilizing car target-modified extracellular vesicles
Experimental Hematology & Oncology (2025)
-
SMART tool builds proteins on cell surfaces
Nature (2025)
