
Abstract
Despite genome-wide association studies (GWAS) of late-onset Alzheimer’s disease (LOAD) having identified many genetic risk loci1,2,3, the underlying disease mechanisms remain largely unclear. Determining causal disease variants and their LOAD-relevant cellular phenotypes has been a challenge. Here, using our approach for identifying functional GWAS risk variants showing allele-specific open chromatin, we systematically identified putative causal LOAD-risk variants in human induced pluripotent stem (iPS)-cell-derived neurons, astrocytes and microglia, and linked a PICALM LOAD-risk allele to a microglial-specific role of PICALM in lipid droplet (LD) accumulation. Allele-specific open-chromatin mapping revealed functional risk variants for 26 LOAD-risk loci, mostly specific to microglia. At the microglial-specific PICALM locus, the LOAD-risk allele of the single-nucleotide polymorphism rs10792832 reduced transcription factor (PU.1) binding and PICALM expression, impairing the uptake of amyloid beta (Aβ) and myelin debris. Notably, microglia carrying the PICALM risk allele showed transcriptional enrichment of pathways for cholesterol synthesis and LD formation. Genetic and pharmacological perturbations of microglia further established a causal link between reduced PICALM expression, LD accumulation and phagocytosis deficits. Our work elucidates the selective LOAD vulnerability in microglia at the PICALM locus through detrimental LD accumulation, providing a neurobiological basis that can be exploited for developing clinical interventions.
Similar content being viewed by others


Main
GWAS analyses have identified 75 genome-wide significant (P < 5 × 10−8) loci for LOAD1,2,3. Aside from the well-established apolipoprotein E (APOE) locus that contains a protein-coding risk variant (that is, APOE4) with a large effect size, most LOAD GWAS risk variants reside in the non-coding regions of the genome without clear functional links to disease pathophysiology. Studies of human brain microglia (hMGs) have identified many LOAD-risk variants associated with gene expression quantitative trait loci (eQTL)4,5,6,7,8,9. However, such association-based mapping approaches do not directly implicate functional or causal variants. Moreover, LDs—cellular organelles containing lipids such as glycerolipids and cholesterol—were recently found to have important roles in ageing and Alzheimer’s disease (AD)10,11,12,13,14,15. APOE4, the strongest genetic risk factor for LOAD, impairs neuron–glia lipid metabolism14,16, and the APOE4-associated glial LD accumulation modulates neuron function and brain memory17,18,19. However, whether other LOAD GWAS risk loci may similarly affect lipids and cholesterol accumulation remains unclear.
We recently developed an allele-specific open chromatin (ASoC) mapping approach that enables the comparison of differential chromatin accessibility of the two alleles of a heterozygous risk variant in the same sample, therefore directly identifying probably functional disease variants affecting chromatin accessibility and gene expression20,21. Here, using the ASoC approach, we systematically identified functional LOAD-risk variants in human iPS-cell-derived MGs (iMGs), astrocytes (iAst) and neurons. For a strong LOAD-risk locus encoding phosphatidylinositol binding clathrin assembly protein (PICALM), we identified a potential causal link between the LOAD-risk allele of rs10792832 and altered chromatin accessibility to transcription factor (TF) PU.1 (SPI1) binding, leading to reduced PICALM expression, increased LD accumulation and phagocytosis deficits in MGs.
Mapping functional LOAD-risk variants
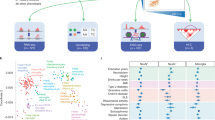
To systematically identify functional LOAD GWAS risk variants, we performed an assay for transposase-accessible chromatin using sequencing (ATAC–seq) and ascertained the allelic imbalance of chromatin accessibility (that is, ASoC)20,21 at heterozygous LOAD-risk single-nucleotide polymorphism (SNP) sites (Fig. 1a). We analysed human iMG and iAst cells (Extended Data Fig. 1a,b), and iPS-cell-derived glutamatergic (iGlut), GABAergic (iGABA) and dopaminergic (iDN) neurons (Fig. 1a, Extended Data Fig. 1c–e and Supplementary Table 1). We identified between 202,019 and 267,119 open-chromatin regions (OCRs) for each cell type. We confirmed cell-type-specific OCRs of MG (for example, SPI1) and Ast (for example, VIM) cells, and clear separation of iMG and iAst cells from neuronal samples (Extended Data Fig. 1f–i). Our iMG and iAst cells were epigenetically similar to hMGs6 and other reported iMGs22 and Ast (hAst)6 cells (Extended Data Fig. 1i). We further validated the functional relevance of iMG and iAst cell OCRs to LOAD by demonstrating strong enrichment of LOAD heritability in iMG OCRs (enrichment fold = 13.6, P = 2.1 × 10−15) and, to a lesser extent, for iAst cells (enrichment fold = 6.3, P = 4.9 × 10−10), but not for iPS-cell-derived neurons (Supplementary Fig. 2a–c).

a, Schematic of the experimental design for ASoC mapping of functional non-coding GWAS risk variants of LOAD in human iMGs and other cell types. The number of donor lines (n) for each cell type is indicated. The diagram was created using BioRender. b, Enrichment of ASoC SNPs (current study) and the reported hMG caQTL or eQTL for GWAS risk of LOAD, neuropsychiatric disorders and other complex traits. GWAS datasets are listed in Supplementary Table 9. A Bayesian hierarchical model (TORUS) was used for the enrichment analysis. ADHD, attention deficit hyperactivity disorder; BMI, body mass index; T2D, type 2 diabetes. c, rs10792832 shows ASoC in an iMG-specific OCR peak. Only iMG samples heterozygous for rs10792832 were used. Note that the ASoC SNP showing the strongest GWAS association is in an upstream enhancer linked to PICALM by ABC analysis. Chr., chromosome. d, Different ATAC–seq reads of the two alleles of the ASoC SNP rs10792832. Plot range, 86156600–86157000 bp. Blue, allele A; red, allele G. Two-sided binomial tests were used for ASoC analysis.
We next identified functional ASoC SNPs in OCRs for each cell type and evaluated their genetic relevance to LOAD. We identified between 15,693 and 72,291 ASoC SNPs, a large proportion of which were cell type specific (Extended Data Fig. 2a, Supplementary Tables 2–6 and Supplementary Fig. 3a). Similar to the genomic characteristics of neuronal ASoC SNPs20,21, 25–30% of ASoC SNPs in iMG and iAst cells were proximal to gene promoters (within 5 kb), while most ASoC SNPs were in distal enhancers (>50 kb) (Supplementary Fig. 3b,c). As expected, ASoC SNPs in iMGs were near genes involved in immune activation and phagocytosis (Supplementary Fig. 3d,e). iMG ASoC SNPs were highly enriched for hMG chromatin accessibility QTLs (caQTL) and eQTLs (Supplementary Tables 2 and 8 and Supplementary Fig. 3f–h), suggesting that they probably alter chromatin accessibility and influence gene expression. Notably, neuronal ASoC SNPs showed greater enrichment for GWAS variants of schizophrenia, neuroticism and intelligence, while iMG ASoC SNPs exhibited the strongest LOAD GWAS risk enrichment (35-fold, P = 1.04 × 10−33) (Fig. 1b, Extended Data Fig. 2b–f and Supplementary Table 9).
To assess the degree to which ASoC SNPs could help to prioritize GWAS risk variants for LOAD and other neuropsychiatric disorders, we colocalized ASoC SNPs with GWAS index SNPs and their linkage-disequilibrium proxies (r2 ≥ 0.8) (Supplementary Tables 2–7). In total, 29–37% of GWAS risk loci contained at least one disease-associated ASoC SNP (Extended Data Fig. 2g–k and Supplementary Table 7). For LOAD, out of the 26 risk loci that could be functionally interpreted by ASoC SNPs, 20 loci with 38 ASoC/LOAD-risk SNPs were accounted for by iMGs (Extended Data Fig. 2g,l and Supplementary Table 2). To identify the target genes for these ASoC/LOAD-risk SNPs in iMGs, we integrated our iMG ASoC data with the hMG enhancer–promoter linkages defined by activity-by-contact (ABC) analysis6 (Supplementary Table 10). We found that 13 ASoC/LOAD-risk SNPs of 9 risk loci could be assigned to a target gene (Extended Data Fig. 2l), including the two loci with the strongest associations with LOAD: the BIN1 locus for which the functional linkage between rs6733839 and BIN1 has been well established7,23 (Supplementary Fig. 3i) and the PICALM locus with an unknown functional link between rs10792832 and its target gene PICALM (Fig. 1c,d and Extended Data Fig. 2l).
LOAD-risk allele reduces PICALM expression
Among multiple SNPs near PICALM showing the strongest GWAS association with LOAD (Fig. 1c), only rs10792832 showed strong ASoC in an iMG-specific OCR (Fig. 1c,d). Brain hMG24 displayed a similar OCR peak pattern to iMGs at the PICALM locus, with rs10792832 also showing ASoC in hMG but not in other cell types (Extended Data Fig. 3a). To investigate the function of rs10792832, we performed CRISPR–Cas9 editing in human iPS cell lines from two non-AD donors (CD04, CD09), changing the risk genotype (G/G) to the non-risk genotype (A/A) (Fig. 2a), and differentiated them into iMGs (Extended Data Fig. 3b–d). We found that the risk allele reduced PICALM expression by about 50% (Fig. 2b,c and Extended Data Fig. 3e) in iMGs but not in iAst cells (Extended Data Fig. 3f).

a, Schematic of CRISPR–Cas9 editing of the ASoC SNP rs10792832 in iPS cells (two donor lines CD04 and CD09), iMG differentiation and cellular phenotypic assays. The diagram was created using BioRender. b,c, Representative western blot image (b) and quantification (c) of the risk allele reduced PICALM protein expression. Protein quantity was normalized to β-actin. Gel source data are provided in Supplementary Fig. 1. Each data point represents the measurement of cell lysates of 12 wells of cultures from 1 experiment; for each donor line (CD04 and CD09) and each condition (risk and non-risk), data are from 2 clones, collected from 4 independent experiments for clone 1 and 3 independent experiments for clone 2 (that is, n = 7). A linear mixed model (LMM) was used to test the fixed effect of the risk allele, with the experimental round and clone identity used as nested random factors; two-sided test, nominal P values are shown; *P < 0.05, **P < 0.01, ***P < 0.001. Data are mean ± s.e.m. d, The LOAD-risk allele of rs10792832 is predicted to disrupt the PU.1-binding motif (encoded by SPI1; MA0080.2). e, Heterozygous iMGs (A/G) show a higher Sanger-sequencing peak of allele A (non-risk) than G (risk) for the ChIP-assay product of PU.1 binding (top), as opposed to the equal allelic peak height for genomic DNA (bottom).
Next, we examined whether PICALM expression was altered in human LOAD. We first confirmed PICALM expression in IBA1+ MGs in the post-mortem human brain (Extended Data Fig. 4a). At the mRNA level, patients with AD showed a reduction in PICALM in the grey matter (Extended Data Fig. 4b and Supplementary Table 11). RNA-sequencing (RNA-seq) analysis of hMGs6 (Supplementary Table 12) also revealed a reduction in PICALM in AD, with lower PICALM expression significantly associated with the risk allele G of rs10792832 in patients with AD (P = 0.004) (Supplementary Fig. 5a–c). The directional effect of the AD risk allele of rs10792832 on PICALM expression was consistent with other hMG eQTL findings8,9,25. Notably, data mining of single-nucleus RNA-seq (snRNA-seq) datasets from the human prefrontal cortex demonstrated a reduced PICALM expression in MGs (MG0 state) during early AD but increased PICALM expression in MG states during late AD (versus non-AD)26; there was no change in PICALM expression in other brain cell types6 (Supplementary Fig. 5d–l), suggesting possible context-dependent effects (MG state, cell type and AD stage) of the LOAD-risk allele on PICALM expression. Moreover, by recapitulating snRNA-seq analysis of AD brain MG states19, we found lower PICALM expression in disease-associated MGs (DAMs) and LD-accumulating MGs (LDAMs) (versus homeostatic MGs) (P = 0.016) (Extended Data Fig. 4c). Although we did not find a difference in brain microglial PICALM protein levels between patients with AD and controls in a small ROSMAP cohort (Extended Data Fig. 4d–g and Supplementary Table 11), a large proteomics dataset of the dorsolateral prefrontal cortex (n = 516) showed a significant reduction in PICALM in AD27 (Extended Data Fig. 4h). Overall, these results support a brain MG-specific reduction in PICALM expression that may be influenced by rs10792832, consistent with our observed transcriptional effect of the LOAD-risk allele of rs10792832 in iMGs.
The PICALM SNP rs10792832 lies within a predicted binding site of PU.1, a key myeloid-specific TF encoded by a strong LOAD-risk gene, SPI13,28, with the risk allele G predicted to disrupt PU.1’s consensus binding motif (Fig. 2d). To assess how allele G affects PU.1 binding, we performed chromatin immunoprecipitation (ChIP) to enrich for PU.1-bound OCRs in iMGs (Extended Data Fig. 3g–k). We found that the risk allele G decreased PU.1 binding at the SNP site (Extended Data Fig. 3i). We also performed ChIP for PU.1 in iMGs heterozygous for rs10792832 to compare the PU.1 binding between the two alleles within the same sample (Fig. 2e and Extended Data Fig. 3g–k). We found that the risk allele G retained about 40% of the PU.1-binding ability of allele A. Together, these data provide compelling evidence that the LOAD-risk allele of rs10792832 reduces PICALM expression specifically in MGs by disrupting PU.1 binding.
PICALM risk allele impairs iMG phagocytosis
Considering the importance of MG phagocytosis in AD29,30 and the role of PICALM in clathrin-mediated endocytosis in non-MG cells31,32, we hypothesized that the LOAD-risk allele of rs10792832 influences iMG phagocytosis by reducing PICALM expression. We examined the phagocytosis of pHrodo-labelled Aβ aggregates in CRISPR-engineered iMGs and found significantly decreased (40–50%) Aβ–pHrodo fluorescence intensity in iMGs carrying the LOAD-risk (versus non-risk) allele (Fig. 3a and Extended Data Fig. 5a). iMGs from a second clone of each iPS cell line also exhibited similar allelic difference in phagocytic activity (Extended Data Fig. 5b,c).

a, iMGs carrying LOAD-risk allele of PICALM show reduced phagocytosis of Aβ–pHrodo (compared with non-risk-allele iMGs), which can be rescued by PICALM-CRISPRa. The normalized Aβ–pHrodo intensity per iMG over a period of 180 min for both the CD04 and CD09 donor lines is shown. For each condition (genotype), data at each assay timepoint are from two independent experiments each with three single-well measurements (that is, differentiations). One clone per line. b,c, Representative western blot image (b) and quantification (c) of PICALM protein expression in iMGs of non-risk, risk and risk-CRISPRa for both lines. Protein quantity was normalized to β-actin. Gel source data are provided in Supplementary Fig. 1. Each data point represents a measurement of cell lysates from 12 wells of cultures from 1 experiment; for each donor line (CD04 and CD09) and each condition (non-risk, risk and risk-CRISPRa), data were collected from 3 independent experiments. One clone per line. d,e, PICALM KO by CRISPR–Cas9 editing C20 cells. The gRNA targeted at exon 1 depleted PICALM expression in immunoblot (d) and immunofluorescence labelling (e). Three experiments were performed. Scale bar, 50 μm. Gel source data are provided in Supplementary Fig. 1. f,g, Representative images (f) and quantification (g) of the phagocytosis of pHrodo-conjugated myelin in PICALM-KO C20 and control cells. Scale bar, 50 μm. In g, each data point represents a single-well measurement from one experiment (n = 4 wells in each group). Three experiments were performed with similar results. Cytochalasin D (10 µM) was used to show the specificity of phagocytosis. For all comparisons, analysis was performed using one-way analysis of variance (ANOVA) with Dunnett’s correction with the experimental round as the random factor. *P < 0.05; **P < 0.01; ***P < 0.001. Data are mean ± s.e.m.
To evaluate whether the diminished phagocytosis in LOAD-risk-allele iMGs resulted from PICALM haploinsufficiency, we used CRISPRa to overexpress PICALM in iMGs carrying the LOAD-risk allele (Fig. 3b and Extended Data Fig. 5d,e). PICALM-CRISPRa fully rescued the Aβ–pHrodo phagocytic deficit of risk-allele iMGs (Fig. 3a and Extended Data Fig. 5a). Moreover, we performed CRISPRoff in the non-risk-allele iMGs to knockdown PICALM (Extended Data Fig. 5f–e). We found that PICALM-CRISPRoff reduced iMG capacity for phagocytosis of myelin-pHrodo and Aβ–pHrodo compared with the non-risk-allele iMGs (Extended Data Fig. 5h–l). These results support a direct link between reduced PICALM expression and iMG phagocytosis.
To further corroborate the effect of PICALM expression on MG phagocytosis, we conducted independent CRISPR–Cas9 editing on an immortalized hMG cell line (C20)33 to knockout (KO) PICALM (Fig. 3d,e). The loss of PICALM did not perturb the Golgi apparatus (Giantin), the distribution of early endosomes (EEA1), AP-4 containing secretory and endocytic vesicles or the internalization of transferrin and cholera toxin through endocytosis (Supplementary Fig. 6). However, PICALM-KO cells exhibited reduced (about 50%) phagocytosis of myelin-pHrodo compared with the wild-type (WT) cells (Fig. 3f,g), matching the phagocytosis deficit that we observed in risk-allele iMGs (Fig. 3a and Extended Data Fig. 5). These results suggest that impaired MG phagocytosis of fragmented myelin and Aβ aggregates could be a causal mechanism for the LOAD-risk allele at the PICALM locus.
Dysregulated lipid genes in PICALM-deficit iMGs
To investigate the molecular mechanism underlying the observed phagocytosis deficit in iMGs carrying the PICALM risk allele, we performed differential expression analysis in iMGs with the LOAD-risk allele (Fig. 4a). We identified 257 upregulated and 244 downregulated genes (false-discovery rate (FDR) < 0.05) in the LOAD-risk-allele iMGs (Fig. 4b and Supplementary Table 13). The risk allele cis-regulated only PICALM (within 250 kb) with similar effects on major PICALM isoforms (Supplementary Tables 13 and 14). The downregulated genes were enriched for Gene Ontology (GO) terms (such as MHC class II protein complex, antigen presentation and clathrin-coated endocytic vesicle membranes) that are important for MG phagocytosis34 (Supplementary Table 15 and Supplementary Fig. 7a). Notably, among the top 10 enriched GO terms for upregulated genes, four were associated with lipid/cholesterol biosynthesis or metabolism. Notably, more than half of the top 20 differentially expressed genes (DEGs) were related to lipid metabolism (for example, DHCR7, HPGD, FDFT1, HMGCR, PLBD1, IMPA2, MTMR1, RETN, ATP6AP2, LRP5 and CRABP2) (Fig. 4b, Extended Data Fig. 6a–d, Supplementary Fig. 7b and Supplementary Table 13). Among the most enriched canonical pathways (Fig. 4c, Supplementary Fig. 8 and Supplementary Table 16), activated pathways were linked to lipid metabolism, such as cholesterol biosynthesis and gene activation by SREBF (encoding SREBP)35. All genes in the activated cholesterol-biosynthesis pathway were upregulated (Supplementary Figs. 7b and 8) in LOAD-risk-allele iMGs.

a, Principal component analysis (PCA) of RNA-seq samples of iMGs derived from CRISPR-engineered iPS cell lines carrying the PICALM risk or non-risk alleles. Samples are from 2 experiments each with 2–3 wells of differentiations for CD04 and CD09 lines, 1 clone per line. Expression of 13,947 genes was used for PCA. The sample labels describe the cell line, clone number, replicates, and batch number in sequential order, divided by the hyphen (-) symbol. b, DEGs in iMGs carrying the LOAD-risk allele. FC, fold change. c, Enriched Ingenuity canonical pathways for all DEGs (FDR < 0.05). Significantly enriched pathways (one-sided Fisher’s exact test, FDR < 0.05) are ranked by their activated or inactivated (inact.) z scores. TH1, T helper type 1. d, Significant correlation (Pearson’s R2) of the expression changes (−log2[fold change]) in iMGs carrying PICALM risk (versus non-risk) allele and in previously reported LD accumulated microglia (LDAM) of the ageing mouse10. Plotted are 56 DEGs (FDR < 0.1) in both RNA-seq datasets.
Next, we compared the transcriptomic similarity of LOAD-risk-allele iMGs to the LDAMs from ageing mice10. For the overlapping DEGs (n = 56) between the two datasets, the expression changes in the LOAD-risk-allele iMGs and the ageing LDAMs were significantly correlated (r2 = 0.22) (Fig. 4d and Supplementary Table 13). The enrichment of lipid/cholesterol metabolism pathways along with the transcriptomic similarity of PICALM-risk-allele iMGs to mouse LDAMs suggests a possible functional link between PICALM dysregulation and LD formation in MGs.
LD accumulation in PICALM-deficit iMGs
Although PICALM has been shown to be involved in lipid/cholesterol metabolism in Drosophila glia and HEK293 cells13,36, its potential role in LD formation in MGs, especially its link to the LOAD-risk allele, has not been demonstrated. We first used filipin to stain intracellular free cholesterol in iMGs differentiated from the isogenic CRISPR-edited pairs of iPS cell lines (Extended Data Fig. 7a,b). We found stronger filipin fluorescence in iMGs carrying the LOAD-risk (versus non-risk) allele. Cholesterol esters, derived from cellular cholesterol, are often stored in LDs along with triacylglycerols (TGs)37. Although filipin only stains free (unesterified) cholesterol, cellular cholesterol accumulation is associated with excess LD10,37,38. Thus, we treated iMGs with BODIPY to stain lipids in LDs10,14,18,19. We observed a twofold to sevenfold increase in LDs in iMGs carrying the PICALM risk allele (Fig. 5a,b and Extended Data Fig. 7c,d). Treating iMGs with triacsin C (TrC), an inhibitor of long-chain acyl-CoA synthetase that reduces LD formation10,18,19,39,40, effectively decreased BODIPY-labelled LDs (Extended Data Fig. 7e–i). The LD accumulation in PICALM risk-allele iMGs was independently confirmed by flow cytometry analysis of BODIPY+ iMGs (Extended Data Fig. 7j,k) and by staining for the LD-associated protein PLIN2 (Extended Data Fig. 7l–n).

a, Immunofluorescence staining of LD (BODIPY+) in iMGs carrying risk or non-risk allele. C1, clone 1; C2, clone 2. Scale bars, 50 µm. b,c, Quantification of a shows increased LD (BODIPY+) puncta density (b) and area per cell (c) in LOAD-risk-allele iMGs (versus non-risk). Each data point represents a single-well measurement from 1 experiment; for each donor line (CD04 and CD09) and each condition (risk and non-risk), data are from 2 clones, collected from 2 independent experiments each with 3 wells of differentiations (n = 12). d, Dysfunctional lysosomes may contribute to LD accumulation in LOAD-risk-allele iMGs. The log2[FC] of known lysosomal genes and LD suppressor genes in LOAD-risk-allele iMGs or with iMGs with PICALM-CRISPRoff (versus non-risk) is shown. e, Representative images of iMG staining for LD (BODIPY+) and lysosomes (LysoTracker+) in iMGs carrying PICALM non-risk or risk alleles. Scale bars, 50 µm. f,g, Quantification of e for lysosome puncta per iMG (f; LysoTracker+) and colocalized lysosome–LD puncta per iMG (g; BODIPY+LysoTracker+). Each data point represents a single-well measurement from one experiment; for each donor line (CD04 and CD09) and each condition (risk and non-risk), data are from 2 clones, collected from 2 independent experiments each with 3 wells of differentiations (n = 12). In all comparisons, an LMM was used to test the fixed effect of risk allele, with the experimental round and clone identity as nested random factors; two-sided test, nominal P values are shown. *P < 0.05; **P< 0.01; ***P < 0.001. Data are mean ± s.e.m.
To examine whether LD accumulation in the LOAD-risk-allele iMGs was due to decreased PICALM expression, we compared the LD staining in iMGs carrying either the LOAD non-risk or risk allele, as well as PICALM-risk-CRISPRa (Extended Data Figs. 5d,e and 8a,b). PICALM-CRISPRa iMGs fully restored LD levels to the levels of those of non-risk iMGs (Extended Data Fig. 8b). Conversely, PICALM knockdown in non-risk CRISPRoff iMGs (Extended Data Fig. 5f,g) significantly increased LD levels (Extended Data Fig. 9a–e). Independent LD staining using LipidTOX produced similar results (Extended Data Fig. 9f–j). These findings in iMGs were further corroborated by detecting LD accumulation in PICALM-KO C20 cells (Extended Data Fig. 9k,l).
TG elevation is a major characteristic of LDAMs10 and APOE4-associated LDs19. To better understand the PICALM-related LD accumulation, we conducted lipidomic profiling on non-risk, risk and risk-CRISPRa iMGs. Among the 55 lipid species altered in risk-allele iMGs (versus non-risk) (FDR < 0.05), 32 were TGs (5.6-fold enrichment, Fisher’s exact test, P < 2.6 × 10−16) and all were increased (Extended Data Fig. 10a,b and Supplementary Table 17). Total TGs (n = 94 species) displayed an approximately twofold increase in iMGs carrying the risk allele, which was completely reversed by PICALM-CRISPRa (Extended Data Fig. 10b,c). Our results align with previously reported TG increases in LDAMs10 and APOE4-associated LDs19, further supporting our observed PICALM-risk-allele-associated LD accumulation in iMGs.
PICALM has been shown to promote neuron–astrocyte lipid transfer in Drosophila and rat astrocytes13. To test whether lipid accumulation in LOAD-risk-allele iMGs could also be attributed to lipid uptake, we performed a lipid-transfer assay41 in which we co-cultured iMGs with the iGlut neurons that were prelabelled with Red-C12 (Extended Data Fig. 10d). We observed similar levels of Red-C12 staining between iMGs with the LOAD-risk and non-risk alleles (Extended Data Fig. 10e–g). By contrast, consistent with the previously observed effect of PICALM on neuron–astrocyte lipid transfer in rat astrocytes13, iAst cells with PICALM-CRISPRoff exhibited around a 50% reduction in BODIPY-labelled LD and Red-C12-labelled lipids (Extended Data Fig. 10h–l). These results indicate cell-type-specific effects of PICALM on lipid transfer and LD formation in two very different types of glial cells, MG and Ast cells, warranting further studies. These findings also support that reduced PICALM expression by the LOAD-risk-allele leads to MG-specific LD accumulation.
PICALM risk allele increases ROS in iMGs
Elevated reactive oxygen species (ROS) levels in mouse LDAMs are associated with cellular oxidative stress and age-related neurodegeneration10. The PICALM LOAD-risk allele iMGs exhibited transcriptional activation of the SERBP pathway (Fig. 4c and Supplementary Fig. 8), indicating increased ROS levels42. To determine whether the LOAD-risk allele also raises ROS in iMGs, we treated iMG cultures with CellROX, a dye for detecting ROS10, and co-stained the cells with BODIPY (for LDs) (Extended Data Fig. 7e). We observed that LOAD-risk-allele iMGs had a greater than twofold increase in CellROX staining (Extended Data Fig. 7f–i), with largely colocalized CellROX and LD staining (Extended Data Fig. 7e). As seen in mouse microglia10, we further confirmed that LD accumulation influences ROS levels in iMGs by using TrC to block LD formation (Extended Data Fig. 7f–i).
Lipids are often peroxidated in the presence of ROS, mediating cellular oxidative stress42. To examine whether lipids in the LOAD-risk-allele iMGs were also peroxidated, we stained iMGs with BODIPY-C11(581/591) (Extended Data Fig. 11a), a fluorescent lipid peroxidation sensor that shifts its fluorescence emission from red to green when peroxidated lipids are present. We observed that lipids in many LDs were peroxidated (Extended Data Fig. 11b). The LOAD-risk-allele iMGs exhibited threefold more peroxidated lipids compared with non-risk iMGs (Extended Data Fig. 11b–d). The ratio of peroxidated to non-peroxidated lipids in risk-allele iMGs was also higher than in non-risk iMGs (Extended Data Fig. 11e). These results indicate a stress state in iMGs carrying the LOAD-risk allele of PICALM.
LD accumulation impairs iMG phagocytosis
LD buildup impairs phagocytosis in mouse microglia10,43. To validate whether the PICALM-associated LD accumulation also affects phagocytosis in iMGs, we first performed a co-staining of LD-containing (BODIPY+) and phagocytic (Aβ–pHrodo+) iMGs of the non-risk, risk and PICALM-CRISPRa lines (Extended Data Fig. 8a,b). We observed that only 2.4–3.2% of iMGs were BODIPY+Aβ–pHrodo+ across all conditions and an increase in BODIPY+ cells in risk-allele iMGs was proportionally accompanied by a decrease in BODIPY−Aβ–pHrodo+ iMGs (Extended Data Fig. 8c). This inverse relationship between BODIPY+ and Aβ–pHrodo+ signals in iMGs suggests a possible mechanistic link between PICALM-associated phagocytosis deficiency and LD accumulation.
We next conducted a phagocytosis assay for LOAD-risk-allele iMGs in the presence or absence of the LD blocker (TrC)10,18,19,39,40 to confirm whether LD accumulation impairs phagocytosis as seen in mouse microglia10,43. LOAD-risk-allele iMGs showed reduced phagocytosis of Aβ and myelin (Extended Data Fig. 12). As expected, TrC treatment of LOAD-risk-allele iMGs restored phagocytosis to levels seen in non-risk iMGs (note that TrC treatment itself did not significantly increase phagocytosis of non-risk iMGs; Supplementary Fig. 9), which was accompanied by a decrease in LD levels in risk-allele iMGs (Extended Data Fig. 12b,c,f,g). Like in mouse microglia10, there was an inverse relationship between BODIPY+ and Aβ–pHrodo+ signals in iMGs, and TrC treatment of the LOAD-risk-allele iMGs shifted the proportions of cells with varying BODIPY/pHrodo combinations to resemble non-risk iMGs (Extended Data Fig. 12d). Colocalization analysis of LD and phagocytosed myelin in iMGs yielded similar results (Extended Data Fig. 12h). Collectively, these results support the notion that the PICALM-risk-allele-induced LD accumulation in iMGs impairs phagocytosis.
Lysosomal abnormality ties to LD increase
APOE4 dysregulates lysosomal function, contributing to glial lipid/cholesterol accumulation19,38. PICALM also has a role in autophagy and lysosomal function in some non-brain cell lines44,45,46. Given the link between lysosome-based autophagy and LD breakdown47, we predicted that LD accumulation in PICALM LOAD-risk-allele iMGs may result from compromised lysosomal function. We first examined the potential dysregulation of lysosomal genes in LOAD-risk-allele iMGs. Of the 15 lysosomal genes downregulated in APOE4 glia38 (Fig. 5d and Supplementary Table 18), 13 were also downregulated in LOAD-risk-allele iMGs. A consistent expression change was seen in iMGs with PICALM-CRISPRoff (versus LOAD-risk-allele iMGs) (Fig. 5d). Moreover, the downregulated genes in LOAD-risk-allele iMGs were enriched for the lysosomal GO terms (Supplementary Fig. 7a), which shared some downregulated lysosomal genes (VAMP1, CD74, TCRIG1) with mouse LDAMs (Figs. 4d and 5d and Supplementary Fig. 7c). Notably, ATP6AP2, one of the most downregulated genes in risk-allele iMGs (Fig. 4b and Supplementary Table 13), encodes a protein that is essential for lysosomal degradative functions; Atp6ap2-mutant mice show increased triglycerides and LD in fat body cells48. The reduced expression of ATP6AP2, VAMP1 and CD74 in LOAD-risk-allele iMGs was further confirmed independently by immunofluorescence staining (Extended Data Fig. 6c,d). Notably, three known LD suppressor genes (SLC33A1, MCOLN1 and GRN)10 all exhibited reduced expression in LOAD-risk-allele iMGs (Fig. 5d), two of which are related to lysosomal function: MCOLN1 is a ROS sensor in lysosomes regulating autophagy49, and GRN encodes the lysosomal protein progranulin related to neurodegenerative diseases50. The findings suggest that lysosomes in LOAD-risk-allele iMGs may be functionally compromised.
We next assayed whether risk-allele iMGs indeed show accumulation of lysosomes that colocalize with LDs. We co-stained non-risk and risk-allele iMGs with LysoTracker (for lysosomes) and BODIPY (for LDs) (Fig. 5e). We found that risk-allele iMGs had a 2.7–4.2-fold increase of lysosomes, and an even larger increase (4.3 to 5.7-fold) of lysosomes that colocalized with LDs (LysoTracker+BODIPY+) (Fig. 5f,g). Together, these data suggest a potential link between altered lysosomal dynamics and LD accumulation in iMGs carrying the LOAD-risk allele of PICALM (Extended Data Fig. 11f).
Discussion
Our integrative analyses of chromatin accessibility in iPS-cell-derived major brain cell types, brain QTLs, chromatin interactions (ABC) and LOAD GWAS data identified functional LOAD-risk variants that account for about one-third of the known LOAD GWAS risk loci, providing a rich resource for prioritizing functional LOAD-risk variants/genes for biological follow-up. Given that around 70% of ASoC SNPs are in distal enhancers and are likely to be functional, our study presents a framework for prioritizing functional non-coding risk variants through combinatorial analyses of whether a SNP is predicted to alter TF-binding sites and/or is a brain eQTL. Moreover, we mechanistically link the putative LOAD causal SNP at the PICALM locus to its MG-specific effect on PICALM expression, resulting in LD accumulation in iMGs partially through lysosomal dysregulation (Extended Data Fig. 11f). Pharmacological perturbation further validated a causal link between LD accumulation and the impaired phagocytosis in iMGs (Extended Data Fig. 11f). While PICALM is known to be expressed in brain MGs51, and its role in lipid/cholesterol metabolism has been reported in Drosophila glia and HEK293 cells13,36, our work demonstrates a human-MG-specific role for PICALM in regulating LD accumulation and phagocytosis, which potentially contribute to LOAD pathophysiology.
In Drosophila and rat astrocytes, PICALM is required for storing peroxidated lipids sourced from neurons13. Our study indicates that PICALM also regulates MG lipid/cholesterol homeostasis, independent of interactions with neurons. The de novo cholesterol and fatty acid synthesis in LOAD-risk-allele iMGs may contribute to LD accumulation: genes in the cholesterol-biosynthesis pathway, including TFs (such as SREBP) that regulate lipids synthesis35, were all upregulated (Fig. 4b and Supplementary Fig. 8). Lipid processing in PICALM-risk-allele iMGs may be exacerbated by lysosomal dysregulation (Fig. 5d–g). Notably, increased lipid synthesis and lysosomal dysregulation in MGs and Ast cells have also been reported for APOE419,38.
As reported in BV2 microglia10, we confirmed that LD accumulation in PICALM-LOAD-risk-allele iMGs mediates phagocytic deficits and ROS elevation. However, many LDs in iMGs were also peroxidated (Extended Data Fig. 11b–e), suggesting that ROS may also occur first and the peroxidized lipids are sequestered in LDs16,42,52. In glia, the main sources of ROS include peroxisomes and mitochondria that perform lipid β-oxidation52. Notably, PICALM and some other clathrin adaptor proteins are reported to protect against ROS production partially through iron regulation53.
We therefore propose a model that connects the PICALM risk allele to LD accumulation and phagocytosis deficits (Extended Data Fig. 11f): (1) partial loss of PICALM in MGs exacerbates ROS levels (Extended Data Fig. 7e), leading to increased lipid/cholesterol synthesis due to increased SREBP; (2) the lipids are peroxidated and sequestered in LDs, which are initially protective against ROS elevation; (3) ongoing lipid build-up causes cellular stress, further elevating ROS and LD levels; and (4) PICALM loss also impairs proper lysosomal autophagy and LD degradation (Fig. 5d–g), resulting in LD accumulation that hinders phagocytosis. Alternatively, the PICALM risk allele might impair phagocytosis early in ageing, causing reduced cellular lipids/cholesterol, which then activates de novo lipid/cholesterol synthesis. We acknowledge a lack of strong correlation between the magnitude of PICALM alteration and its functional effects in CRISPR-engineered iMGs, probably due to technical variations in iMG differentiation and/or the nonlinear relationship between gene expression and cellular phenotypes due to epistasis and systems buffering54,55. However, we cannot formally exclude a possible long-range cis-effect of the AD-risk variant on genes other than PICALM. Future research will help to clarify how the PICALM risk allele compromises MG function.
Although our study indicates a reduced PICALM expression in LOAD-risk-allele iMGs, PICALM expression in patients with AD may also be influenced by cell type, MG state and disease stage31,56. Moreover, despite the epigenomic similarities between iMGs and hMG, the functional effects of PICALM risk allele in iMGs may also be present in myeloid cells/macrophages, which warrants further investigation. Finally, the impaired phagocytosis in PICALM-risk-allele iMGs implies a neuroprotective role of phagocytosis in clearing Aβ or damaged myelin, but MG phagocytosis in vivo may also be detrimental through excessive synaptic pruning57. Nonetheless, given that the APOE4 allele was recently found to induce LD accumulation in MGs and impair neuronal function18,19, our findings with PICALM further strengthen the notion of a causal role of LDs in mediating LOAD genetic risk factors, providing potential mechanistic targets for AD treatments. Given the reported genetic interaction between the PICALM risk-allele and APOE419,58, it will be interesting to empirically examine possible epistasis between these two AD risk factors.
Methods
Human iPS cell lines and culture
The human iPS cell lines used for ATAC–seq (Supplementary Table 1) were derived at Rutgers University Cell and DNA Repository (RUCDR). The human iPS cell lines were generated using the Sendai virus method to ensure that they are integration-free and underwent the following quality-control procedures: immunofluorescence staining for pluripotency, mycoplasma contamination test, in-house RNA-seq-based pluripotency test (Pluritest) and eSNP-karyotyping20,21 or G-band karyotyping at RUCDR. All donors were of European ancestry and the samples were previously used for schizophrenia GWAS studies59,60. All donors were also analysed for copy-number variants, and none had large copy-number variants (>100 kb)61. There are 29 schizophrenia cases and 33 controls, of which 37 are male with an average age of 49.5 years (schizophrenia case–control status or age does not affect ASoC mapping20,21). Two control-donor human iPS cell lines homozygous for APOE3, CD04 and CD09 (abbreviated from the full cell line IDs: CD0000004 and CD0000009) were used in CRISPR–Cas9 editing. Human iPS cells were cultured using a feeder-free method on Matrigel-coated (Thermo Fisher Scientific) plates in mTeSR plus medium (100-0276, StemCell). The media were changed every other day, and cells were passaged as clumps every 4–6 days using ReLeSR (100-0483, StemCell). All cell cultures were confirmed to be mycoplasma-free using the PCR detection kit (ab289834, Abcam). Human iPS cell lines were obtained from the RUCDR NIMH Stem Cell Center (www.nimhgenetics.org). These iPS cell lines were generated from cryopreserved lymphoblasts deposited by the Molecular Genetics of Schizophrenia (MGS) consortium59,60, which had collected the biomaterials with informed consent as approved by the Endeavor Health (formerly NorthShore University HealthSystem) institutional review board (IRB), which also approved the current study.
PICALM RNA expression in human post-mortem brains
Frozen human brain samples (frontal cortex BA10 region) were received through the NIH biobank from Harvard Brain Tissue Resource Center and the University of Miami Brain Endowment Bank. For the PICALM expression assay, the grey matter was dissected from brain blocks on dry ice. RNA was isolated using the Direct-zol RNA MiniPrep Kit (Zymo), and reverse-transcribed into complementary DNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the manufacturer’s instructions. Quantitative PCR (qPCR) reactions were set up using PowerUp SYBR Green Master Mix for qPCR (Applied Biosystems) and run on the QuantStudio Real-Time PCR System (Applied Biosystems). The data were analysed using the \({2}^{-\Delta \Delta {C}_{t}}\) method and normalized to COTL1. Primer sequences were as follows: PICALM-exon-1-F, TCTGCCGTATCCAAGACAGT; PICALM-exon-2-R, AAGACCACCACCCAACTACT; COTL1-F, CCAAGATCGACAAAGAGGCTT; COTL1-R, CGATGGTGGAGCCGTCATATTT.
PICALM staining of hMGs
Paraffin sections (6 μm) from post-mortem human brain tissue were obtained from UCLA or RUSH Alzheimer’s Disease Center (RADC) repositories. The RADC brains came from the Religious Orders Study and Rush Memory and Aging Project (ROSMAP)62. All ROSMAP participants enrolled without known dementia and agreed to detailed clinical evaluation and brain donation at death. Both studies were approved by the Rush University Medical Center IRB. The sections were incubated at 60 °C for 60 min, deparaffinized in xylene and rehydrated through a series of increasingly dilute ethanol solutions. Epitope retrieval was performed using decloaking buffer (Biocare Medical) at 95 °C for 30 min. Subsequently, freshly prepared 0.1% sodium borohydride was added to the sections and incubated at 4 °C for 30 min. Peroxidase blocking (3% H2O2) was carried out at room temperature for 30 min. The sections were then treated with permeabilization buffer (0.2% Triton X-100 in 1× TBS) at room temperature for 45 min. Non-specific epitopes were blocked by incubating the sections for 60 min in blocking buffer-I (10% donkey serum, 1% BSA, 0.3 M glycine, 0.1% Triton X-100) followed by a 60 min incubation with background punisher (Biocare Medical). Antibody staining was performed at room temperature using the intelliPATH FLX system and reagents provided by the manufacturer (Biocare Medical). Rabbit anti-PICALM antibodies (Sigma-Aldrich, HPA019061, 1:200) and goat anti-IBA1 (Abcam, ab5076, 1:1,000) were incubated with the sections for 48 h at 4 °C. After washing, secondary antibodies were applied and incubated overnight at 4 °C. Autofluorescence was quenched using 0.1% Sudan Black B in 70% ethanol, and nuclei were stained with Hoechst 33342. Finally, coverslips were mounted using VectorShield mounting medium. Images were acquired on an automated Nikon Eclipse Ti2 microscope fitted with the Yokogawa spinning-disk field-scanning confocal system and Photometrics PRIME 95B sCMOS camera, using a ×20 objective. z stack images were deconvolved using Nikon NIS-Elements AR5.20.01 software and processed with Fiji/ImageJ (1.54f, 64 bit). For quantification of PICALM in IBA1+ microglia, all processing was conducted by a researcher blinded to individual diagnoses. Images were acquired on the SLIDEVIEW VS200 slide scanner (Olympus) using ×20 magnification. Individual microglia were cropped using OlyVIA software (Olympus). All further processing was conducted in FiJi/ImageJ (1.54f, 64 bit). Background fluorescence was measured from non-specific tissue staining and normalized by subtracting 50% of the measured values. Binary masks of microglia were generated by thresholding the IBA1 signal, and manually inspected to remove non-specific signals or adjacent cells. PICALM signal was multiplied by the microglial masks, and fluorescence intensities (integrated densities) were measured. Data were plotted and analysed using Prism 10 (GraphPad).
iMG differentiation from human iPS cells
iMGs were generated from human iPS cell lines as described in our previous study63 using the Brownjohn’s method64. At least 2 days after passaging, when human iPS cells reached about 80% confluency, they were dissociated with Accutase (07920, StemCell) and plated at 10,000 cells per well in 96-well round-bottom ultra-low-attachment plates (7007, Corning) in 100 µl embryoid body (EB) medium (complete mTeSR with 50 ng ml−1 BMP-4 (120-05, PeproTech), 20 ng ml−1 SCF (300-07, PeproTech), 50 ng ml−1 VEGF-121 (100-20 A, PeproTech) and ROCK inhibitor (1254/1, R&D Systems)). Haematopoietic medium was prepared by adding to the X-VIVO 15 (BE08-879H, Lonza), 1% GlutaMax (35050061, Thermo Fisher Scientific), 1% penicillin–streptomycin (10378016, Thermo Fisher Scientific), 55 µM β-mercaptoethanol (21985023, Thermo Fisher Scientific), 100 ng ml−1 M-CSF (300-25, PeproTech) and 25 ng ml−1 IL-3 (200-03, PeproTech). After 5 days of culturing EBs in haematopoietic medium, primitive macrophage progenitors (PMPs) started appearing in the suspension and were produced continuously in suspension for 34 days. After 10 days of culturing EBs, PMPs were collected from suspension and plated in RPMI 1640 medium (21870076, Thermo Fisher Scientific) at 180,000 cells per cm2 in 6- or 12-well plates. Complete iMG medium was as follows: RPMI 1640 with 10% FBS (S11150H, R&D Systems), 1% penicillin–streptomycin, 1% GlutaMax, 100 ng ml−1 IL-34 (200-34, PeptroTech) and 10 ng ml−1 GM-CSF (300-03, PeproTech). The final differentiation of PMPs into iMGs occurred over 25 days.
iAst cell differentiation from NPCs
iPS-cell-derived neural progenitor cells (NPCs) were prepared using PSC neural induction medium (A1647801, Thermo Fisher Scientific). NPCs were differentiated to astrocytes by seeding dissociated single cells at 15,000 cells per cm2 density on Matrigel-coated plates in astrocyte medium (1801, ScienCell: astrocyte medium, 2% FBS (0010), astrocyte growth supplement (1852) and 10 U ml−1 penicillin–streptomycin solution (0503)). The initial NPC seeding density and single-cell dissociation are critical, particularly during the first 30 days of differentiation, to efficiently generate a homogenous population of astrocytes. On day −1, NPCs were pipetted with a p1000 pipette 3–5 times to yield a single-cell suspension and inhibit cell death. The NPC medium was switched to the astrocyte medium on day 0. From day 2, cells were fed every 48 h for 20–30 days. After 30 days of differentiation, astrocytes were split 1:3 weekly with Accutase and expanded for up to 120 days (15–17 passages) in the astrocyte medium. The final differentiation of iAst cells occurred over 30 days.
Differentiation of glutamatergic neurons
We followed an established protocol to differentiate iPS cells into glutamatergic neurons (iN-Glut)65. In brief, iPS cells were dissociated into single cells using Accutase (07920, StemCell) and replated at 7.5 × 105 cells per well in a six-well plate in mTeSR plus medium (100-0276, StemCell) with 5 μM ROCK inhibitor (1254/1, R&D Systems) on day −1. On day 0, cells were infected with 200 μl per well lentivirus cocktail containing 100 μl NGN2 virus and 100 μl rtTA virus65. After a two-day puromycin selection, iGlut cells on day 5 were dissociated with Accutase and plated as a 100 μl blob on glass coverslips (GG-12-15-Pre, Neuvitro). From day 6 onwards, 500 μl of neuronal culture medium was added into each well with a half-volume medium change every 3 days. Doxycycline was withdrawn on day 21 of differentiation. The final differentiation of iN cells occurred over 30 days.
Differentiation of dopaminergic neurons
The protocol for the differentiation of dopaminergic neurons (iDNs) was adapted from a previous study66. In brief, dopaminergic priming medium was added to the cells at 50% confluence on day 0. On day 7, the cells were replated onto six-well plates coated with Matrigel at 5 × 105 cells per well and switched to dopaminergic differentiation medium. The medium was changed every other day. On day 30, dopaminergic neurons were collected using Accutase (07920, StemCell) for ATAC–seq.
Differentiation of GABAergic neurons
We generated GABAergic neurons (iN-GA) from NPCs using the protocol described previously67 but with NPCs as the source cells. NPCs were seeded at 200,000 cells per cm2 on day 0. Virus neural expansion medium cocktail was added on day 1 with ASCL1-puro and DLX2-hygro virus and replaced by the expansion medium cocktail containing 2 µg ml−1 doxycycline (D9891, Sigma-Aldrich) the same day. Puromycin and hygromycin selection were conducted between day 2 and day 6. On day 7, we switched the medium to conditioned NeuralbasalPlus medium and changed the medium every 3 days. Doxycycline was withdrawn on day 16, and 50 nM Ara-C was included in the medium if non-neuronal cells were observed. On day 28, neurons were collected using Accutase for ATAC–seq.
Immunofluorescence staining of iMG and iAst cells
For characterizing iMG and iAst cells, cells were fixed in 4% PFA (P6148, Sigma-Aldrich) for 10 min at room temperature, followed by incubation with primary antibodies at 4 °C overnight in 3% BSA containing 0.3% Triton X-100. Cells were washed three times in PBS, and then incubated with secondary antibodies at room temperature for 1 h in 3% BSA containing 0.3 % Triton X-100. Next, the cells were washed another three times with PBS and incubated in 0.5 μg ml−1 4′,6-diamidino-2-phenylindole (DAPI) at room temperature for 10 min. Images were acquired using the Nikon ECLIPSE TE2000-U microscope.
Primary antibodies used for microglial immunofluorescence and their dilutions for incubation were as follows: rat anti-TREM2 (MABN755, Sigma-Aldrich, 1:100), rabbit anti-CD45 (SAB4502541, Sigma-Aldrich, 1:200), mouse anti-PU1 (89136, Cell Signalling, 1:100), mouse anti-IBA1 (MA5-27726, Thermo Fisher Scientific, 1:100), rabbit anti-P2Y12 (702516, Invitrogen, 1:200), rabbit anti-TMEM119 (AB209064, Abcam, 1:100), rabbit anti-PLIN2 (15294-1-AP, Proteintech, 1:200), rabbit anti-ATP6AP2 (SAB2702080, Sigma-Aldrich, 1:100), rabbit anti-VAMP1 (702787, Thermo Fisher Scientific, 1:100), rabbit anti-HMGCR (SAB4200528, Sigma-Aldrich, 1:100) and mouse anti-CD74 (14-0747-82, Thermo Fisher Scientific, 1:100). Secondary antibodies were Alexa 488 donkey anti-rat (A21208, Invitrogen, 1:1,000), Alexa 594 anti-rabbit (A21207, Invitrogen, 1:1,000), Alexa 647 anti-mouse (A32787, Invitrogen, 1:1,000), Alexa donkey 594 anti-mouse (A21203, Invitrogen, 1:1,000) and Alexa donkey 647 anti-rabbit (A32795, Invitrogen, 1:1,000). Primary antibodies used for iAst immunofluorescence and their dilutions for incubation were rabbit anti-vimentin (3932, Cell Signaling, 1:200), mouse anti-GFAP (G3893, Sigma-Aldrich, 1:100) and mouse anti-s100β (S2532, Sigma-Aldrich, 1:100). Secondary antibodies were Alexa 488 donkey anti-rabbit (A21206, Invitrogen, 1:1,000) and Alexa 594 anti-mouse (A21203, Invitrogen, 1:1,000).
RNA isolation and sequencing
Cells from human iPS, iAst and iMG cell cultures were dissociated using Accutase (07920, StemCell). Total RNAs were extracted using the RNeasy Plus Kit (74134, Qiagen). cDNAs were reverse transcribed from RNAs using a high-capacity cDNA reverse transcription kit (4368814, Applied Biosystems). RNA-seq was performed by Novogene on the Illumina NovaSeq 2000 platform with targeted 30 million paired-end reads (2 × 150 bp) per sample.
RNA-seq data and differential expression analyses
Raw FASTQ files were aligned to the human hg38 genome GRCh38.p14 using STAR v.2.7.2 and counted according to GENCODE annotation release version 35 on the fly. The ComBat-seq function from the R package sva was applied to remove batch effects between the two experiments (that is, rounds of iMG differentiation). We subsequently used the R package EdgeR (v.4.0.16) to calculate counts per million values from sva-corrected read counts for PCA analysis and plotting. Differential gene expression analysis was performed by applying general linear models and F tests between different groups (risk versus non-risk) (glmQLFit and glmQLFtest functions). When constructing the generalized linear models, we considered cell line ID (CD04 and CD09) as a coefficient to further remove line-specific effects. DEGs were defined as their Benjamini–Hochberg-adjusted P values (FDR) < 0.05.
ATAC–seq
ATAC–seq sample preparation was performed as previously described20,21. In brief, 75,000 viable cells were used for each transposition mixture reaction. The samples were then incubated at 37 °C for 30 min on a thermomixer at 1,000 rpm. The eluted DNA was shipped to the University of Minnesota Genomic Center for library preparation and ATAC–seq.
ATAC–seq data analysis and peaking calling
All raw sequence reads generated by Illumina NextSeq were demultiplexed at the University of Minnesota Genomics Center and provided as 2 × 75 bp paired-end FASTQ files (targeting 60 million reads per sample). Only paired-end reads that survived Trimmomatic processing v.0.39 (ILLUMINACLIP:NexteraPE-PE.fa:2:30:7, SLIDINGWINDOW:3:18, MINLENGTH:26) were retained. The FASTQ files were individually mapped against the human genome reference file including decoy sequences (GRCh38p7.13/hg38, 1000 Genome Project) using bowtie2 (-x 2000, -mm --qc-filter --met 1 --sensitive --no-mixed -t) and subsequently merged and sorted as BAM-formatted files using samtools v.1.14, with only uniquely high-quality mapped reads (MAPQ > 30, SAM flags 0×1, 0×2) retained. Picard tools MarkDuplicate was then used to remove all PCR and optical duplicated reads from the BAM file.
To further eliminate allelic bias towards reference alleles during the alignment step, we performed WASP (v.0.3.4) calibration on the generated raw BAM files68. In brief, we first called the VCF file profiles on all SNP variants per sample individually using GATK HaplotypeCaller to generate cell-line-specific VCF files. The cell-line-specific VCFs were used as the basis of WASP calibration and realignment, and new WASP-calibrated BAM file sets were collected as the final output for the ATAC–seq peak calling and ASoC SNP calling (see below). All analysed ATAC–seq samples passed standard quality control based on the characteristic nucleosomal periodicity of the insert fragment size distribution and high signal-to-noise ratio around transcription start sites (TSSs). For peak calling, MACS269 was used to generate peak files (narrowPeak format) with the recommended settings at FDR = 0.05 (-f BAMPE, --nomodel, --call-summits --keep-dup-all -B). Peaks that fell within the ENCODE blacklisted regions were removed. We also removed peaks within chromosomes X and Y, mitochondrial genome and decoy regions.
ASoC mapping
The ASoC approach20,21 was used to identify putative functional variants that showed differential chromatin accessibility between the two alleles of a SNP in ATAC–seq samples heterozygous for the tested SNP, with the assumption that a functional common GWAS SNP does not display monoallelic chromatin accessibility. In brief, GATK (v.4.1.8.1) was used for SNP calling as recommended by the GATK Best Practices (https://gatk.broadinstitute.org/hc/en-us/sections/360007226651-Best-Practices-Workflows)70. As noted above, WASP-calibrated BAM files were used as input and variants were called against the human GRCh38.p14 (hg38) reference genome and the corresponding dbSNP version 154, and only reads with MAPQ score ≥ 30 were used (-stand_call_conf 30). Subsequently, recalibration of SNPs and indels was performed in tandem using the VariantRecalibrator function (-an DP -an QD -an FS -an SOR -an MQ -an ReadPosRankSum -mode SNP -tranche 100.0 -tranche 99.5 -tranche 95.0 -tranche 90.0) and scores were recalibrated using reference database including HapMap v.3.3 (priority = 15), 1000G_omni v2.5 (priority = 12), Broad Institute 1000G high-confidence SNP list phase 1 (priority = 10), Mills 1000G golden standard INDEL list (priority = 12) and dbSNP v154 (priority = 2). Heterozygous SNP sites with tranche level >99.5% were extracted. To reduce bias introduced by any acquired (or de novo) mutations during cell growth, only SNPs with corresponding rs# records found in dbSNP v154 were retained. Biallelic SNP sites (GT: 0/1) with minimum read depth count (DP) ≥ 20 and minimum reference or alternative allele count ≥ 2 were retained. The binomial P values (non-hyperbolic) were calculated using binom.test(x, n, P = 0.5, alternative = “two.sided”, conf.level = 0.95) from the R package, and Benjamini–Hochberg correction was applied to all qualified SNPs as the correcting factor of the R function p.adjust(x, method = “fdr”). We set the threshold of ASoC SNP at an FDR value of 0.05. To ascertain whether rs10792832 also exhibits ASoC in human brain MG, we first converted the snATAC–seq data of hMG24 into pseudo-bulk ATAC–seq data for each sample. We then used GATK for SNP calling to identify individuals heterozygous (A/G) for rs10792832 in hMG, followed by ASoC testing as described above.
The read pileup statistics proximal to SNP sites were generated using samtools mpileup function, and differential allele-specific reads was performed using the SNPsplit Perl package (v.0.3.2) (www.bioinformatics.babraham.ac.uk/projects/SNPsplit). The final readouts from both read pileup and SNP-specific reads were visualized using the R package Gviz. Moreover, when comparing the changes in chromatin accessibility caused by genotypes across samples or between different cell types, read counts were scaled and normalized using the deepTools package (v.2.0) bamCoverage function and re-scaled to reads per genomic content as the base unit71. We confirmed there was no obvious mapping bias to reference alleles by visualizing the volcano plots that graph the allelic read-depth ratios against −log2[P] values in scatter plots.
sLDSC analysis of GWAS enrichment
Stratified linkage disequilibrium score regression (sLDSC)72 analysis was performed using the hg38 version of European genotype data (SNPs) from 1000 Genomes Phase 3 and v.2.2 baseline linkage disequilibrium/weights as previously described20. In brief, linkage disequilibrium score estimations were precalculated from the hg38 version of the 1000 Genomes EUR file set (w_hm3_no_hla.snplist), with a window size of 1 cM (ld-wind-cm 1). We used the GWAS summary statistics of major psychiatric disorders and non-psychiatric diseases (Supplementary Table 9) for partition heritability, with several datasets lifted over from hg19 to hg38 when necessary. Disease-specific regressions were performed using hm3 SNP weights against each disease independently for cell-type-specific analysis.
Torus GWAS enrichment analysis
Bayesian hierarchical model (TORUS) was applied to perform an SNP-based enrichment analysis to explore whether ASoC SNPs are enriched in any of the diseases73 as previously described20. For the GWAS enrichment test, ASoC SNPs derived from each cell type were applied independently. The annotations are encoded as Boolean (true if an SNP has an annotation). The GWAS datasets used for enrichment/TORUS analysis were consistent with the diseases analysed in sLDSC. A univariate analysis was performed to assess the enrichment of ASoC SNPs in each GWAS dataset.
CRISPR–Cas9 editing of human iPS cells
CRISPR guide RNA (gRNA) sequences were designed as described74, and we selected the gRNAs with the highest scores for specificity (Supplementary Table 19). The gRNAs were cloned into the pSpCas9(BB)-2A-Puro vector (Addgene, 62988) for co-expression with Cas9 based on an established protocol75. For transfection, 3 μg of CRISPR–Cas9–gRNA construct was combined with 3 μg of ssODNs (1:1 ratio) in Opti-MEM medium (31985062, Thermo Fisher Scientific) and Lipofectamine stem reagent (STEM00001, Thermo Fisher Scientific) was used for transfection. Puromycin selection was performed to eliminate untransfected cells and was withdrawn after 72 h of transfection. Resistant colonies were collected 14 days after transfection and a small amount of DNA from each colony was used for Sanger sequencing to verify editing. The purity of the selected clones was confirmed for on-target editing and the absence of off-target editing (see below).
Quality control of the CRISPR-edited iPS cell lines
Primers were designed to amplify regions corresponding to the four top-ranking predicted off-targets to check on-target and off-target editing. A list of all primer and oligo sequences is provided in Supplementary Table 19. To confirm the pluripotency of CRISPR–Cas9-edited human iPS cell lines, the cells were stained with pluripotency marker antibodies: rabbit anti-OCT4 (ab181557, Abcam, 1:250), goat anti-NANOG (AF1997-SP, R&D Systems, 1:50) and mouse anti-SSEA4 (ab16287, Abcam, 1:250). Images were taken using a Nikon ECLIPSE TE2000-U microscope.
eSNP-karyotyping was performed for all cell lines used to eliminate potential chromosomal abnormalities, as previously described20,21. RNA-seq data were processed using the eSNP Karyotyping package76 rewritten for GATK v.4 and R v.4.2, using raw FASTQ files as the input. Alignment to the human hg38 genome was performed by Bowtie2 v.2.5.1, and only common SNPs (MAF > 0.05) from dbSNP 154 were retained for zygosity block analysis. The plotted zygosity block size was 1.0 Mb.
CRISPRoff epigenome editing of human iPS cells
CRISPRoff77 was used to repress PICALM expression in iMGs. The gRNA sequences were designed using Benchling (https://benchling.com; Supplementary Table 19), and cloned into the CROPseq-Guide-Puro vector (Addgene, 86708) for co-expression with CRISPRoff v.2.1 (Addgene, 167981) as described previously77. After 72 h of drug selection, transduced cells were sorted using a BD FACSAria II, and the sorted cells were passaged twice and then differentiated into iMGs.
CRISPRa to overexpress PICALM
We used CRISPR-ERA78 to design PICALM activation gRNA. A total of four gRNA candidates close to the TSS of PICALM with low predicted off targets and E scores were selected and cloned into the lentiviral gRNA vector lentiGuide-Hygro-mTagBFP2 (Addgene, 99374) through Gibson assembly. We then co-transfected gRNA plasmids with lenti-EF1a-dCas9-VPR-Puro (Addgene, 99373)79 into HEK293T cells using Fugene HD (Promega) to evaluate the PICALM activation efficiency of each CRISPRa gRNA and selected one gRNA with the highest activation efficiency for further establishing inducible CRISPRa iPS cell line that can overexpress PICALM. To establish inducible CRISPRa iPS cell line on risk allele background, we first transduced lentivirus for the lentiGuide-Hygro-mTagBFP2 carrying the selected PICALM gRNA and sorted BFP-positive cells to enrich the cells with high expression of gRNAs. After sorting, iPS cells were maintained in 100 µg ml−1 hygromycin to enrich cell stably expressing the gRNAs. Then cells were transduced with the lentivirus of inducible dxCas9-VPR expressing vector pLenti-tetON-dxCas9(3.7)-VPR-EF1a-TagRFP-2A-tet3G (Addgene, 167937). BFP- and RFP-positive cells were sorted to enrich iPS cells with high expression of dxCas9-VPR. The sorted iPS cells were maintained with hygromycin and expanded for iMGs differentiation, followed by CRISPRa induction of PICALM expression by treating cells with 2 µg ml−1 doxycycline for 25 days before collecting iMGs for analyses. The four tested gRNAs are gRNA 3-GAGTTCCATCACGTAACGCG, gRNA 4-GCCTCAGGCGACCTGTTGGC, gRNA 6-GCAGTGTCAACGTCTTTCCA, and gRNA 7-GGGCGGGCGTCGAAGAGGAA (the best-performing one used in CRISPRa in iMGs).
Gene expression analysis by qPCR
For qPCR, reverse transcription was performed using the Thermo Fisher Scientific High-capacity RNA-to-cDNA reverse transcription kit (4366596, Applied Biosystems) with random hexamers according to the manufacturer’s protocol. qPCR was performed using the TaqMan Universal PCR Master Mix (4364338, Applied Biosystems) on the Roche 480 II instrument (with Roche LightCycler 480 1.5.1), using gene-specific FAM-labelled TaqMan probes or custom-designed probes from IDT (Supplementary Table 19). GAPDH was used as the control.
Myelin isolation from mouse brains for phagocytosis
Myelin was isolated from mouse brains by homogenization in 0.32 M sucrose buffer (0.32 M sucrose and 2 mM EGTA). The samples were then further homogenized using a Dounce homogenizer, layered on top of 0.85 M sucrose buffer (0.85 M sucrose and 2 mM EGTA), and centrifuged at 75,000g at 4 °C for 30 min. Crude myelin was collected from the interface, resuspended in Tris-Cl Buffer (0.2 M Tris-HCl, pH 7.5), and homogenized using a Dounce homogenizer. The samples were centrifuged at 75,000g at 4 °C for 15 min. The pellets were resuspended in Tris-HCl solution (20 mM Tris-HCl, 2 mM EDTA, 1 mM DTT, pH 7.5) and homogenized using a Dounce homogenizer. The samples were then centrifuged at 12,000g at 4 °C for 15 min, and the pellets were resuspended in Tris-Cl Solution. The samples were then centrifuged at 12,000g at 4 °C for 10 min. Pellets were resuspended in 0.32 M sucrose buffer, layered on top of 0.85 M sucrose buffer and centrifuged at 75,000g at 4 °C for 30 min. Purified myelin was collected from the interface, resuspended in Tris-HCl buffer, and homogenized using a Dounce homogenizer. The samples were next centrifuged at 75,000g at 4 °C for 15 min, and the pellets were resuspended in Tris-HCl solution and homogenized using a Dounce homogenizer. The samples were then centrifuged at 12,000g at 4 °C for 15 min, and the pellets resuspended in Tris-HCl solution and centrifuged at 12,000g at 4 °C for 10 min. The pellets were resuspended in Tris-HCl Solution, aliquoted and stored at −80 °C. The protein content of isolated myelin was determined using the BCA protein assay kit.
Phagocytosis assay for iMGs
iMGs were grown on MatTek 96-well plates with a glass bottom (NC1844174, Thermo Fisher Scientific) until day 25. For Aβ phagocytosis, the β-amyloid (1-42) aggregation kit was used (A-1170-025, rPeptide). The peptide was resuspended in 5 mM Tris at 1 mg ml−1 concentration. Myelin and Aβ peptides were labelled using the pHrodo Red Microscale Labelling Kit (P35363, Thermo Fisher Scientific) and pHrodo Deep Red Labelling Kit (P35355, Thermo Fisher Scientific) according to the vendor’s protocol.
For pHrodo phagocytosis experiments, pHrodo-labelled myelin or Aβ was diluted to 15 µg ml−1 in RPMI 1640 medium (21870076, Thermo Fisher Scientific), bath sonicated for 1 min and added to the iMGs along with CellMask Green Plasma Membrane Stain (C37608, Thermo Fisher Scientific, 1:1,000) and NucBlue Live ReadyProbes Reagent (R37605, Thermo Fisher Scientific, 2 drops per ml), mixed gently and incubated at 5% CO2 and 37 °C for 30 min. As a negative control, 10 µM cytochalasin D (PHZ1063, Thermo Fisher Scientific) was added to cells along with pHrodo-labelled protein and retained throughout uptake assays. Live imaging (5% CO2, 37 °C) was performed for 3 h using the Nikon ECLIPSE TE2000-U microscope at 45 min intervals.
For the pHrodo phagocytosis experiment that included LD staining, the iMGs were treated with 1 µM TrC (10007448, Cayman Chemical) in complete iMG medium for 18 h. Next, BODIPY 493/503 (D3922, Thermo Fisher Scientific, 1:1,000) and CD45 antibodies (14-0451-82, Thermo Fisher Scientific, 1:500) were added to cells with and without TrC and incubated for 30 min and quickly washed twice with RPMI 1640. Then pHrodo-labelled myelin or Aβ was diluted to 15 µg ml−1 in RPMI 1640 medium, bath sonicated for 1 min and added to the iMGs along with NucBlue Live ReadyProbes Reagent and incubated at 5% CO2, 37 °C for 30 min. As a negative control, 10 µM cytochalasin D was added to cells along with pHrodo-labelled protein and retained throughout uptake assays. Live Imaging (5% CO2, 37 °C) was performed for a total of 3 h using Nikon ECLIPSE TE2000-U microscope at 45 min intervals. Fiji/ImageJ (v.1.54f, 64 bit) software was used to quantify pHrodo fluorescence intensity (https://fiji.sc).
ChIP
The ChIP–qPCR assay was performed by combining two protocols from the Simple ChIP Enzymatic Chromatin IP kit (91820, Cell Signaling) and the Magna ChIP A/G Chromatin Immunoprecipitation kit (17-10085, Sigma-Aldrich). In total, 107 cells were used for each reaction with 1% formaldehyde (28908, Thermo Fisher Scientific) cross-linking in 20 ml of cell suspension. The Cell Signaling IP kit was used for nuclei preparation and subsequent recovery reactions according to the vendor’s protocols. For chromatin digestion, 1.25 µl of micrococcal nuclease was used and incubated for 20 min at 37 °C to digest DNA to the length of approximately 150–900 bp. To break the nuclear membrane, lysate was sonicated for three sets of 20 s pulses with a 1/8-inch probe.
The Sigma-Aldrich ChIP Assay kit was used for the reaction according to the vendor’s instructions. Normal rabbit IgG (2729, Cell Signaling) was used as negative control. In total, 1 µl of proteinase K was used for reverse cross-linking of protein–DNA complexes to free DNA at 62 °C for 2 h with shaking, followed by incubation at 95 °C for 10 min. For each reaction, DNA was eluted into 50 µl of elution buffer C. qPCR was performed using TaqMan Universal PCR Master Mix (4364338, Applied Biosystems) on the Roche 480 II instrument, using IDT custom probe for detection PICALM/PU.1 ratio (Supplementary Table 19). The ChIP DNA product for the heterozygous site rs10792832 was also subjected to Sanger sequencing (Thermo Fisher Scientific, Sequencing Analysis Software 7.0.0). Genomics DNAs for the heterozygous site were sequenced by using primers used for on-target CRISPR–Cas9 editing of rs10792832.
Fatty acid (Red-C12) transfer assay
iMG and iAst cells were grown on coverslips until day 25. Neurons (iNs) were grown on coverslips until day 30. Cells were incubated with 8 μM BODIPY 558/568 (Red-C12, D3835, Thermo Fisher Scientific) for 16 h in neuronal growth medium, washed twice with warm PBS and incubated with fresh medium for 1 h. Red-C12 labelled neurons and unlabelled astrocytes/microglia were washed twice with warm PBS, and the RedC12 intensity was examined using fluorescence microscopy.
LD staining with BODIPY for iMGs
iMGs were grown on glass coverslips until day 25. Cells were then fixed for 30 min at room temperature with 4% PFA (P6148, Sigma-Aldrich) in PBS, briefly washed in PBS twice and incubated in PBS with BODIPY 493/503 (D3922, Thermo Fisher Scientific, 1:1,000 from 1 mg ml−1 stock solution in DMSO) and DAPI for 10 min at room temperature. The BODIPY intensity was examined by fluorescence microscopy.
LD staining with LipidTOX for iMGs
iMGs were grown on glass coverslips until day 25. Cells were then fixed for 30 min at room temperature with 4% PFA (P6148, Sigma-Aldrich) in PBS, briefly washed in PBS twice and incubated in PBS with LipidTOX (H34476, Thermo Fisher Scientific, 1:1,000) and DAPI for 1 h at room temperature. The LipidTOX intensity was examined using fluorescence microscopy.
ROS staining
iMGs were grown on glass coverslips until day 25. Cells were treated with 1 µM TrC (10007448, Cayman Chemical) in complete iMG medium for 18 h. Cells were subsequently incubated in complete iMG medium with CellROX Deep Red (C10422, Invitrogen, 1:500) for 30 min at 37 °C. Next, the cells were stained with BODIPY to detect LDs. The CellROX intensity was examined by fluorescence microscopy.
Lipid peroxidation assay using BODIPY C11
iMGs were grown on glass coverslips until day 25. Cells were treated with 1 µM TrC as described in ROS staining. Next, cells were incubated in complete iMG medium with BODIPY 581/591 C11 (D3861, Thermo Fisher Scientific, 1:1,000) for 15 min at 37 °C, then fixed for antibody staining by rat anti-TREM-2 (MABN755, Sigma-Aldrich, 1:100). Five hundred and sixty-eight nm excitation wavelength was applied to excite reduced BODIPYC11 and 488 nm excited oxidized BODIPYC11. Fiji/ImageJ (v.1.54f 64 bit) software was used to quantify BODIPYC11 fluorescence intensity (https://fiji.sc).
Filipin staining for iMGs
iMGs were grown on glass coverslips until day 25. Cells were then fixed for 10 min at room temperature with 4% PFA (P6148, Sigma-Aldrich) in PBS. Cells were incubated with a solution of filipin (0.1 mg ml−1, F‐9765, Sigma-Aldrich) for 30 min. After staining with were rat anti-TREM-2 (MABN755, Sigma-Aldrich, 1:100), cells were washed and counterstained with propidium iodide (0.35 μg ml−1; P4170, Sigma-Aldrich) for 10 min at room temperature. Four hundred and five nm excitation wavelength was used to excite filipin. For each field of view (FOV), filipin fluorescence intensity was calculated by dividing the number of blue puncta by the number of microglia. The values were then normalized to the filipin fluorescence intensity value of the risk allele and used for statistical analysis. The Fiji software was used to quantify filipin fluorescence intensity (https://fiji.sc).
Lysosomal staining for iMGs
iMGs were grown on glass coverslips until day 25. Cells were incubated with LysoTracker Red DND-99 (L7528, Invitrogen, 100 nM) in complete iMG medium for 30 min at 37 °C. Cells were then fixed for 30 min at room temperature with 4% methanol-free PFA (28906, Pierce) for 10 min at room temperature. The cells were then stained with BODIPY as described above (LD staining with BODIPY) to detect LDs. The images were taken on a Nikon ECLIPSE TE2000-U microscope.
FACS sorting of iMGs
iMGs were grown on six-well plates until day 25. iMGs were then dissociated with Accutase (07920, StemCell) and stained with CD45-PE (12-0451-82, Invitrogen, 1:300) for 30 min at 37 °C and washed with RPMI medium three times by centrifugation at 300g for 5 min. Next the iMGs were stained with BODIPY 493/503 (D3922, Thermo Fisher Scientific, 1:1,000 from 1 mg ml−1 stock solution in DMSO) for 10 min at 37 °C with after washing twice with RPMI medium by centrifugation at 300g for 5 min. Cells were resuspended in PBS and sorted using BD FACsAria Fusion. Data were collected using BD FACSDiva software (v.9.0.1) and analysed using FlowJo (v.11.0).
PICALM KO by CRISPR–Cas9 editing C20 cells
C20 cells were maintained in DMEM/F12 medium containing 10% FBS and 1% penicillin–streptomycin. PICALM exon 1 was targeted using the following oligonucleotide sequences—sgRNA F, CACCGGCCGGTGACACTGTGCTGGG; and R, AAACCCCAGCACAGTGTCACCGGCC. Control oligo sequences were generated using sequences not specific to the human genome. Recombinant lentiviruses were generated in HEK293T cells using MISSION Lentiviral Packaging Mix (Sigma-Aldrich, SHP001). C20 cells were transduced with filtered virus-containing medium, and stable pools were selected in blasticidin (20 µg ml−1).
Immunoblots for iMG and C20 cells
For iMG, cells were lysed in phosphosafe buffer (71296, EMD Millipore) with protease inhibitor (04693132001, Roche) and PhosphoSTOP tablet (4906845001, Roche). Next, 50 μg of protein lysates were run on TGX Stain-Free Precast Gel (4–15%) (4561083, Bio-Rad) in Tris/glycine/SDS running buffer (1610732, Bio-Rad). The gel was then transferred onto a 0.45-μm PVDF membrane (88585, Thermo Fisher Scientific). The membrane was blocked with 5% skimmed milk and probed with rabbit anti-PICALM (HPA019061, Sigma-Aldrich, 1:1,000) or mouse anti-β-actin (A5316, Millipore, 1:10,000) at 4 °C overnight. Secondary antibodies, anti-rabbit-HRP (7074, Cell Signaling, 1:20,000) and anti-mouse-HRP (7076, Cell Signaling, 1:20,000), were then added. The blots were imaged and quantified using Bio-Rad Image Lab v.6.1.0.
For C20 line, cells were lysed in RIPA buffer (50 mM Tris, 50 mM NaCl, 0.5% sodium deoxycholate, 1% Triton X-100, 0.1% SDS, 5 mM EDTA, pH 8) containing complete protease inhibitors (Roche), sonicated and lysates cleared by centrifugation (21,000g for 2 min). Fifty μg of lysate was used per lane on 4–20% SDS PAGE gels and transferred onto nitrocellulose membranes. Non-specific sites were blocked with PBS containing 1% BSA and 1% fish gelatin at room temperature for 1 h. Membranes were incubated with primary antibodies rabbit anti-PICALM (Sigma-Aldrich, HPA019061, 1:500) and mouse anti-β-actin (Proteintech, 66009-1-lg, 1:50,000) in at 4 °C for 16 h. Secondary antibodies IRDye 680RD donkey anti-rabbit IgG and IRDye 800CW donkey anti-mouse IgG (Li-COR) were incubated at room temperature for 2 h. Blots were imaged and quantified on a Li-COR Odyssey infrared imaging system.
Myelin phagocytosis for C20 cells
Myelin isolated from the mouse brain (as described above) was conjugated to pHrodo-Green (Thermo Fisher Scientific, 35369) or pHrodo-Red (Thermo Fisher Scientific, P36600). C20 cells were incubated with pHrodo-conjugated myelin (20 µg ml−1) at 37 °C (5% CO2) for 1 h. Negative controls were treated with cytochalasin D (10 nM, Invitrogen). After fixation, cells were stained with rabbit anti-BIN1 antibody (Proteintech, 14647-1-AP, 1:500) for 2 h. Images were acquired on a Nikon Eclipse Ti2 (Yokogawa spinning-disk field-scanning confocal) microscope at ×20 magnification and captured using a Photometrics PRIME 95B sCMOS camera. Single-plane images were processed using Fiji/ImageJ (v.1.54f, 64 bit) software to threshold whole-cell masks (created from BIN1 staining). The integrated density of pHrodo-myelin within each cell was measured from five random fields of view (per biological replicates) and the median values from each replicate were used for statistical analysis.
LD staining for C20 cells
Live C20 cells were labelled with BODIPY (2 µM in PBS) at 37 °C (5% CO2) for 15 min. At room temperature, cells were fixed with 4% PFA for 30 min, and nuclei were labelled with Hoechst 33342. The volumes of BODIPY+ droplets and the number of nuclei was quantified from deconvolved image stacks using ImageJ (v.1.54f, 64 bit)/Fiji software. Five random fields of view (per biological replicate) were acquired for quantification, and the median values from each biological replicate were used for statistical analysis.
Organelle marker staining for C20 cells
For transferrin endocytosis, C20 cells were washed and incubated in uptake medium (DMEM containing 25 mM HEPES) for 1 h. Cells were treated with 10 µg ml−1 transferrin Alexa Fluor 555 conjugate (Invitrogen, T35352) in DMEM/HEPES containing 1 mg ml−1 BSA at 37 °C for 30 min. Cells were then chilled to 4 °C, and non-internalized transferrin was washed from cell surfaces with acid wash (0.5 M NaCl, 0.2 M acetic acid, pH 2.8) before fixing the cells in PFA. For cholera toxin internalization, cells were washed with labelling medium (1 mg ml−1 BSA in serum-free DMEM containing 25 mM HEPES), chilled to 4 °C and treated with 100 nM cholera toxin subunit B Alexa Fluor 647 conjugate (Invitrogen, C34778) in labelling medium at 4 °C for 10 min. Cells were washed with ice-cold PBS and fixed with 4% PFA (in PBS) at room temperature for 15 min. Nuclei were labelled with Hoechst 33342.
For immunofluorescence staining with antibodies, C20 cells grown on glass coverslips were fixed with 4% PFA and blocked (3% BSA, 50 mM NH4Cl, 10 mM glycine, PBS) at room temperature for 30 min. The cells were incubated with rabbit anti-Giantin (Covance, PRB-114C, 1:5,000), mouse anti-EEA1 (BD Transduction Laboratories, 610457, 1:500) or mouse anti-AP-4ε (BD Transduction, 612018, 1:100) antibodies at room temperature for 2 h. All C20 cell images were acquired on a Nikon Eclipse Ti2 spinning-disk field-scanning confocal microscope at ×60 magnification. z stacks (100 nm step size) were processed to generate maximum-intensity projections using Fiji/ImageJ (1.54f, 64 bit).
Lipid extraction from iMGs
iMGs carrying the PICALM risk allele, non-risk allele or risk-CRISPRa were grown in six-well plates until day 25. iMGs were dissociated with Accutase (07920, StemCell), collected and washed with PBS three times by centrifugation at 300g for 5 min. Lipids were extracted from iMGs as described previously80,81. In brief, a deuterated lipid standard mixture (330820, LM6003, 860739, 330727, 860657, 860658, Avanti Research) was added to iMGs equally and organic solvent (chloroform:methanol, 1:2 (v/v)) was added to iMG pellets and subjected to tip sonication, followed by vortexing and centrifugation to extract neutral lipid. The organic layer was collected and saved for later use and acidic extraction was performed on the remaining pellet by a mixture of chloroform:methanol:37% 1 M HCl (40:80:1, v/v/v). After the addition of chloroform and 0.1 M HCl to induce phase separation, the samples were vortexed and centrifuged. The bottom organic layer was combined with the previously collected organic layer and dried using vacuum centrifuge. The dried lipid extract was reconstituted in methanol:chloroform (1:3, v/v) for further analysis.
Lipidomics analysis of iMGs
Lipid extracts were subjected to global lipidomics analysis using liquid chromatography–tandem mass spectrometry (LC–MS/MS). LC–MS-grade methanol (A456, Thermo Fisher Scientific), acetonitrile (A955, Thermo Fisher Scientific), isopropanol (A461, Thermo Fisher Scientific), water (W71, Thermo Fisher Scientific), formic acid (A117, Thermo Fisher Scientific) and ammonium formate (78314, Millipore Sigma) were used. The samples from two different iMG differentiations were analysed, with lipidomics data acquired in two technical replicates for each sample. Reconstituted lipid extracts were separated on a Hypersil GOLD Vanquish C18 UHPLC column (15 cm × 2.1 mm, C18 1.9 μm and 175 Å) using an Orbitrap Tribrid IQ-X mass spectrometer (Thermo Fisher Scientific) coupled to Vanquish Horizon UHPLC (Thermo Fisher Scientific). A binary gradient was used at a flow rate of 300 µl min−1, using mobile phase A (water:acetonitrile, 6:4, with 10 mM ammonium formate and 0.1% formic acid) and organic phase (isopropanol:methanol:acetonitrile, 8:1:1, with 10 mM ammonium formate and 0.1% formic acid). In brief, mobile phase B increased from 20% to 95% over 17 min and was maintained at 95% for 5 min for washing. It was ramped down to 20% over 0.1 min and equilibrated for 5 min for the next injection. The analytical column was maintained at 50 °C. A full-scan MS was performed using the Orbitrap with a resolution of 60,000 at m/z 200. MS/MS spectra were acquired at a resolution of 15,000 at m/z 200. MS/MS scans were acquired for 1.5 s, followed by an MS scan. In positive-ion mode, MS/MS fragmentation using a stepped collision energy of 30%, 35% and 40% in higher-energy collisional dissociation (HCD) was performed with a spray voltage of 3.5 kV. In negative-ion mode, a spray voltage of 3 kV and a stepped collision energy of 30%, 35% and 40% in MS/MS acquisition using HCD were used. The acquired tandem mass spectra were processed using LipidSearch v.5.1.6 (Thermo Fisher Scientific) for lipid annotation and quantification. Lipids were identified based on precursor ion masses and their corresponding MS/MS spectra were matched against the database. Annotated lipids were quantified by calculating their peak areas using LipidSearch. All lipids were normalized to the peak area of an internal standard with the same head group as the target lipids. Total lipid levels were determined by summing the peak areas of individual lipid species sharing the same head group.
Imaging quantification and statistical analyses
For image analyses, we assayed fluorescence intensity, puncta density, puncta area and/or puncta size using the number of samples specified in the corresponding figures. For each FOV, the fluorescence intensity was calculated by dividing the number of fluorescent puncta by the number of microglia. The puncta number and the total area of puncta were acquired by applying a threshold to the respective fluorescent areas and performing the Analyze particles function. The number of microglia was calculated by applying a threshold to the DAPI fluorescence signal and measuring the number of nuclei through the same function. The images were acquired in a manner that the operator was blinded to the sample and genotype identification. For testing statistical differences between two groups, we used a two-tailed unpaired Student’s t-test when the experimental design did not involve multiple batches (for example, different rounds of experiments); otherwise, we used the R packages lme4 and lmerTest to fit data into an LMM to account for potential random effects from different experimental rounds and clones. For statistical tests involving more than two groups, we applied a one-way ANOVA with the appropriate post hoc test (Dunnett or Tukey), while accounting for any possible random effects from experimental rounds when applicable. The data analysis was performed using R v.4.3.2, GraphPad Prism 9 and Microsoft Excel. The results were considered to be significant if P < 0.05. All data with error bars were presented as the mean ± s.e.m. For ATAC–seq analysis of ASoC SNPs, the two-sided binomial test in R package was used to test allelic bias, and the Benjamini–Hochberg correction was applied to all qualified ASoC SNPs. For RNA-seq DEG analysis, limma (v.3.58.1)/EdgeR (v.4.0.16) with a generalized linear model (GLM) F test was used, and the Benjamini–Hochberg procedure was used to adjust P values accounting for multiple testing.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
ATAC–seq data for iMG, iAst and neurons are available at Gene Expression Omnibus under accession codes GSE263804 and GSE188491. RNA-seq data are available under accession code GSE263809. The ASoC SNP list and the functionally interpreted GWAS risk loci are available at Zenodo82 (https://zenodo.org/records/14853787). Other publicly available datasets used in our study include: Alzheimer’s disease GWAS Summary Statistics (https://www.ebi.ac.uk/gwas/studies/GCST90027158); human fresh microglia regulome study (https://www.synapse.org/Synapse:syn26207321); Human iMG study (GSE164315); cell-type-specific cis-eQTLs in eight human brain cell types83 (https://doi.org/10.5281/zenodo.5543734); a meta-analysis of single-nucleus eQTLs linking genetic risk to brain disorders84 (https://zenodo.org/records/14908182); human microglial state dynamics in Alzheimer’s disease progression (GSE227223); hMG scRNA-seq dataset at different microglia states (homeostatic, LDAM and DAM) (GSE254205); the human GRCh38 reference genome, Ensembl GRCh38.v84 genome annotations and JASPAR transcription-factor-binding profiles as a pre-compiled 10x Genomics Cell Ranger reference set (https://www.10xgenomics.com/support/software/cell-ranger-arc/downloads#reference-downloads).
Code availability
The codes and scripts (R and bash) used in ATAC–seq, RNA-seq, GWAS enrichment, TORUS and circus plot have been deposited at Zenodo85 (https://doi.org/10.5281/zenodo.15238434).
References
Lambert, J. C. et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458 (2013).
Jansen, I. E. et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413 (2019).
Bellenguez, C. et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022).
Raj, T. et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet. 50, 1584–1592 (2018).
Young, A. M. H. et al. A map of transcriptional heterogeneity and regulatory variation in human microglia. Nat. Genet. 53, 861–868 (2021).
Kosoy, R. et al. Genetics of the human microglia regulome refines Alzheimer’s disease risk loci. Nat. Genet. 54, 1145–1154 (2022).
Yang, X. et al. Functional characterization of Alzheimer’s disease genetic variants in microglia. Nat. Genet. 55, 1735–1744 (2023).
Lopes, K. P. et al. Genetic analysis of the human microglial transcriptome across brain regions, aging and disease pathologies. Nat. Genet. 54, 4–17 (2022).
Bryois, J. et al. Cell-type-specific cis-eQTLs in eight human brain cell types identify novel risk genes for psychiatric and neurological disorders. Nat. Neurosci. 25, 1104–1112 (2022).
Marschallinger, J. et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci. 23, 194–208 (2020).
Lindner, K. et al. Isoform- and cell-state-specific lipidation of ApoE in astrocytes. Cell Rep. 38, 110435 (2022).
Sienski, G. et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.aaz4564 (2021).
Moulton, M. J. et al. Neuronal ROS-induced glial lipid droplet formation is altered by loss of Alzheimer’s disease-associated genes. Proc. Natl Acad. Sci. USA https://doi.org/10.1073/pnas.2112095118 (2021).
Qi, G. et al. ApoE4 impairs neuron-astrocyte coupling of fatty acid metabolism. Cell Rep. 34, 108572 (2021).
Zhao, X., Zhang, S., Sanders, A. R. & Duan, J. Brain lipids and lipid droplet dysregulation in Alzheimer’s disease and neuropsychiatric disorders. Complex Psychiatry 9, 154–171 (2023).
Liu, L., MacKenzie, K. R., Putluri, N., Maletić-Savatić, M. & Bellen, H. J. The glia-neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in Glia via APOE/D. Cell Metab. 26, 719–737 (2017).
Blanchard, J. W. et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 611, 769–779 (2022).
Victor, M. B. et al. Lipid accumulation induced by APOE4 impairs microglial surveillance of neuronal-network activity. Cell Stem Cell 29, 1197–1212 (2022).
Haney, M. S. et al. APOE4/4 is linked to damaging lipid droplets in Alzheimer’s disease microglia. Nature 628, 154–161 (2024).
Zhang, S. et al. Allele-specific open chromatin in human iPSC neurons elucidates functional disease variants. Science 369, 561–565 (2020).
Zhang, S. et al. Multiple genes in a single GWAS risk locus synergistically mediate aberrant synaptic development and function in human neurons. Cell Genom. 3, 100399 (2023).
Novikova, G. et al. Integration of Alzheimer’s disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat. Commun. 12, 1610 (2021).
Nott, A. et al. Brain cell type-specific enhancer–promoter interactome maps and disease-risk association. Science 366, 1134–1139 (2019).
Xiong, X. et al. Epigenomic dissection of Alzheimer’s disease pinpoints causal variants and reveals epigenome erosion. Cell 186, 4422–4437 (2023).
Jang, B. et al. SingleBrain: a meta-analysis of single-nucleus eQTLs linking genetic risk to brain disorders. Preprint at medRxiv https://doi.org/10.1101/2025.03.06.25323424 (2025).
Sun, N. et al. Human microglial state dynamics in Alzheimer’s disease progression. Cell 186, 4386–4403 (2023).
Johnson, E. C. B. et al. Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat. Neurosci. 25, 213–225 (2022).
Andrews, S. J. et al. The complex genetic architecture of Alzheimer’s disease: novel insights and future directions. eBioMedicine https://doi.org/10.1016/j.ebiom.2023.104511 (2023).
Romero-Molina, C., Garretti, F., Andrews, S. J., Marcora, E. & Goate, A. M. Microglial efferocytosis: diving into the Alzheimer’s disease gene pool. Neuron 110, 3513–3533 (2022).
Salter, M. W. & Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 23, 1018–1027 (2017).
Ando, K. et al. PICALM and Alzheimer’s disease: an update and perspectives. Cells https://doi.org/10.3390/cells11243994 (2022).
Zhang, B. et al. Synaptic vesicle size and number are regulated by a clathrin adaptor protein required for endocytosis. Neuron 21, 1465–1475 (1998).
Rai, M. A. et al. Comparative analysis of human microglial models for studies of HIV replication and pathogenesis. Retrovirology 17, 35 (2020).
Bö, L. et al. Detection of MHC class II-antigens on macrophages and microglia, but not on astrocytes and endothelia in active multiple sclerosis lesions. J. Neuroimmunol. 51, 135–146 (1994).
Rong, S. et al. Expression of SREBP-1c requires SREBP-2-mediated generation of a sterol ligand for LXR in livers of mice. eLife https://doi.org/10.7554/eLife.25015 (2017).
Mercer, J. L. et al. Modulation of PICALM levels perturbs cellular cholesterol homeostasis. PLoS ONE 10, e0129776 (2015).
Olzmann, J. A. & Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 20, 137–155 (2019).
Tcw, J. et al. Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell 185, 2213–2233 (2022).
Namatame, I., Tomoda, H., Arai, H., Inoue, K. & Omura, S. Complete inhibition of mouse macrophage-derived foam cell formation by triacsin C. J. Biochem. 125, 319–327 (1999).
Khatchadourian, A., Bourque, S. D., Richard, V. R., Titorenko, V. I. & Maysinger, D. Dynamics and regulation of lipid droplet formation in lipopolysaccharide (LPS)-stimulated microglia. Biochim. Biophys. Acta 1821, 607–617 (2012).
Ioannou, M. S. et al. Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity. Cell 177, 1522–1535 (2019).
Liu, L. et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160, 177–190 (2015).
Wu, X., Miller, J. A., Lee, B. T. K., Wang, Y. & Ruedl, C. Reducing microglial lipid load enhances beta amyloid phagocytosis in an Alzheimer’s disease mouse model. Sci. Adv. 11, eadq6038 (2025).
Tian, Y., Chang, J. C., Fan, E. Y., Flajolet, M. & Greengard, P. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc. Natl Acad. Sci. USA 110, 17071–17076 (2013).
Moreau, K. et al. PICALM modulates autophagy activity and tau accumulation. Nat. Commun. 5, 4998 (2014).
Hattersley, K. J., Carosi, J. M., Hein, L. K., Bensalem, J. & Sargeant, T. J. PICALM regulates cathepsin D processing and lysosomal function. Biochem. Biophys. Res. Commun. 570, 103–109 (2021).
Schulze, R. J. et al. Direct lysosome-based autophagy of lipid droplets in hepatocytes. Proc. Natl Acad. Sci. USA 117, 32443–32452 (2020).
Hirose, T. et al. ATP6AP2 variant impairs CNS development and neuronal survival to cause fulminant neurodegeneration. J. Clin. Invest. 129, 2145–2162 (2019).
Zhang, X. et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 7, 12109 (2016).
Simon, M. J., Logan, T., DeVos, S. L. & Di Paolo, G. Lysosomal functions of progranulin and implications for treatment of frontotemporal dementia. Trends Cell Biol. 33, 324–339 (2023).
Ando, K. et al. Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer’s brains. Acta Neuropathol. 125, 861–878 (2013).
Sies, H. & Jones, D. P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 21, 363–383 (2020).
Dubreuil, M. M. et al. Systematic identification of regulators of oxidative stress reveals non-canonical roles for peroxisomal import and the pentose phosphate pathway. Cell Rep 30, 1417–1433 (2020).
Green, R. M. et al. Developmental nonlinearity drives phenotypic robustness. Nat. Commun. 8, 1970 (2017).
Lotfollahi, M. et al. Predicting cellular responses to complex perturbations in high-throughput screens. Mol. Syst. Biol. 19, e11517 (2023).
Baig, S. et al. Distribution and expression of picalm in Alzheimer disease. J. Neuropathol. Exp. Neurol. 69, 1071–1077 (2010).
Hong, S. et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016).
Jun, G. et al. Meta-analysis confirms CR1, CLU, and PICALM as alzheimer disease risk loci and reveals interactions with APOE genotypes. Arch. Neurol. 67, 1473–1484 (2010).
Shi, J. et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 460, 753–757 (2009).
Ripke, S. et al. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 43, 969–976 (2011).
Levinson, D. F. et al. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. Am. J. Psychiatry 168, 302–316 (2011).
Bennett, D. A. et al. Religious Orders Study and Rush Memory and Aging Project. J. Alzheimers Dis. 64, S161–S189 (2018).
Butler Iii, R. R. et al. The genetic relevance of human induced pluripotent stem cell-derived microglia to Alzheimer’s disease and major neuropsychiatric disorders. Mol. Neuropsychiatry 5, 85–96 (2020).
Brownjohn, P. W. et al. Functional studies of missense TREM2 mutations in human stem cell-derived microglia. Stem Cell Rep. 10, 1294–1307 (2018).
Zhang, Y. et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798 (2013).
Gonzalez, R. et al. Deriving dopaminergic neurons for clinical use. A practical approach. Sci. Rep. 3, 1463 (2013).
Yang, N. et al. Generation of pure GABAergic neurons by transcription factor programming. Nat. Methods 14, 621–628 (2017).
van de Geijn, B., McVicker, G., Gilad, Y. & Pritchard, J. K. WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods 12, 1061–1063 (2015).
Zhang, Y. et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Bulik-Sullivan, B. K. et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Wen, X. Molecular QTL discovery incorporating genomic annotations using Bayesian false discovery rate control. Ann. Appl. Stat. 10, 1619–1638 (2016).
Joung, J. et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 12, 828–863 (2017)
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Weissbein, U., Schachter, M., Egli, D. & Benvenisty, N. Analysis of chromosomal aberrations and recombination by allelic bias in RNA-seq. Nat. Commun. 7, 12144 (2016).
Nunez, J. K. et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell 184, 2503–2519 (2021).
Liu, H., Wang, X. & Qi, L.S. Bio. Protoc. 7, e2522 (2017).
Ho, S. M. et al. Evaluating synthetic activation and repression of neuropsychiatric-related genes in hiPSC-derived NPCs, neurons, and astrocytes. Stem Cell Rep. 9, 615–628 (2017).
Jung, J. H. et al. Characterization of lipid alterations by oncogenic PIK3CA mutations using untargeted lipidomics in breast cancer. OMICS 27, 327–335 (2023).
Byeon, S. K. et al. High-resolution mass spectrometric analysis of cardiolipin profiles in Barth syndrome. Mitochondrion 60, 27–32 (2021).
Kozlova, A. et al. Data for ‘PICALM Alzheimer’s risk allele causes aberrant lipid droplets in microglia’. Zenodo https://doi.org/10.5281/zenodo.14853786 (2025).
Bryois, J. Summary statistics of cell-type specific cis-eQTLs in eight brain cell-types. Zenodo https://doi.org/10.5281/zenodo.5543734 (2021).
Jang, B., Won, H.-H. & Raj, T. SingleBrain: single-nucleus expression QTL meta-analysis summary statistics across multiple human brain cohorts. Zenodo https://doi.org/10.5281/zenodo.14907598 (2025).
Kozlova, A. et al. Code for ‘PICALM Alzheimer’s risk allele causes aberrant lipid droplets in microglia’. Zenodo https://doi.org/10.5281/zenodo.11105250 (2025).
Acknowledgements
We thank the staff at the NIH NeuroBioBank for providing post-mortem human brain tissues from the Harvard Brain Tissue Resource Center and the University of Miami Brain Endowment Bank; the members of the Goizueta Alzheimer’s Disease Research Center (ADRC) and Rush ADRC for providing human brain tissue sections; and M. Moulton and L. Goldman for comments on the manuscript. This work was supported by National Institute on Aging grants R01AG063175 (J.D. and G.T.), R01AG081374 (J.D.), RF1AG079141 (G.T.), R01 AG073260 (H.J.B.) and U01 AG072439 (H.J.B.). A.S. is supported by a BrightFocus fellowship. ROSMAP is supported by P30AG10161 (D.A.B.), P30AG72975 (J.A.S.), R01AG15819 (D.A.B.), R01AG17917 (D.A.B.), U01AG46152 (D.A.B.) and U01AG61356 (D.A.B.). A.P. is supported by NIH grants U01CA271410 and P30CA15083. The work is also partially supported by National Institute of Mental Health grants R01MH116281 and R01MH106575 (J.D.).
Author information
Authors and Affiliations
Contributions
A.K. performed iPS cell differentiation, CRISPR editing and iMG assays, and wrote the manuscript. S.Z. analysed ATAC–seq and RNA-seq data and other genomic data and wrote the manuscript. A.S. performed experiments with C20 cells and brain histology and wrote the manuscript. H.Z. performed iPS cell differentiation, CRISPRa and ATAC–seq. S.S. contributed to experiments with C20 cells, and M.P. performed human brain staining. S.K.B. and A.P. performed lipidomic profiling and wrote the manuscript. C.T. helped with iAst cell differentiation and snRNA-seq analysis. X.S. performed GWAS enrichment analysis that was supervised by X.H., K.S. and X.Z., and B.J. helped with CRISPR editing, iPS cell culture and DNA sequencing. Z.P.P. helped with the data interpretation and neuron differentiation. J.A.S. and D.A.B. provided ROSMAP human brain sections and interpreted the results. A.R.S. and H.J.B. helped with design and data interpretation and edited the manuscript. G.T. supervised the experiments with C20, brain histology and wrote the manuscript. J.D. conceived and supervised the study and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 ATAC-seq quality control (QC) and OCR peak calling.
(a) Representative images of IF staining (left) of iMG (TREM2+CD45+PU1+) used for ATAC-seq and iMG purity quantification (right). Each datapoint represents one single-well measurement from one experiment of one donor line (n = 5). Scale bar, 50 μm. (b) Representative images of IF staining (left) of iAst (S100B+, VIM+, or GFAP+) used for ATAC-seq and iAst purity quantification (right). Note that GFAP staining and imaging was performed independently from S100B/VIM staining and imaging. Each datapoint represents one single-well measurement from one experiment of one donor line (n = 5 lines). Scale bar: 50 μm. (c) ATAC-seq fragment size distribution plots show periodical nucleosome-free region patterns that are similar between samples and different iPSC-derived cell types. (d) and (e) show comparable and acceptable according to the ENCODE ATAC-seq guideline version 4: FRiP (fragment reads in peaks) score > 0.3, TSS (transcription start site) annotation > 5 across cell types. (f) iMG-specific OCR peaks for a known MG-specific gene SPI1 (also known as PU.1) and much stronger peaks for VIM in iAs than in other cell types. (g) PCA clustering of human iPSC-derived MG, Ast, NGN2-Glut, GABAergic, and dopaminergic neurons. Normalized ATAC-seq reads derived from a 501-bp union peak set from all five cell types (622,987 peaks in total) were used for PCA. (h) UpSet plot showing the cell type-specific ATAC-seq peaks and shared peaks across different cell types. (i) PCA of ATAC-seq peak accessibility of iMG and iAst samples from the current study in comparison to previously reported datasets of human brain MG and Ast (circled areas) or iAst as well as other brain cell types6,22. 210,833 peaks from the previous studies6,22 were used for PCA. GABA, GABAergic neurons; hOlig, human oligodendrocytes; MGAS, the mixture of microglia and astrocytes.
Extended Data Fig. 2 Enrichment of ASoC SNPs for GWAS risk variants in LOAD and major NPD.
(a) Venn diagram showing ASoC SNPs identified in each cell type. (b) to (f) Dot plots showing the fold of GWAS enrichment (log2) and P-value (-log10) for each disorder or trait across different cell types. The analysis was conducted using Torus (see Methods). (g) to (k) Number of GWAS risk loci with at least one of the GWAS index risk SNPs or their proxies demonstrating ASoC in different cell types for LOAD, SZ, BP (bipolar), MDD (major depressive disorder), and PD. Note the LOAD GWAS risk loci with functional LOAD risk SNPs showing ASoCs are predominantly in iMG (19/26 loci). (l) Circos plot depicting 19 LOAD risk loci with 38 GWAS risk SNPs also ASoC SNPs in iMG. Tracks from the innermost to the outermost circles: LOAD risk loci (n = 9) with ABC enhancer/ASoC SNP links (purple) to at least one target gene; FDR (-log10) of ASoC SNPs, each dot (orange) representing a SNP; LOAD risk SNPs that are also ASoC SNPs (with rs numbers); LOAD GWAS association p-values (-log10), each dot (red) representing a SNP; index LOAD risk genes at each locus, where target genes link to an ABC enhancer/ASoC SNP (purple); chromosomal regions of LOAD risk loci. PICALM region is highlighted in yellow.
Extended Data Fig. 3 The effects of the LOAD risk-allele on chromatin accessibility, PICALM expression and PU.1 binding.
(a) Human brain OCR peaks in different cell types in comparison with our iMG at the PICALM locus. Note the rs10792832-flanking peak (highlighted in the vertical yellow box) identified in our iMG is only present in brain microglia but not in other cell types. The adjacent ATAC-seq read pileup plot (right) for rs10792832 shows significantly more ATAC-reads of allele A than allele G (i.e., ASoC) in brain microglia. Except for our iMG, data of all other cell types are from a snATAC-seq study24 and reanalysed by using 10x Genomics Cell Ranger ARC 2.0.2. Ex = excitatory neurons, In = inhibitory neurons, Ast = astrocytes, oligo = oligodendrocytes, OPC=oligodendrocyte precursor cells, Vas = vascular cells. (b) IF staining of the CRISPR-engineered isogenic iMG (left panels) shows high and comparable purity (TREM2+/CD45+/PU.1+) (right panel). Each datapoint represents single-well measurement; for each condition (risk and non-risk), data are from two independent donor lines each with one clone from one experiment (n = 2). Scale bar: 50 µm. (c) The representative image of iMG stained with P2RY12+ and IBA1+ (left) and quantification of iMG purity (right). (d) The representative image of iMG stained with TMEM119+ and IBA1+ (left) and quantification of iMG purity (right). In (c) and (d), each datapoint represents a single-well measurement from one experiment; for each condition (risk and non-risk), data are from two independent donor lines (CD04 and CD09) each with 2 clones from one experiment with 3 wells of differentiations (n = 12). Scale bar, 50 µm. Two-sided unpaired Student’s t-test. ns = not significant. (e) qPCR shows reduced PICALM expression in risk-allele iMG. Each datapoint represents a single-well measurement from one experiment; for each donor line (CD04 and CD09) and each condition (risk and non-risk), data are from 2 clones, collected across 3 independent experiments each with 3 wells of differentiations for clone 1 and 2 experiments each with 3 wells of differentiations for clone 2 (n = 15). (f) qPCR result showing that the risk allele of rs10792832 does not affect PICALM expression in iAst. Each datapoint represents a single-well measurement from one experiment; for each donor line (CD04 and CD09) and each condition (risk and non-risk), data are from 2 clones, collected from 2 independent experiments each with 3 wells of differentiations (n = 12). In both (e) and (f), expression was normalized to GAPDH. Linear Mixed Model (LMM) was applied to test the fixed effect of genotype, with experimental round and clone identity as nested random factors; two-sided test. (g) PU.1 antibody validation in PMP cells from which iMG were further differentiated. (h) Schematic design of ChIP-qPCR to assay PU.1 binding. The illustration was created using BioRender. Sudwarts, A. (2025) https://BioRender.com/nux3l58. (i) The LOAD risk-allele G of rs10792832 shows reduced PU.1 binding in qPCR of ChIP products. n = 2 experiments for each line. Homozygous iMG was used to compare the allelic effect. (j) The allele A of rs10792832 exhibits higher Sanger sequencing peaks for PU.1 ChIP-seq PCR products of iMG from two iPSC lines heterozygous for rs10792832. Note the equal peak height of the two alleles for genomic PCR products (sequenced by using primers for on-target editing confirmation) of the same heterozygous samples as in the top panels. (k) Quantification of the allelic ratios of rs10792832 for Sanger sequencing peaks in (j). In all comparisons. * P < 0.05, ** P < 0.01, ***, P < 0.001; mean ± s.e.m.
Extended Data Fig. 4 PICALM expression in human postmortem brains.
(a) Human post-mortem brain sections from an asymptomatic patient were stained with antibodies against PICALM (green) and IBA1 (magenta). White arrows indicate PICALM expressed in MG, and yellow arrows indicate PICALM expressed in a blood vessel. Line-scan (kymography) analysis (right) demonstrates the overlapping expression. The white line in IBA1 composite image shows the location of the line scan. Scale bar, 50 µm. (b) Reduced PICALM expression in the grey matter of AD patients’ postmortem brains (n = 20) compared to controls (n = 19). Two-sided unpaired Student’s t-test was used. (c) Comparison of brain MG state-specific marker gene expression between different human brain MG states. MG states were defined as described in the snRNA-seq study19. Z-scored expression values of MG state-specific markers and PICALM are shown. HOMEO, homeostatic MG including cell clusters 1,2,3,5 and 8; DAM, disease-associated MG including clusters 0 and 7; LDAM, LD-accumulative MG including clusters 4 and 6. Note lower PICALM expression in DAM and LDAM cell clusters. (d) Representative immunofluorescence staining images of post-mortem brain sections from unaffected control (CERAD 0) and AD (CERAD 3) patients. Green arrows in single-channel images indicate IBA1+ microglia which are also positive for PICALM. CERAD = Consortium to Establish a Registry for AD. (e) Representative immunofluorescence staining of PICALM in individual microglia from Braak stages 1–5 and CERAD scores 0–3, quantified in e-f. Scale bar 20 µm. (f) Quantification of PICALM expression in MG of AD and non-AD controls. AD, n = 15 with the diagnosis of “definitely AD” with a CERAD score of 3; the rest include control (n = 16), probable AD and possible AD (n = 9) (Supplementary Table 11). The grey background dots represent individual MG. ns, not significant in the two-sided unpaired Student’s t-test. (g) Quantification of PICALM expression in MG of AD and non-AD controls stratified by Braak Stage (0-5) (left) and by CERAD score (0 = control; 1-2, probable or possible AD; and 3, definitely AD) (right). Not significant in one-way ANOVA. (h) PICALM protein abundance in postmortem brain prefrontal cortex between AD, asymptomatic AD (AsymAD), and non-AD individuals. Data are from a published dataset27. One-way ANOVA; Dunnett’s correction. In all comparisons. * P < 0.05, ** P < 0.01, ***, P < 0.001; ns=not significant; mean ± s.e.m.
Extended Data Fig. 5 The LOAD risk-allele of PICALM impairs MG phagocytosis.
(a) Representative fluorescence images of iMG showing time-dependent phagocytosis of Aβ-pHrodo (green). RFP+, iMG infected with pLenti-CRISPRa. Time indicates minutes after adding Aβ-pHrodo. Scale bar, 100 μm. (b) Representative images showing time-dependent iMG phagocytosis of Aβ-pHrodo (red) for a second clone of both isogenic pairs CD04 and CD09. Scale bar, 100 μm. (c) Quantification of Aβ-pHrodo phagocytosis in iMG of CD04 and CD09 lines. Data are from 2 experiments, each with 3 wells of differentiations, one clone (clone 2) per line. LMM was used with experimental round as the random factor; two-sided test, nominal p-value; (d) Schematics of CRISPRa design to increase PICALM expression in risk-allele iMG (non-risk and risk control iMG also contain pLenti-CRISPRa encoding RFP). Illustrations were created using BioRender. Sudwarts, A. (2025) https://BioRender.com/vveaczo. (e) qPCR results showing normalized PICALM expression in iMGs with non-risk, risk, and risk-CRISPRa lines. Expression was normalized to GAPDH. Each datapoint represents a single-well measurement from one experiment; for each line (CD04 and CD09) and each condition (non-risk, risk, and risk-CRISPRa), data are from one clone, collected from 2 experiments each with 3 wells of differentiations (n = 6). (f) Schematics of CRISPRoff to knockdown PICALM expression for CD04 and CD09 lines with PICALM non-risk alleles. (g) qPCR results show CRISPRoff-induced reduction of PICALM in iMG for both lines. Expression was normalized to GAPDH. Each datapoint represents a single-well measurement from one experiment; for each line (CD04 and CD09) and each condition (risk, non-risk, and non-risk-CRISPRoff), data are from one clone from one experiment with 3 wells of differentiations (n = 3). (h) Representative fluorescence images of iMG showing time-dependent phagocytosis of myelin-pHrodo (red). Cell mask stains for the plasma membrane and NucBlue stains the nucleus in live cells. Time indicates minutes after adding myelin pHrodo. Scale bar, 100 μm. (i) Quantifying myelin-pHrodo phagocytosis in iMG carrying non-risk, risk, and non-risk-CRISPRoff for CD04 and CD09 lines. Scale bar, 100 μm. (j) Representative images showing time-dependent iMG phagocytosis of Aβ-pHrodo (red). (k) Quantification of Aβ-pHrodo phagocytosis in iMG carrying non-risk, risk, and non-risk-CRISPRoff for both CD04 and CD09 lines. (l) Cytochalasin D (10 µM) nearly completely inhibits phagocytosis. Cell mask stains for the plasma membrane and NucBlue stains nuclei in live cells. Time indicates minutes after adding myelin pHrodo. Scale bar, 100 μm. From (h) to (k), for each donor line (CD04 and CD09) and each condition (non-risk, risk, and non-risk-CRISPRoff), data are from one clone of one experiment with 3 wells of differentiations (each with 2-3 FOV). For all comparisons except in (c), one-way ANOVA was used; Dunnett’s correction. * P < 0.05, ** P < 0.01, ***, P < 0.001; mean ± s.e.m.
Extended Data Fig. 6 Validation of the selected DE genes by qPCR and IF staining.
(a) and (b) qPCR results for NEAT1, ATP6AP2, LDLR, HMGCS1, and DHCR7 in iMG (PICALM risk allele vs. non-risk). Expression was normalized to GAPDH. Each datapoint represents a single-well measurement from one experiment; for each gene and each condition (risk and non-risk), data are from 2 donor lines (CD04 and CD09; one clone per line), collected in one experiment with 2 wells of differentiations (n = 4). (c) IF staining and (d) quantification of the mean fluorescence intensity of selected DE genes in day-25 iMG (non-risk vs risk alleles). Scale bar, 50 µm. Each datapoint represents a single-well measurement from one experiment; for each gene and each condition (risk and non-risk), data are from 2 donor lines (CD04 and CD09; one clone per line), collected in one experiment with 2 wells of differentiations (n = 4). For all comparisons, LMM was used to test the fixed effect of genotype (risk vs. non-risk), with cell line identify (CD04 or CD09) as the random factor; two-sided test, nominal p-value; * P < 0.05, ** P < 0.01, ***, P < 0.001; mean ± s.e.m.
Extended Data Fig. 7 LOAD risk-allele of PICALM causes LD accumulation in iMG.
(a) IF filipin staining in day-25 iMG (TREM2+) carrying the LOAD risk or non-risk allele of rs10792832. (b) Quantification of filipin fluorescence intensity in (a). PI, propidium iodide for nucleus staining. Scale bar, 50 µm. Each datapoint represents a single-well measurement field of view (FOV) of one experiment; for each condition (risk and non-risk), data are from two donor lines (CD04 and CD09), collected from one experiment with two wells of differentiations each with 2-3 FOV (n = 12 for risk group and n = 8 for non-risk group). (c) and (d) Quantification of LD staining shows increased LD (BODIPY+) size and fluorescence intensity in LOAD risk-allele iMG (vs. non-risk), respectively. (e) Fluorescence staining of LD (BODIPY+) and ROS (CellRox+) with or without pretreatment of Triacsin C (TrC) in iMG carrying risk or non-risk allele. Scale bar, 50 µm. (f) to (i) Quantification of (e) shows increased LD and CellRox density and area in iMG carrying LOAD risk-allele (vs. non-risk). Each datapoint represents a single well measurement (FOV) from one experiment; data are from a single clone of CD04 line, collected from one experiment with 2 wells of differentiations each with 4 FOV (n = 8). One-way ANOVA with Tukey’s correction. (j) Representative histogram of flow cytometry analysis of LD (BODIPY+) in iMG. Gating strategy is shown in Supplementary Fig. 10. (k) Quantification of the proportion of BODIPY+ cells from (j). Each datapoint represents a single-well measurement from one experiment; for each donor line (CD04 and CD09) and each condition (risk and non-risk), data are from single clone, collected from 3 experiments (n = 3). (l) Representative fluorescence images of LD staining with PLIN2. Scale bar, 50 µm. (m) and (n) Quantification of PLIN2+ LD area per iMG and LD fluorescence intensity per iMG, respectively. For (c), (d), (m), and (n), each datapoint represents a single-well measurement from one experiment; for each donor line (CD04 and CD09) and each condition (risk and non-risk), data are from 2 clones, collected from 2 independent experiments each with 3 wells of differentiations (n = 12). LMM was used to test the fixed effect of genotype (risk vs. non-risk), with clone identify and experimental rounds as the random factors; two-sided test, nominal P-values. For (b) and (k), two-sided unpaired Student’s t-test was used. For all comparisons, * P < 0.05, ** P < 0.01, ***, P < 0.001; mean ± s.e.m.
Extended Data Fig. 8 PICALM-CRISPRa rescues the LD accumulation and phagocytosis-deficiency in LOAD risk-allele iMG.
(a) Representative fluorescence images show time-dependent phagocytosis of Aβ-pHrodo and LD accumulation (BODIPY+) in iMG carrying the LOAD risk-allele or non-risk allele and in risk-CRISPRa of PICALM. RFP+, iMG infected with pLenti-CRISPRa (note that both non-risk and risk control iMG also contain pLenti-CRISPRa that encodes RFP). Scale bar, 100 μm. (b) PICALM-CRISPRa rescues the LD accumulation in iMG carrying the LOAD risk-allele to a level similar to that in MG carrying the non-risk allele for both CD04 and CD09. Each datapoint represents a single-well measurement from one experiment; for each cell line (CD04 and CD09) and condition (non-risk, risk-allele and risk-CRISPRa), data are from one clone (per line), collected from 2 independent experiments each with 3 wells of differentiations (n = 6). One-way ANOVA with experimental round as the random factor; Dunnett’s correction. * P < 0.05, ** P < 0.01, ***, P < 0.001; mean ± s.e.m. (c) Pie charts depict the proportion of iMG stained positive for Aβ-pHrodo, BODIPY, or both from co-localization analysis of the fluorescence images in (a). Note the Aβ-pHrodo+/BODIPY+ iMG are rare, and PICALM-CRISPRa rescues the phagocytosis deficit in LOAD risk-allele iMG by mainly converting BODIPY+ iMG to phagocytic cells without LD (Aβ-pHrodo+/BODIPY−).
Extended Data Fig. 9 PICALM knockdown (KD) or knockout (KO) causes LD accumulation.
(a) Representative fluorescence staining of LD (BODIPY+) in iMG carrying risk or non-risk allele and in non-risk-CRISPRoff iMG. Scale bar, 50 μm. (b) to (e) Increased LD (BODIPY+) area, density per cell, size, and fluorescence intensity in iMG carrying the LOAD risk-allele (vs. non-risk) for both CD04 and CD09 lines. Note iMG with PICALM-CRISPRoff also show increased LD accumulation (vs. non-risk allele), mimicking the effect of the PICALM risk allele. (f) LD staining with LipidTox. Scale bar, 50 µm. (g) to (j) Increased LD density, size, area, and fluorescence intensity in iMG carrying the LOAD risk-allele (vs. non-risk). PICALM-CRISPRoff reduces LD density, size, area, and fluorescence intensity (vs. non-risk allele). For (b) to (e) and (g) to (j), each datapoint represents a single-well measurement (FOV) from one experiment; for each donor line (CD04 and CD09) and each condition (risk, non-risk, and CRISPRoff), data are from one experiment with 3 wells of differentiations each with 3-4 FOV. One-way ANOVA; Dunnett’s correction. (k) LD (BODIPY+) staining in PICALM-KO C20 cells and (l) quantification of LD number per cell. Scale bar, 25 µm. Each datapoint represents a single-well measurement (averaged from 5 FOV) from one experiment; data are from 3 experiments (n = 3). Two-sided unpaired Student’s t-test was used. For all comparisons, * P < 0.05, ** P < 0.01, ***, P < 0.001; mean ± s.e.m.
Extended Data Fig. 10 Lipidomics profiling in iMG and the effect of PICALM on neuron-to-glia lipid transfer.
(a) Volcano plot showing the log2 (FC) of the 901 detected lipid species in risk iMG (vs. non-risk iMG; CD04 and CD09 lines) and the -log10 (P-value) from one-way ANOVA. The top 20 increased lipid species are highlighted, most of which are TG. (b) Heatmap of row Z-scored lipid abundances across non-risk, risk-allele, and risk-CRISPRa iMG for the lipid species increased in risk-allele iMG (vs. non-risk iMG) at FDR < 0.05. (c) Normalized abundance of total TG in non-risk, risk-allele, and risk-CRISPRa iMG for CD04 and CD09 lines. For each cell line (CD04 and CD09) and each condition (non-risk, risk, risk-CRISPRa), data for (a) to (c) are from one experiment with 2 wells of differentiations each with 2 technical replicates (n = 4). One-way ANOVA; Dunnett’s correction. (d) Diagram of the lipid (Red-C12 is a fatty acid analogue) transfer system between neurons (iN-Glut) and iMG or iAst. iN-Glut was labelled with Red-C12 for 18 hr, then co-cultured with iMG or iAst for 4 hr before staining iMG or iAst with BODIPY. Illustrations were created using BioRender. Sudwarts, A. (2025) https://BioRender.com/flsko8h. (e) Red-C12 imaging in the co-cultured iMG carrying LOAD risk-allele or non-risk allele. Scale bar, 50 µm. (f) and (g) Quantification of the transferred Red-C12 area and density in iMG. Each datapoint represents single-well measurement (FOV) of an experiment; for each condition (risk and non-risk), data are from line CD04 (one clone), collected from one experiment with 3 wells of differentiations each with 3-4 FOVs (n = 9-12). (h) Red-C12 imaging and LD (BODIPY+) staining of the co-cultured iAst with PICALM-CRISPRoff (over the genetic background of non-risk allele since the LOAD risk-allele of PICALM does not affect its expression in iAst). Note the partial overlap of Red-C12 and BODIPY staining, suggesting the transferred Red-C12 partially contributes to LD formation in iAst. Scale bar, 50 µm. (i) and (j) Quantifying LD density and area in the co-cultured iAst. (k) and (l) Quantifying the Red-C12 density and area in iAst. Each datapoint represents single-well measurement (FOV) of an experiment; for each condition (non-risk and CRISPRoff), data are from line CD04 (one clone), collected from one experiment with 3 wells of differentiations each with 3-4 FOVs (n = 9-12). (f) to (l), two-sided unpaired Student’s t-test was used. For all comparisons, * P < 0.05, ** P < 0.01, ***, P < 0.001; mean ± s.e.m.
Extended Data Fig. 11 iMG carrying the LOAD risk-allele of rs10792832 show increased lipid peroxidation.
(a) The schematics show C11-BODIPY (581/591), a fluorescent lipid peroxidation sensor, shifts its fluorescence from red to green in the presence of ROS. Illustrations were created using BioRender. Sudwarts, A. (2025) https://BioRender.com/21bufax. (b) Fluorescence staining with peroxidation sensor C11-BODIPY in iMG (TREM2+) carrying the LOAD risk or non-risk allele in the presence or absence of TrC. Scale bar, 50 µm. (c) and (d) Increased LD (C11-BODIPY green fluorescence) density and area in iMG carrying the LOAD risk-allele (vs. non-risk). Note the peroxidized LD density and area are reduced in iMG treated with TrC. (e) The ratio of peroxidized vs. non-peroxidized (BD.11 Ox/BD.11 non−ox) lipids in iMG carrying the LOAD risk-allele or non-risk allele, with or without TrC treatment. Each datapoint represents a single-well measurement (FOV) of one experiment; for each condition (risk, risk-TrC, non-risk, and non-risk-TrC), data are from line CD04 (one clone), collected from one experiment with 3 wells of differentiations each with 2-3 FOV. For all comparisons, one-way ANOVA with Tukey’s correction; * P < 0.05, ** P < 0.01, ***, P < 0.001; mean ± s.e.m. (f) Mechanistic insight on the link between the LOAD GWAS SNP risk allele and the reduced PICALM expression, lysosomal dysfunction, LD accumulation, LD peroxidation, cellular ROX level, and phagocytosis deficits in iMG. (-), inhibition; FA: fatty acids; Px: peroxisome; M: mitochondria; sER: smooth ER; Ly: lysosome; Chl: cholesterol. Illustrations were created using BioRender. Sudwarts, A. (2025) https://BioRender.com/bmv8afh.
Extended Data Fig. 12 LD accumulation impairs iMG phagocytosis.
(a) Representative fluorescence images show time-dependent phagocytosis of Aβ-pHrodo and LD accumulation (BODIPY+) in iMG (CD45+) carrying the LOAD risk-allele or non-risk allele of PICALM in the presence or absence of TrC. Scale bar, 100 μm. (b) Reduced Aβ-pHrodo intensity in iMG carrying the LOAD risk-allele (vs. non-risk) can be rescued by TrC treatment. (c) TrC treatment rescues the LD in iMG carrying the LOAD risk-allele to a level similar to that in MG with the non-risk allele. (d) Pie charts show the proportion of iMG (CD45+) stained positive for Aβ-pHrodo, BODIPY, or both from co-localization analysis of the fluorescence images in (a). Note that TrC treatment rescues the phagocytosis deficit in iMG carrying the LOAD risk-allele by mainly converting BODIPY+ iMG to phagocytic cells without LD (Aβ-pHrodo+/BODIPY-). (e) to (h) The same as in (a) to (d) except that the phagocytosis assay was for myelin-pHrodo. In (a) to (h), each datapoint represents a single-well measurement (FOV) from one experiment; for each condition (risk, risk-TrC, and non-risk), data are from CD09 line (one clone), collected from one experiment with 2-3 differentiations each with 2–4 FOV. One-way ANOVA with Dunnett’s correction; * P < 0.05, ** P < 0.01, ***, P < 0.001; mean ± s.e.m.
Supplementary information
Supplementary Information
Supplementary Figs. 1–10, the legends for Supplementary Tables 1–19 and Supplementary References.
Supplementary Tables
Supplementary Tables 1–19.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kozlova, A., Zhang, S., Sudwarts, A. et al. PICALM Alzheimer’s risk allele causes aberrant lipid droplets in microglia. Nature (2025). https://doi.org/10.1038/s41586-025-09486-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41586-025-09486-x
