
Abstract
Oxygen redox chemistry is central to life1 and many human-made technologies, such as in energy storage2,3,4. The large energy gain from oxygen redox reactions is often connected with the occurrence of harmful reactive oxygen species3,5,6. Key species are superoxide and the highly reactive singlet oxygen3,4,5,6,7, which may evolve from superoxide. However, the factors determining the formation of singlet oxygen, rather than the relatively unreactive triplet oxygen, are unknown. Here we report that the release of triplet or singlet oxygen is governed by individual Marcus normal and inverted region behaviour. We found that as the driving force for the reaction increases, the initially dominant evolution of triplet oxygen slows down, and singlet oxygen evolution becomes predominant with higher maximum kinetics. This behaviour also applies to the widely observed superoxide disproportionation, in which one superoxide is oxidized by another, in both non-aqueous and aqueous systems, with Lewis and Brønsted acidity controlling the driving forces. Singlet oxygen yields governed by these conditions are relevant, for example, in batteries or cellular organelles in which superoxide forms. Our findings suggest ways to understand and control spin states and kinetics in oxygen redox chemistry, with implications for fields, including life sciences, pure chemistry and energy storage.
Similar content being viewed by others

Main
More than half a century after the discovery that singlet oxygen forms from superoxide8, what governs the evolution of singlet or triplet oxygen under many conditions relevant to life and human-made oxygen redox systems remains unknown. Oxygen redox chemistry is crucial to life1 and encompasses some of the most fundamental and widespread chemical reactions, including those in batteries2,3,4,9, fuel cells and electrocatalysis10, and organic chemistry11. The oxidation states that are accessed in these reactions range from 0 (dioxygen) to –2 (oxide), with intermediate redox states of –½ and –1, pertaining to superoxide (O2−) and peroxide (O22−) or the oxide radical (O•–). Superoxide is pivotal in oxygen reduction and evolution reactions because it is the closest oxidation state to dioxygen. Dioxygen appears either in its ground state as triplet oxygen (3O2 or 3Σg–) or as singlet oxygen (1O2 or 1Δg) in its first electronically excited state. Whereas 3O2 is relatively unreactive, 1O2 is highly reactive with most organic matter11,12. This is readily evidenced in metal-ion batteries6,7 and metal-O2 batteries3,4, in which 1O2 is the primary source of degradation, causing decomposition of organic electrolytes and conductive carbon additives, which ultimately degrades the overall device function. Moreover, 1O2 and (su)peroxide are well-known reactive oxygen species in biological systems and are involved in several processes from signalling to cell damage1,5.
Superoxide liberates oxygen under a broad range of oxidizing conditions. A prevalent process is disproportionation due to the instability of superoxides in most of the environments3,9,13. This reaction occurs in both protic (aqueous)14,15 and aprotic environments with relatively strong Lewis acids such as Li+ and Na+ (refs. 6,9,13,16). During disproportionation (2O2− → O2 + O22−), one superoxide molecule is reduced to peroxide, whereas the other is oxidized to form either 3O2 or 1O2. Examples can be found in both cellular respiration1,5 and batteries13,16,17,18,19,20. Relative 3O2 and 1O2 yields and kinetics of superoxide disproportionation, and superoxide oxidation more generally, are therefore fundamental to these systems, but underlying mechanisms are still unknown.
Here we examined 1O2 evolution from the oxidation of superoxide through both chemical reaction and disproportionation over a wide range of driving forces. On the basis of this, we observe distinct Marcus normal and inverted region behaviour for 3O2 and 1O2 evolution, which explains 1O2 formation over a broad range of scenarios in which superoxide oxidation occurs, for example, in biological systems and energy storage.
Excited species through electron transfer
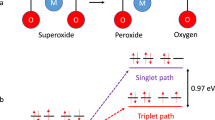
Superoxide oxidation may form both 3O2 and 1O2 according to Wigner–Witmer spin conservation rules21. This applies to disproportionation, in which two doublet superoxides can produce triplet or singlet products17, and to oxidation by a redox mediator (RMox) to form one excited (1O2) and one ground state molecule (the reduced mediator, RMred) (ref. 22).
We previously investigated the mediated oxidation of superoxide through the reaction KO2 + RMox → O2 + RMred + K+ in the search for possible 1O2 evolution18. The potential \({E}_{{\text{RM}}^{\text{ox}/\text{red}}}^{\circ }\) was limited to moderate values because of the stability of the ether electrolyte used. We measured 3O2 and 1O2 yields using mass spectrometry and a chemical trap, respectively, and tracked the reaction kinetics using ultraviolet–visible (UV–Vis) spectroscopy. The data shown in Extended Data Fig. 1 supported two main conclusions: first, 1O2 was observed with RMs with \({E}_{{\text{RM}}^{\text{ox}/\text{red}}}^{\circ }\gtrsim {E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\text{KO}}_{2}}^{\circ }+1\,{\rm{V}}\), which could be in accordance with the frequently quoted threshold3,23,24,25,26 for 1O2 evolution based on the energy difference of 0.97 eV between 3O2 and 1O2; and second, the kinetics of superoxide oxidation over the measured range of driving forces exhibited Marcus normal and inverted region behaviour. This behaviour is characterized by a parabolic relationship between the logarithm of the kinetic constant k and the driving force (free energy change) \(-\Delta {G}^{\circ }=\left({E}_{{\text{RM}}^{\text{ox}/\text{red}}}^{\circ }-{E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\text{KO}}_{2}}^{\circ }\right)F\):
where Zel is the collision factor, λ is the reorganization energy and F is the Faraday constant22,27,28. The kinetic constant k reaches a peak when –ΔG° = λ and electron transfer becomes barrierless. The inverted region results from growing barriers at even higher driving forces. The data suggested that superoxide oxidation kinetics follow a single parabola and that some 1O2 is generated for –ΔG° ≳ 0.97 eV. However, what controls the extent to which 1O2 or 3O2 evolve is yet unknown.
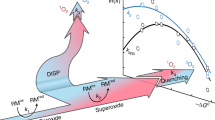
1O2 evolution should be a distinct elementary step both thermodynamically and kinetically. We, therefore, propose that the observed parabola in Extended Data Fig. 1 describes oxidation to 3O2, and a second parabola should appear at sufficiently large driving forces, representing oxidation to 1O2 (Fig. 1a). The intersection of these two parabolas would indicate the transition from 3O2 to 1O2. This follows classical work by Marcus on the electrogeneration of electronically excited species22,27. However, this hypothesis has not yet been experimentally evaluated for 1O2 generation.

a, Hypothesis for how the driving force could govern 3O2 and 1O2 formation kinetics from superoxide oxidation. The left parabola results from previously measured rate constants for mediated superoxide oxidation with driving forces up to –ΔG° ≈ 1.2 eV as shown in Extended Data Fig. 1. However, a single kinetic parabola could not conclusively explain why 1O2 formed for –ΔG° ≳ 1 eV. Based on the considerations in b, individual kinetic parabolas (k3 and k1) for the reactions yielding 3O2 and 1O2 can be constructed with the full line showing their sum. The maxima are shifted by \({\Delta G}_{1\leftarrow 3}^{\circ }\approx 0.97{\rm{eV}}\) (see text), and equal prefactors and reorganization energies are assumed. The blue- and red-shaded area shows the transition from k3/(k1+3) = 0.99 to k1/(k1+3) = 0.99. b, Potential energy surfaces for mediated superoxide oxidation for different driving forces. Black, blue and red parabolas denote reactants (KO2 + RMox) and 3O2 or 1O2 in the products (3O2 + RMred or 1O2 + RMred), respectively. The cases shown are for barrierless reactions to 3O2 (i) and 1O2 (iii) and equal barriers (ii). The subscripts 3 and 1 denote triplet and singlet states, respectively.
Based on equation (1), a kinetic expression with separate terms for 3O2 and 1O2 evolution can be given as
The subscripts 3 and 1 on k, Zel and λ denote values for 3O2 and 1O2, respectively. The blue- and red-dashed parabolas in Fig. 1a represent the terms for 3O2 and 1O2, whereas the solid line shows their sum. The k3 parabola results from the transition from the black to the blue potential energy surfaces in Fig. 1b, which are shown for –ΔG° ≈ λ3 for case (i). The vibrational ground states of 3O2 and 1O2 differ by \({\Delta H}_{1\leftarrow 3}^{\circ }=\Delta {H}^{\circ }\left(\genfrac{}{}{0ex}{}{1}{}\,{\Delta }_{\text{g}}\leftarrow \genfrac{}{}{0ex}{}{3}{}\,{\Sigma }_{\text{g}}^{-}\right)=0.97\,{\rm{eV}}\), where ΔH is the enthalpy change12. Assuming, for now, a vanishing entropy change ΔS as typically done in electrochemiluminescence literature28, \({\Delta G}_{1\leftarrow 3}^{\circ }=\Delta {G}^{\circ }\left(\genfrac{}{}{0ex}{}{1}{}\,{\Delta }_{\text{g}}\leftarrow \genfrac{}{}{0ex}{}{3}{}\,{\Sigma }_{\text{g}}^{-}\right)\approx {\Delta H}_{1\leftarrow 3}^{\circ }\), by which the singlet potential energy surface shifts vertically (Fig. 1b, red). For sufficiently large driving forces, the barriers ΔG‡ and kinetics for crossing to 3O2 and 1O2 become equal (Fig. 1, case (ii)) or even barrierless to 1O2 (case (iii)). Hence, individual kinetic parabolas for superoxide oxidation to 3O2 and 1O2 can be constructed. The hypothetical parabola for 1O2 evolution can be drawn if we further assume, for now, equal collision factors Zel,1 = Zel,1 and equal reorganization energies λ1 = λ3 as fitted to the data in Extended Data Fig. 1. The theory developed initially for homogeneous electron transfer can still be used for semiconductors or insulators, as noted by Marcus22. Therefore, the considerations in Fig. 1 apply to the oxidation of solid superoxides and superoxide solutions.
Kinetics over extended driving forces
To test the hypothesis of individual kinetic parabolas for 3O2 and 1O2 evolution, we selected acetonitrile (MeCN) as the solvent. Acetonitrile is highly stable against oxidation and, therefore, allows for large driving forces (Extended Data Fig. 2a) to be tested. We used a wide range of mediators (Extended Data Fig. 2b) with redox potentials that exceed the expected maximum of the 1O2 parabola. Figure 2a shows the measured kinetics as a function of driving force relative to \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\text{KO}}_{2}}^{\circ }\) (Methods). The measured kinetics can be adequately fitted to equation (2). The blue- and red-dashed parabolas represent the terms for 3O2 and 1O2 evolution, whereas the solid line shows their sum. Evidence for these parabolas corresponding to 3O2 and 1O2 is provided by measured yields (Fig. 2b,c). The 3O2 yields were measured by mass spectrometry and quantified as the molar ratio 3O2/RMox. 1O2 formation was measured by its specific 1,270 nm near-infrared (NIR) radiation (Methods). Absolute quantification of 1O2 is difficult, and values were, therefore, normalized to the maximum observed.

a, We measured the kinetic constants k for mediated KO2 oxidation in MeCN electrolyte with mediators covering a large range of redox potentials. Plot of ln(k) compared with the mediator potential (\({E}_{{\text{RM}}^{\text{ox}/\text{red}}}^{\circ }\), top axis) and driving force (–ΔG°, bottom axis). \(-\Delta {G}^{\circ }=\left({E}_{{\text{RM}}^{\text{ox}/\text{red}}}^{\circ }-{E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\text{KO}}_{2}}^{\circ }\right)\,F\), where \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\text{KO}}_{2}}^{\circ }=2.48\,{\rm{V}}\) on the K/K+ scale. The mediators are shown in Extended Data Fig. 2. The full line best fits equation (2); the broken line parabolas represent the first and second terms in equation (2). The fitted values are Zel,3 = 1.10 × 10–2 cm s–1, Zel,1 = 7.00 × 10–2 cm s–1, λ3 = λ1 = 0.95 eV, \({\Delta G}_{1\leftarrow 3}^{\circ }=0.84\,{\rm{eV}}\) and R2 = 0.998. Based on these fits, the standard potential \({E}_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}/{\text{KO}}_{2}}^{\circ }\) and associated driving force \({\Delta G}_{1\leftarrow 3}^{\circ }\) are marked. They are linked by \({E}_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}/{\text{KO}}_{2}}^{\circ }\,=\,{E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\text{KO}}_{2}}^{\circ }+{\Delta G}_{1\leftarrow 3}^{\circ }/F\). The blue- and red-shaded area indicates the transition from k3/(k1+3) = 0.99 to k1/(k1+3) = 0.99. b, 3O2 yield per mole of RMox (bars) during KO2 oxidation as measured by mass spectrometry. The dashed line and the circular markers show simulated 3O2 yields considering 1O2 quenching by solvent and redox mediator (Methods and Extended Data Fig. 3). The dashed line used the trend line for the mediator quenching rate constant kQ, whereas the markers use the individually measured values (Extended Data Fig. 3c). c, Normalized 1,270 nm NIR emission (bars) during KO2 oxidation. The dashed line and the circular markers show the simulated NIR emission considering 1O2 formation with the kinetics k1 (the right parabola in a) and 1O2 quenching by solvent and redox mediator (Methods and Extended Data Fig. 3). Data are presented as mean ± s.d. (n ≥ 3).
Observable yields of 3O2/RMox and NIR intensities do not simply resemble the relative kinetics of 3O2 and 1O2 formation as obtained in Fig. 2a. Instead, the formed 1O2 undergoes multiple decay pathways, of which some yield 3O2 and only a small fraction emits the NIR radiation12. To rationalize the measured values, we simulated them based on formation rates and 1O2 decay processes (Methods and Extended Data Fig. 3): (1) 3O2 and 1O2 formation rates as given by the parabolas in Fig. 2a; (2) physical and reactive 1O2 quenching by the solvent; (3) physical and reactive 1O2 quenching by the mediators; and (4) 3O2 losses resulting from reactive quenching of 1O2 with solvent or mediator. Simulation results are shown as dashed lines and markers in Fig. 2b,c and resemble the measured data. Deviations result from simplifications of the model, such as not accounting for specific reactivities of the chemically diverse mediators (Methods). Overall, a proper fit of the kinetics to the double parabola in equation (2) (Fig. 2a), the deficiencies in 3O2 yields and the growing NIR signal beyond the predicted onset potential (Fig. 2b,c) all support the proposed hypothesis that individual Marcus parabolas govern 3O2 and 1O2 formation from superoxide.
The adequate fit supports several important conclusions: (1) The prefactors Zel,1 ≈ 6.3 × Zel,3 result in substantially higher maximum kinetics for 1O2 evolution. (2) Driving forces for maximum kinetics are solvent-dependent, as shown by the comparison between values in ether solvent (Fig. 1a; λ3 = 0.61 eV) and acetonitrile (Fig. 2a; λ3 = 0.95 eV). This aligns with the prediction27 of Marcus and work by Miller29. The crossing point of the two parabolas is not at a constant driving force, which benefits 1O2 for solvents with lower λ (Extended Data Fig. 4a). (3) \({E}_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}/{\rm{superoxide}}}^{\circ }\) should not be considered the threshold above which 1O2 rather than 3O2 forms18,24,26 as suggested from the electrochemiluminescence literature (Methods) but as a possible onset of 1O2. Reaching \({E}_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}/{\rm{superoxide}}}^{\circ }\) per se tells little about the relative 3O2 and 1O2 formation kinetics because of the solvent-dependent reorganization energy (Extended Data Fig. 4). Instead, driving forces –ΔG° > λ3 are required to slow 3O2 formation to benefit 1O2. (4) In the electrochemiluminescence literature, the entropy change is unknown and typically neglected28. Having data that show the peaks for 3O2 and 1O2 permits us to determine \({\Delta G}_{1\leftarrow 3}^{\circ }=0.84\,{\rm{eV}}\) and with \({\Delta H}_{1\leftarrow 3}^{\circ }=\,{\Delta G}_{1\leftarrow 3}^{\circ }+T{\Delta S}_{1\leftarrow 3}^{\circ }\) we obtain \(T{\Delta S}_{1\leftarrow 3}^{\circ }=-\,0.13\,{\rm{eV}}\). Consequently, \({E}_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}/{\rm{superoxide}}}^{\circ }={E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\rm{superoxide}}}^{\circ }+0.84\,{\rm{V}}\) can now be given more precisely, rather than with the typically used difference of 0.97 V.
Figure 2a establishes a working curve for an extensive range of ΔG°, facilitating an understanding of the behaviour of various important systems. Lewis-acid- and Brønsted-acid-driven superoxide disproportionation are two widely relevant cases, examined in the following sections. More examples of superoxide oxidation, in which an explanation of whether, or not, 1O2 forms has been unknown, but which can now be explained, are examined in Extended Data Fig. 4 and the Methods. These examples include superoxide in contact with CO2 and organic peroxides, with relevance for energy storage and biological systems1,4,30.
Disproportionation in non-aqueous systems
1O2 is known to cause degradation in non-aqueous alkali metal-O2 batteries3,13,16,17,18,19. The Li–O2 battery, for example, operates by reversibly forming lithium peroxide at the positive electrode, O2 + 2e– + 2Li+ ⇄ Li2O2. Initially formed LiO2 disproportionates, which always results in some 1O2 according to 2LiO2 → Li2O2 + x3O2 + (1 − x)1O2 (refs. 13,16,17,18,19). Weak Lewis acids such as tetrabutylammonium (TBA+), or other similar cations from ionic liquid electrolytes, are often present in these cells. Despite not driving disproportionation themselves, weak Lewis acids were found to raise the 1O2 fraction from about 2% in pure Li+ electrolyte to around 20% for 1/1 Li+/TBA+ electrolyte16. However, to date, it has been unclear why pure Li+ generates 1O2 at all and why weak Lewis acids should increase the 1O2 fraction.
Figure 3a shows the thermodynamics of the relevant redox couples as a function of the Li+ and TBA+ salt fractions in glyme electrolyte. Figure 3b shows the NIR emission intensities on adding KO2 as a superoxide source into these electrolytes. The thermodynamics are explored in further detail in the Methods and Extended Data Fig. 5 and are summarized here. Li2O2 is insoluble, and the potential \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{{\rm{Li}}}_{2}{{\rm{O}}}_{2}({\rm{s}})}\), therefore, fixed (Fig. 3a, black line). However, superoxide is appreciably soluble in non-aqueous electrolytes31,32,33: species include \({({{\rm{Li}}}^{+}{{{\rm{O}}}_{2}}^{-})}_{n\ge 1,({\rm{sln}})}\) clusters or ion pairs, where (sln) denotes solvate species. Solid LiO2(s) has the lowest Gibbs free energy, and increasingly solvated species are increasingly less stable32,33,34. The 3O2/superoxide potentials are, therefore, within the upper limit of \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\rm{Li}}{{\rm{O}}}_{2}({\rm{s}})}\) and the lower limit of \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{({{\rm{Li}}}^{+}{{\rm{O}}}_{2}^{-})}_{({\rm{sln}})}}\) (Fig. 3a, light-blue gradient-coloured box). When TBA+ is present, the lower limit extends to \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{({{\rm{TBA}}}^{+}{{\rm{O}}}_{2}^{-})}_{({\rm{sln}})}}\).

a, Thermodynamics of relevant redox couples for Li+-induced superoxide disproportionation as a function of the fractions of Li+ and TBA+ salt. The gradient boxes and short arrows indicate increasing superoxide solvation with potentials between values relevant for solid LiO2(s) (dark colour), solvated \({({{\rm{Li}}}^{+}{{{\rm{O}}}_{2}}^{-})}_{n\ge 1,({\rm{sln}})}\) clusters, and solvated \({({{\rm{Li}}}^{+}{{{\rm{O}}}_{2}}^{-})}_{({\rm{sln}})}\) (faded colour). LiO2 is in all relevant electrolytes at least somewhat soluble as \({({{\rm{Li}}}^{+}{{{\rm{O}}}_{2}}^{-})}_{n\ge 1,({\rm{sln}})}\) clusters. The nature of superoxide shifts from \({({{\rm{Li}}}^{+}{{{\rm{O}}}_{2}}^{-})}_{n\ge 1,({\rm{sln}})}\) towards \({({{\rm{TBA}}}^{+}{{{\rm{O}}}_{2}}^{-})}_{({\rm{sln}})}\) as the cation changes from pure Li+ towards pure TBA+. The inclined lines indicate the associated shift of the potentials. \({E}_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}/{\rm{superoxide}}}={E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\rm{superoxide}}}+0.84\,{\rm{V}}\). The driving force \(\Delta G=-\,({E}_{{\rm{superoxide}}/{\text{Li}}_{2}{\text{O}}_{2}}-{E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\rm{superoxide}}})F\) grows with the shift of the O2/superoxide and superoxide/Li2O2(s) potentials. Note that the O2 reduction potential changes nonlinearly with the Li+:TBA+ ratio32. b, Normalized 1,270 nm NIR emission as a function of Li+ mole fraction, which determines the driving force. The markers show superoxide disproportionation kinetics as measured by pressure evolution. The dotted line is a guide to the eye. Glyme served as the electrolyte solvent. Data are presented as mean ± s.d. (n ≥ 3).
The superoxide/Li2O2(s) couple acts as the oxidant during superoxide disproportionation. \({E}_{{\rm{superoxide}}/{{\rm{Li}}}_{2}{{\rm{O}}}_{2(\text{s})}}\) exceeds \({E}_{{{}^{1}{\rm{O}}}_{2}/{\rm{superoxide}}}\) already in pure Li+ electrolyte, in which the superoxide species are solvated, and therefore explains why an onset of 1O2 is observed. 1O2 is detected in pure Li+ electrolyte using more sensitive chemical trapping16. Addition of TBA+ favours more solvated species, which also explains why TBA+ increases the fraction of 1O2. In line with this, Fig. 3b shows steeply increasing 1O2 evolution as the TBA+ mole fraction increases, along with faster kinetics.
Proton-induced disproportionation
We next investigated how pH-related changes in driving force affect the 1O2 formation from proton-induced superoxide disproportionation. This is particularly relevant for living organisms, in which superoxide is found in organelles with pH levels ranging between 4.7 and 8 (ref. 35), as well as in aqueous electrocatalysis10. Figure 4a shows the Pourbaix diagram with the potentials of relevant redox couples as a function of pH. Again, the driving force for disproportionation is the difference between the 3O2/superoxide and superoxide/peroxide couple, marked with the vertical arrows. It grows from about 0.5 eV at pH 14 to about 1.5 eV below pH 4.8 (Fig. 4b). The oxidant potential exceeds \({E}_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}/{{\rm{O}}}_{2}^{-}}^{\circ }\) at pH about 10.5, in which an onset of 1O2 formation can be expected.

a, Pourbaix diagram showing the pH-dependent stability range of aqueous electrolyte and the standard potentials \({E}^{\circ }\) of the indicated redox couples on the normal hydrogen electrode (NHE) scale15. The kinks in the curves arise from the pKa values of H2O2 (pKa = 11.7) and HO2 (pKa = 4.8). For simplicity, O2–/H2O2 is written while the pH-dependent (de)protonated species are meant. \({E}_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}/{{{\rm{O}}}_{2}}^{-}}^{\circ }={E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{{\rm{O}}}_{2}^{-}}^{\circ }+0.84\,{\rm{V}}.\) b, The pH-dependent driving force for superoxide disproportionation is \(-\Delta {G}^{\circ }=({E}_{{{\rm{O}}}_{2}^{-}/{\text{H}}_{2}{\text{O}}_{2}}^{\circ }-{E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{{\rm{O}}}_{2}^{-}}^{\circ })F\) as indicated by the vertical arrows in a. c, 1O2-specific NIR emission at 1,270 nm as a function of pH. Each marker represents a single measurement. The dotted line is a guide to the eye. PBS, phosphate-buffered saline.
Figure 4c shows the 1O2-specific NIR signal at 1,270 nm on proton-induced superoxide disproportionation over a pH range from about 1 to 10.8. We exposed KO2 to various buffer solutions under vigorous stirring and recorded the NIR signal (Methods). A strongly increasing NIR signal with decreasing pH is in accord with the increasing driving force. A pH of around 11 giving low but non-negligible 1O2 yields is consistent with reliable theoretical17 and experimental works14 on the reaction \({{\rm{HO}}}_{2}+{{\rm{O}}}_{2}^{-}\to {{\rm{O}}}_{2}+{{\rm{O}}}_{2}^{-}\), which found 1O2 evolution only about 0.3 eV endoergic and around 0.2% 1O2 production on exposing KO2 to H2O2 as the proton source.
Conclusions
We found that the driving force for superoxide oxidation to 3O2 and 1O2 is the common descriptor that determines the spin state, following individual Marcus normal and inverted region behaviour. 1O2 can become significant only because the kinetics for 3O2 evolution slows down in its inverted region. The results help clarify previously inconclusive findings about 1O2 formation from superoxide, including through interaction with chemical oxidants, and proton and Lewis-acid-driven disproportionation. For disproportionation, the results explain the increasing 1O2 formation with stronger Brønsted and weaker Lewis acidity, respectively, because of their impact on driving forces. Increasing 1O2 yield with lower pH aligns with higher driving forces as the pH decreases. This corresponds with higher and lower pH in respiratory (mitochondria) and degenerative organelles (lysosomes)35, respectively, in which 1O2 must be avoided or may even be beneficial. This connection between pH and 1O2 formation may well have been a so far unrecognized evolutionary driver for the pH found in organelles. In human-made redox systems, in which 1O2 is, in most cases, detrimental and damaging, strategies to suppress 1O2 should aim at reducing the driving forces for superoxide oxidation, increasing the reorganization energy, or avoiding situations in which superoxide disproportionates. We discuss the wider relevance for oxygen redox systems in life sciences and energy, and for the electrogeneration of excited species more generally in the Methods. The findings offer insights into understanding and controlling spin states and kinetics in oxygen redox chemistry, with implications for fields such as the life sciences and energy storage.
Methods
Materials
All chemicals were from Sigma Aldrich unless indicated differently. Potassium superoxide (KO2), potassium perchlorate (KClO4, ≥99.99%), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, 99.99%) and tetrabutylammonium bis(trifluoromethanesulfonyl)imide (TBATFSI, ≥99.0%) were dried under reduced pressure for 24 h at 100 °C. Decamethylferrocene (DMFc), tris[4-(diethylamino)phenyl]amine (TDPA), N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD), ferrocene (Fc), ferrocenium tetrafluoroborate (FcBF4), 9,10-diphenylanthracene (DPA), 1,4-bis(diphenylamino)benzene (DPAB), thianthrene (ThA), 1,4-dimethoxyanthracene (DMeOA, Acros Organics), 1,4-di-tert-butyl-2,5-dimethoxybenzene (tMeOB, BLDpharm) and 5,10-dihydro-5,10-dimethylphenazine (DMPZ, TCI chemicals) were used as received. N-methyl phenothiazine (MPT), 1,4-diazabicyclo[2.2.2]octane (DABCO) were sublimated. 18-Crown-6 and tetrafluorobenzoquinone (F4BQ) were recrystallized from ethanol. Tetraethylene glycol dimethyl ether (TEGDME, ≥99%) was dried over lithium, distilled under vacuum and further stored over activated 3 Å molecular sieves. Acetonitrile (MeCN, 99.8% anhydrous) was stored over activated molecular sieves. Both solvents had a water content below 5 ppm as determined by Karl–Fischer titration (Mettler Toledo). All non-aqueous experiments were performed in an Ar-filled glovebox (Vigor) or hermetically sealed setups without air exposure. The structures of the mediators and their redox potentials are shown in Extended Data Fig. 2.
Electrochemistry and mediator oxidation
Electrochemistry experiments were performed using a BioLogic potentiostat (SP-300 and MPG-2). Cyclic voltammetry was performed with a glassy carbon disk as the working electrode and a glassy carbon rod as the counter electrode in a one-compartment glass cell. Partially delithiated Li(1–x)FePO4 (LFP, MTI), separated by a Vycor glass frit, was used as the reference electrode. DMFc/DMFc+ was used as the internal standard and converted using \({E}_{{\rm{DMFc}}/{{\rm{DMFc}}}^{+}}^{\circ }={E}_{{\rm{Li}}/{{\rm{Li}}}^{+}}^{\circ }+3.16\,{\rm{V}}={E}_{{\rm{K}}/{{\rm{K}}}^{+}}^{\circ }\)\(+3.02\,{\rm{V}}.\) DMeOA, tMeOB, DPA and ThA were electrochemically oxidized in an H-cell. The oxidation compartment contained a Pt working electrode, the reference electrode and 5 ml MeCN containing 2 mM RM and 10 mM KClO4. The reduction compartment contained Ni foam as a counterelectrode with 5 ml of 100 mM 1,4-benzoquinone (sublimed) in MeCN. A K+ selective ion-exchange membrane separated the two compartments. RMs were oxidized galvanostatically to 80% of their total capacity. DMFc, TDPA, TMPD, DMPZ, MPT and DPAB were oxidized using 1 equiv. NOBF4 in MeCN. After 3 h stirring, they were precipitated with cold diethyl ether, filtered and dried under a vacuum at 30 °C for 12 h. F4BQ is the oxidized form itself.
Measurements of kinetics
Kinetics of mediated superoxide oxidation were measured using UV–Vis spectroscopy using an Avantes AvaSpec-HSC spectrometer with AVALIGHT-DH-S-BAL light source and fibre optics to perform measurements inside the glove box. Pure KO2 powder was pressed into about 0.5 mm thick pellets using a 7 mm die set and a hand press (PIKE). In a 10-mm quartz cuvette (Hellma), a KO2 pellet was placed in a polytetrafluoroethylene frame for alignment, followed by a magnetic stirring bar and then the cuvette was sealed with a gas-tight injection lid. RMox solution containing 10 mM KClO4 was then injected using a gas-tight syringe (Hamilton). Consumption of RMox was followed except for F4BQ, in which formation of RMred was followed (Extended Data Fig. 7). Data and error bars are presented as mean ± s.d. (n ≥ 3). Error bars appear asymmetric on an ln(k) scale. Repetitions mean that each time a new portion of RMox was produced by electrochemically oxidizing a portion of RMred or by dissolving the chemically produced oxidized form. Differences in the magnitude of the error bars among mediators arise from their large chemical diversity and specific reactivities. For example, Fc+ and reduced quinones react with O2, or high-voltage RMox shows limited long-term stability in the electrolyte. For the latter, it was checked that degradation was at least several times slower than the oxidation of KO2.
Kinetics of Li+-induced superoxide disproportionation in TEGDME were measured by placing KO2 powder in a closed reaction vessel equipped with a pressure sensor (Omega, PAA35X) and injecting the Li+ electrolyte using a syringe through a septum (Extended Data Fig. 8).
3O2 yields and 1,270 nm emission measurements
3O2 yields on mediated superoxide oxidation were measured using mass spectrometry, as detailed previously30. The RMox solutions were injected using a gas-tight syringe, and the measurement continued until the O2 signal ceased. We used the 1O2-specific NIR emission at 1,270 nm from the decay of 1O2 to 3O2 to determine 1O2 yields and lifetimes as detailed previously30. The signal was recorded from the detector using an oscilloscope (Pico Technology) and at a gain of 820 V (control voltage). Extended Data Fig. 6 shows examples of the recorded signal during mediated oxidation, as well as Li+- and H+-induced disproportionation.
From oxidation rates to 3O2 yields and NIR emission intensities
We use the yields of 3O2/RMox and the normalized NIR intensities in Fig. 2b,c to prove that the two kinetic parabolas correspond to 3O2 and 1O2 evolution. To rationalize their assignment, we use a minimal model to calculate expected 3O2/RMox and NIR emission intensities based on formation rates and 1O2 decay processes. We considered the following processes: (1) 3O2 and 1O2 formation rates (k3 and k1) as given by the two kinetic parabolas in Fig. 2a; (2) physical and reactive 1O2 quenching by the solvent; (3) physical and reactive 1O2 quenching by the reduced mediators RMred. Note that the same processes with RMox are typically negligible in comparison because of the electron demand of these processes36; and (4) 3O2 losses (3O2/RMox < 1) resulting from reactive quenching of 1O2 with solvent or mediator.
Losses in 3O2/RMox in Fig. 2b result from incomplete physical quenching of 1O2 to 3O2 due to reactions of 1O2 with electrolyte or RM. Hence, we start by considering the decay routes. There are multiple decay routes, as shown in Extended Data Fig. 3f, and only a small fraction of the total 1O2 can be detected by the NIR emission at 1,270 nm (refs. 12,37). First, interactions of 1O2 with solvent will result in electronic-to-vibrational (e–v) deactivation and reactions with the solvent. The first-order 1O2 decay rate constant kD = kd,r + kd is composed of a reactive fraction kd,r and a non-reactive fraction kd. Second, electron-rich species, such as reduced mediators, exert charge transfer quenching; however, they may also react with the electrophilic 1O2 along pathways depending on the particular chemistry36. The rate constant kQ = kq,r + kq is equally composed of a reactive fraction kq,r and a non-reactive fraction kq. Third, radiative decay of 1O2 to 3O2 with the emission of a 1,270 nm photon.
We measured the rate constants kD and kQ using the luminescence lifetime. An O2-saturated solution containing Rose Bengal (absorbance of about 0.1 in a 1 cm fluorescence cuvette) was illuminated using a pulsed Coherent OBIS 561 nm laser. The lifetime (τ) and the decay constant (k = 1/τ) of 1O2 were measured using the NIR detector. τ was obtained by fitting the decay profile with exp(–t/τ) (Extended Data Fig. 3a). τ in the absence of a quencher relates to the solvent quenching rate constant kD. kQ is obtained by plotting 1/τ against the RMox concentration \({c}_{{{\rm{RM}}}^{{\rm{ox}}}}\) (Extended Data Fig. 3b,c). Similar to previously reported quenchers12, kQ decreases with \({E}_{{\text{RM}}^{\text{ox}/\text{red}}}^{\circ }\) following a trendline with a slope of about –103.5 V−1. The logarithmic dependence of the non-radiative fraction kq is explicable by the required partial charge transfer from the e–-rich quencher to the 1O2, which makes quenchers with lower redox potentials more efficient12. We consider that the reactive fraction kq,r depends similarly on redox potential, given that the addition reactions equally require charge transfer from the substrate to 1O2 (refs. 12,36). The scattering of kQ around the trendline arises from the large chemical diversity of the used mediators. We used both the trendline and the individual values of kQ for the prediction of NIR intensities in Fig. 2c. As for the fraction of reactive deactivation, kd,r/kD and kq,r/kQ, these can be determined from 3O2 consumption12, which we do below when simulating values of 3O2/RMox.
The surface area A of the KO2 powder, needed for the calculations, was determined by analysing optical images (Extended Data Fig. 3d). KO2 powder was dispersed in TEGDME, sonicated, a drop was placed between a microscope slide and a cover slip and sealed air-tight. Images were acquired with transmitted light on a Nikon Ti2E-01 inverted microscope using a Plan Apo λ 40×/0.95 DIC (Differential Interference Contrast) air PFS (Perfect Focus System) objective lens, resulting in a pixel size of 0.183 μm. The images were analysed using the ilastik pixel classification and object classification workflows (https://www.ilastik.org/), resulting in a histogram of particle sizes (Extended Data Fig. 3e). The surface area was calculated by assuming spherical particles, yielding A = 0.23 ± 0.04 m2 g–1.
As quantification of absolute 1O2 yields from NIR emission is not straightforward, we normalized the values. The NIR intensity at time t after bringing KO2 in contact with RMox will be proportional to the 1O2 concentration, \({I}_{1,270{\rm{nm}}}(t)\propto {c}_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}}(t)\). The 1O2 formation rate by mediated KO2 oxidation is \({\nu }_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}}(t)=A\cdot {k}_{1}\cdot {c}_{{{\rm{RM}}}^{{\rm{ox}}}}(t)\), with k1 being the rate constant as given in Fig. 2a. A is constant because the experiments were performed with a large excess of KO2 over RMox. \({c}_{{{\rm{RM}}}^{{\rm{ox}}}}\) and \({c}_{{{\rm{RM}}}^{{\rm{red}}}}\) are given by \({c}_{{{\rm{RM}}}^{{\rm{ox}}}}(t)={c}_{{{\rm{RM}}}^{{\rm{ox}}}}(0)\cdot {{\rm{e}}}^{-{k}_{{\rm{tot}}}\cdot A\cdot t}\) and \({c}_{{{\rm{RM}}}^{{\rm{red}}}}(t)={c}_{{{\rm{RM}}}^{{\rm{ox}}}}(0)-{c}_{{{\rm{RM}}}^{{\rm{ox}}}}(t)+\)\({c}_{{{\rm{RM}}}^{{\rm{red}}}}(0)-{\nu }_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2},{\rm{q}},{\rm{r}}}(t)\cdot t\), where ktot = k1 + k3. The term \({\nu }_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2},{\rm{q}},{\rm{r}}}(t)\) is the rate at which 1O2 reacts with RMred as detailed below. 1O2 formation and decay will balance37, which results in the presence of solvent and mediator quenching in \({c}_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}}(t)={\nu }_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}}(t)/({k}_{{\rm{d}}}+{k}_{{\rm{Q}}}\cdot {c}_{{{\rm{RM}}}^{{\rm{red}}}}(t))\). kQ is either the measured value for the particular mediator or the trendline in Extended Data Fig. 3c. Finally, analogous to the experiment where we integrate the NIR signal until it ceases, we arrive at the expected NIR emission:
To simulate losses of 3O2 (3O2/RMox < 1 in Fig. 2b), we used the rate of reactive 1O2 decay \({\nu }_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2},{\rm{r}}}(t)=({k}_{{\rm{d}},{\rm{r}}}+{k}_{{\rm{q}},{\rm{r}}}\cdot {c}_{{{\rm{RM}}}^{{\rm{red}}}}(t))\cdot {c}_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}}(t)\) to define fr as the reactive fraction of the total 1O2 decay rate
\({\nu }_{{}^{1}{\rm{O}}_{2},{\rm{r}}}(t)\) equals the total mediated 1O2 formation rate times fr: \({\nu }_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2},{\rm{r}}}(t)={f}_{{\rm{r}}}\cdot A\cdot {k}_{1}\cdot {c}_{{{\rm{RM}}}^{{\rm{ox}}}}(t)\). As some of the RMred reacts, its loss is accounted for using \({\nu }_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2},{\rm{q}},{\rm{r}}}(t)={k}_{{\rm{q}},{\rm{r}}}\cdot {c}_{{{\rm{RM}}}^{{\rm{red}}}}(t)\cdot {c}_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}}(t)={f}_{{\rm{q}},{\rm{r}}}\,\cdot A\cdot {k}_{1}\cdot {c}_{{{\rm{RM}}}^{{\rm{ox}}}}(t)\), where \({f}_{{\rm{q}},{\rm{r}}}=\,{k}_{{\rm{q}},{\rm{r}}}\cdot {c}_{{{\rm{RM}}}^{{\rm{red}}}}(t)/({k}_{{\rm{D}}}+{k}_{{\rm{Q}}}\cdot {c}_{{{\rm{RM}}}^{{\rm{red}}}}(t))\). Calculations of fr, fq,r and \({c}_{{{\rm{RM}}}^{{\rm{red}}}}(t)\) were iterated until convergence was achieved. Finally, 3O2 yields are
The fractions of reactive deactivation, kd,r/kD and kq,r/kQ were obtained by fitting the measured values of 3O2/RMox in Fig. 2b with the simulated ones from equation (5). The simulated 3O2/RMox are plotted as the dashed line in Fig. 2b.
Deviations between simulated and measured values may result from the simplicity of the model. The main simplification is as follows: (1) kQ was measured in homogeneous solutions of RMred and the model calculates bulk concentrations \({c}_{{{\rm{RM}}}^{{\rm{ox}}}}\) and \({c}_{{{\rm{RM}}}^{{\rm{red}}}}\), but during the heterogenous KO2 oxidation, bulk and surface concentrations of RMox and RMred differ. (2) We fitted a common fraction of mediator reactivity (kq,r/kQ) for all mediators, causing kq,r and kQ to decrease exponentially with growing \({E}_{{\text{RM}}^{\text{ox}/\text{red}}}^{\circ }\). As a trend, this is justified given that both reaction and charge transferquenching require e– transfer, but individual reactivity will vary because of the large chemical diversity of mediators36. (3) We did not account for quenching by O2 and \({{\rm{O}}}_{2}^{-}\). (4) Some RMs show further reactivities, which can cause 3O2 loss: Fc+ reacts with O2 (ref. 38), TDPA gets in contact with O2 spontaneously oxidized to TDPA+ and TDPA2+ (ref. 18), and reduced quinones bind to O2 (ref. 39).
Proton-induced disproportionation
Six types of buffers were used for proton-induced superoxide disproportionation. Britton–Robinson (BR) buffers were prepared with 0.1 M acetic acid, 0.1 M boric acid and 0.1 M phosphoric acid. NaOH (1 M) and HCl (1 M) were used to adjust the pH. Phosphate-buffered saline (PBS) solution and aqueous PBS powder solution (for pH 7.4) were added and the pH was adjusted using 1 M NaOH or 1 M HCl. Citrate buffers were prepared using citric acid and trisodium citrate dihydrate. To adjust the pH, we varied the concentrations of citric acid and trisodium citrate dihydrate. Acidic buffers of KCl and HCl (pH 1 and 2) were prepared by mixing 0.2 M KCl with 0.2 M HCl and adjusting the volume. Neutral to basic buffers of KH2PO4/NaOH (pH 7.1 and 10.8) were prepared by mixing 0.1 M KH2PO4 with 0.1 M NaOH and adjusting the volume accordingly. To trap the hydrogen peroxide (H2O2) produced, to eliminate the possibility of forming 1O2 by peroxoacids40, titanium(IV)oxysulfate solution (TiOSO4, 15 wt% in dilute sulfuric acid), 1 M NaOH was used to adjust the pH of TiOSO4 solutions to pH 1.2 and 1.55.
Figure 4c shows 1O2 yields still increasing when the pH is below 4.8, the pKa of O2−. KO2 hydrolysis increases the pH in aqueous media according to KO2 + H2O → HO2 + K+ + OH–. Buffer capacities were selected so that the amount of KO2 did not significantly affect the resulting pH after the reaction. However, despite the buffers, the local pH at the reaction site will be higher than the average. The importance of buffering for a local pH close to the average is evident in control experiments without a buffer. Even at a pH of 1.5, we could not detect 1O2 in an unbuffered H2SO4 solution, whereas we could in the buffered one (Extended Data Fig. 6e). Therefore, the 1O2 yields in Fig. 4c result from a higher local pH.
Driving forces on superoxide oxidation
Superoxide experiences a broad range of oxidizing conditions to liberate oxygen, but explanations for why and to what extent certain oxidizing redox couples evolve 1O2 have been unknown. Extended Data Fig. 4 shows the driving forces for superoxide oxidation with various redox couples. The driving forces are shown in comparison with the Marcus kinetic parabola in ether and acetonitrile solvent from Figs. 1 and 2. Li+- and H+-induced disproportionation are shown in Figs. 3 and 4. The other examples we discuss in Extended Data Fig. 4 arise from superoxide in contact with CO2 or organic peroxides, with relevance for energy storage and biology1,30,41.
Considering CO2 first, we have previously shown that CO2 in contact with O2− yields 1O2, but the energetics were unknown30. CO2 in contact with O2− is known to form peroxomonocarbonates and peroxodicarbonates by repeated uptake of CO2 by O2−. Intermediate peroxocarbonate species may be reduced by O2−, which releases O2 (refs. 41,42,43,44). However, the O2 spin state has previously not been considered. Extended Data Fig. 4c shows likely redox couples of oxidized/reduced peroxocarbonate species, but their redox potentials are not established experimentally. A previous study has shown, using density functional theory (DFT), that depending on the cation present and the solvent, the particular peroxocarbonate redox couples that oxidize O2− to O2 differ4, but likely involve CO4•–/CO42–, LiCO4•/LiCO4–, Li2C2O6/Li2C2O6– or Li3C2O6/Li3C2O6–. The calculations have shown reaction energies relative to the O2−/3O2 between about 1 and 1.4 eV. These driving forces, hence, explain 1O2 formation from CO2 in contact with O2− (Extended Data Fig. 4b).
1O2 formation has been examined for exposure of superoxide to organic peroxides, given their occurrence in biological systems45. No reaction was observed with alkyl peroxides, whereas acyl peroxides yielded 1O2 (refs. 45,46); however, this has not been connected with driving forces for superoxide oxidation. Nucleophilic attack of O2− on acyl peroxides forms an acyl radical and carboxylate (Extended Data Fig. 4d). The potential for the acyl radical/carboxylate redox couple has been reported between 1.5 V and 1.7 V compared with SHE (ref. 23) (1.95–2.05 V compared with O2/O2−), which explains 1O2 formation according to Extended Data Fig. 4a. Alkyl peroxides (ROOR) such as di-tert-butyl, dicumyl and di-n-butyl peroxide have been reported not to form 1O2 (ref. 45). This can be understood by ROOR only breaking down to RO– and RO• at low potentials (–1.31 V to –1.15 V compared with SHE, that is, –0.86 V to –0.70 V compared with O2/O2−) (ref. 47). The RO•/RO– couple (–0.06 V to 0.04 V compared with SHE, that is, 0.39–0.49 V compared with O2/O2−) could, if formed, oxidize O2−, but not to 1O2.
Proton-induced disproportionation that yields low but non-negligible 1O2 yields at a high pH of about 11 is consistent with reliable theoretical and experimental works. Using a high-level ab initio method, a previous study17 found that the reaction HO2 + O2− → O2 + HO2 can proceed by both the singlet and triplet pathways, with the singlet pathway being only about 0.3 eV endoergic. Equivalent experiments exposing KO2 to H2O2 in toluene found about 0.2% 1O2 production, as measured using a chemical trap14. In this experiment, H2O2 was the proton source to first form HO2 (H2O2 + O2− → HO2− + HO2). The conditions in these theoretical and experimental studies correspond to a pH of about 11 (pKa = 11.7 for H2O2), in which the reaction to 1O2 is only weakly driven. Conversely, 1O2 was not detected with alkyl hydroperoxides ROOH as the proton source (pKa ≈ 12.6) (ref. 45,48), a result that insufficient driving forces can now explain.
Relation between \({{\boldsymbol{E}}}_{{\genfrac{}{}{0ex}{}{1}{}{\bf{O}}}_{2}/{{\bf{O}}}_{{\bf{2}}}^{-}}^{\circ }\) and the 1O2 fraction
The electrochemiluminescence literature refers to energy-sufficient processes to form the electronically excited species23,28,49. For example, consider the generic redox couples R/R•– and M•+/M (note that these could be, for example, 3O2/O2− and RMox/RMred). The process R•– + M•+ → 1R* + M is considered energy-sufficient to form the excited species 1R*, if \(({E}_{{{\rm{M}}}^{\cdot +}/{\rm{M}}}^{\circ }-{E}_{{\rm{R}}/{{\rm{R}}}^{\cdot -}}^{\circ })F\ge \,-\Delta {H}^{\circ }(\,\genfrac{}{}{0ex}{}{1}{}\,{{\rm{R}}}^{* }\leftarrow {\rm{R}})\). This condition is fulfilled if the potential of the oxidizing redox couple exceeds the redox potential of the excited species: \({E}_{{{\rm{M}}}^{\cdot +}/{\rm{M}}}^{\circ }\ge {E}_{\genfrac{}{}{0ex}{}{1}{}{{\rm{R}}}^{* }/{{\rm{R}}}^{\cdot -}}^{\circ }={E}_{{{\rm{R}}/{\rm{R}}}^{\cdot -}}^{\circ }+\Delta {H}^{\circ }(\,\genfrac{}{}{0ex}{}{1}{}\,{{\rm{R}}}^{* }\leftarrow {\rm{R}})/F\). The connotation of energy-sufficient processes led to the interpretation that \({E}_{\genfrac{}{}{0ex}{}{1}{}{{\rm{R}}}^{* }/{{\rm{R}}}^{\cdot -}}^{\circ }\) or \({E}_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}/{{\rm{O}}}_{2}^{-}}^{\circ }\) establishes a threshold above which the excited species rather than ground state species forms18,24,26. Extended Data Fig. 4a shows that reaching this threshold potential (or the driving force for which this is exceeded) gives no indication about the extent to which 1O2 rather than 3O2 forms. An onset of 1O2 may be expected at \({\Delta G}_{1\leftarrow 3}^{\circ }\), but its formation will become significant only for driving forces –ΔG° > λ3, for which 3O2 formation slows down to benefit 1O2 formation.
Thermodynamics in mixed alkali metal/TBA+ electrolytes
Superoxide disproportionation in Li+ and Na+ containing glyme electrolytes was found to always yield some 1O2 according to 2MO2 → M2O2 + x3O2 + (1 − x)1O2 (refs. 13,16,18,19). Li+ yielded small fractions (about 2%) at large kinetics, and Na+ yielded larger fractions (around 12%) at slow kinetics16. Often, electrolytes for non-aqueous metal-O2 batteries contain weakly Lewis acidic cations, such as tetrabutylammonium (TBA+) or other cations from ionic liquid electrolytes. In mixed Li+/TBA+ and Na+/TBA+ (1/1) electrolytes, the 1O2 yields increased to about 20 and 18%, respectively. The reasons for this behaviour must, hence, lie in (1) already sufficient driving forces for 1O2 formation in pure Li+ and Na+ electrolytes; and (2) increasing driving forces on adding TBA+.
M2O2 (M = Li+, Na+) are insoluble50,51 and the potential \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{{\rm{M}}}_{2}{{\rm{O}}}_{2}({\rm{s}})}^{\circ }\,=\) \({\Delta }_{{\rm{f}}}{G}^{\circ }({{\rm{M}}}_{2}{{\rm{O}}}_{2(\text{s})})/F=2.96\,{\rm{V}}\) compared with M+/M, therefore, fixed to the value obtained using the formation energy –ΔfG° of solid Li2O2(s). Given that the superoxide/M2O2(s) couple acts as the oxidant during superoxide disproportionation, the driving force is given by \(-\Delta G=({E}_{{\rm{superoxide}}/{{\rm{M}}}_{2}{{\rm{O}}}_{2(\text{s})}}-{E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\rm{superoxide}}})F\). Note that here superoxide does not denote a particular species, but it could be anything, including solid MO2(s), solvated \({({{\rm{M}}}^{+}{{\rm{O}}}_{2}^{-})}_{n\ge 1,({\rm{sln}})}\) clusters and ion pairs, or the weakly coordinated \({({{\rm{TBA}}}^{+}{{\rm{O}}}_{2}^{-})}_{({\rm{sln}})}\). \({E}_{{\rm{superoxide}}/{\text{M}}_{2}{\text{O}}_{2(\text{s})}}\) cannot be directly measured but can be inferred from \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{{\rm{M}}}_{2}{{\rm{O}}}_{2(\text{s})}}\) and \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\rm{superoxide}}}\). Using ΔG = –zFE, where z is the number of transferred electrons, it can be derived that \({E}_{{\rm{superoxide}}/{{\rm{M}}}_{2}{{\rm{O}}}_{2(\text{s})}}=2{E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{{\rm{M}}}_{2}{{\rm{O}}}_{2(\text{s})}}-{E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\rm{superoxide}}}\). For the stable solid compounds (Li2O2, Na2O2, NaO2, K2O2 and KO2), tabulated formation energies ΔfG° can be found and the \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{{\rm{M}}}_{2}{{\rm{O}}}_{2}({\rm{s}})}^{\circ }\) and \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\rm{M}}{{\rm{O}}}_{2}({\rm{s}})}^{\circ }\) be calculated as shown in Extended Data Fig. 5a. On the basis of this, KO2 is not expected to disproportionate to K2O2 and the K+-case, hence, not be further considered.
\({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\rm{superoxide}}}\) require further consideration given the electrolyte-dependent solubilities of superoxide. Theoretical work shows that solvated \({({{\rm{Li}}}^{+}{{\rm{O}}}_{2}^{-})}_{n\ge 1,({\rm{sln}})}\) species are less stable in terms of Gibbs free energy than the bulk solid LiO2(s), but as the cluster size n grows, the structure approaches bulk MO2(s) and free energy approaches a constant value16,50,51. Aggregation into \({({{\rm{Li}}}^{+}{{\rm{O}}}_{2}^{-})}_{n > 1,({\rm{sln}})}\) clusters stabilizes the solvated species relative to separated \({({{\rm{Li}}}^{+}{{\rm{O}}}_{2}^{-})}_{({\rm{sln}})}\) species16,34. The Gibbs free energy grows, therefore, in the order of increasing solvation: LiO2(s) < (Li+O2–)n>1,(sln) < (Li+O2–)(sln). Accordingly, the potentials in pure M+ electrolyte are in the order and within the limits \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{{\rm{LiO}}}_{2(\text{s})}} > {E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{({{\rm{Li}}}^{+}{{\rm{O}}}_{2}^{-})}_{n > 1({\rm{sln}})}} > {E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{({{\rm{Li}}}^{+}{{\rm{O}}}_{2}^{-})}_{({\rm{sln}})}}\). If TBA+ is present, the even weaker association in \({({{\rm{TBA}}}^{+}{{\rm{O}}}_{2}^{-})}_{({\rm{sln}})}\) extends the lower potential limit to \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{({{\rm{TBA}}}^{+}{{\rm{O}}}_{2}^{-})}_{({\rm{sln}})}}\). The values for these potential limits, as shown in Fig. 3a, Extended Data Fig. 5c,d, were estimated from cyclic voltammograms with the salt shifting from pure M+ to pure TBA+ (Extended Data Fig. 5b). The largely solvation-independent DMFc/DMFc+ redox couple was used as internal standard. ΔfG° of solid LiO2(s) is not available, but \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{\rm{Li}}{{\rm{O}}}_{2}({\rm{s}})}\) may be estimated from cyclic voltammograms in poorly solvating electrolytes, in which large \({({{\rm{Li}}}^{+}{{\rm{O}}}_{2}^{-})}_{n,({\rm{sln}})}\) clusters approach the thermodynamics of LiO2(s). The shift in the onset of O2 reduction in Li+ compared with TBA+ electrolyte at slow scan rates was taken as the difference between \({E}_{{{}^{3}{\rm{O}}}_{2}/{{\rm{LiO}}}_{2(\text{s})}}\) and \({E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{({{\rm{TBA}}}^{+}{{\rm{O}}}_{2}^{-})}_{({\rm{sln}})}}.{E}_{{\genfrac{}{}{0ex}{}{3}{}{\rm{O}}}_{2}/{({{\rm{Li}}}^{+}{{\rm{O}}}_{2}^{-})}_{({\rm{sln}})}}\) as the lower limit of potentials in pure Li+ electrolyte was estimated from the potential shift between pure Li+ and pure TBA+ electrolytes with the highly solvating solvent 1-methylimidazole. These were taken from ref. 32 and show a shift of 33 mV.
Extended Data Fig. 5d shows the analogous thermodynamics for Na+/TBA+ mixtures as shown in Fig. 3 for Li+/TBA+. Considerations as above for the relative stabilities of the Li superoxides apply analogously to the relative stability of Na superoxide clusters compared with the NaO2(s) bulk. Theoretical work has similarly shown the stabilization by forming \({({{\rm{Na}}}^{+}{{{\rm{O}}}_{2}}^{-})}_{n > 1,({\rm{sln}})}\) clusters52,53. Extended Data Fig. 5e shows the NIR emission on adding KO2 into glyme electrolyte containing various Na+/TBA+ ratios. The data show that, even in a pure Na+ electrolyte, the driving force is sufficient for 1O2 formation and that added TBA+ increases the driving force and formation.
More generally, the electrolyte properties (solvent(s), salts(s) and their concentrations) will affect the reorganization energy and hence the maxima and crossing point of the two kinetic parabolas. The classical approach to accounting for this is the equation given by Marcus54, which connects the reorganization energy with the effective dielectric properties of the electrolyte and the separation of the redox centres. A lower dielectric constant and smaller separation will result in a larger reorganization energy. A refined equation by Marcus55 further takes into account the ionic environment. These considerations apply well, for example, to aqueous anionic redox couples and the series of alkali metal cations from Li+ to Cs+ as spectator cations, in which λ decreases56. However, caution is required with non-aqueous, low dielectric constant media, in which strong ion pairing occurs. Some works suggest an inverse trend for λ among the alkali metals19,57. Ion pairing and even clustering is particularly severe for (su)peroxide as the redox anions as discussed above. Superoxide forms in non-aqueous Li+ and Na+ electrolytes clusters32,33 and the peroxides are practically insoluble51. The order and extent to which the reorganization energy changes for superoxide oxidation in non-aqueous media among the alkali cations may, therefore, not be predicted straightforwardly and would merit further investigation. As we observe 1O2 at low driving force for the Na+ electrolyte, the reorganization energy appears sufficiently low therein.
Wider relevance for life sciences and energy
Our study contributes to understanding how the pH affects the link between the four important reactive oxygen species (ROS) superoxide, peroxide, 3O2 and 1O2. Disproportionation is notably the pathway to maintain a low superoxide concentration. However, detoxification from superoxide produces the harmful 1O2. Superoxide occurs in cells in several organelles with different pH levels between 4.7 and 8, but the superoxide-degrading enzyme superoxide dismutase occurs only in neutral to basic organelles35. In pathological situations, the pH balance is known to be affected (typically towards lower pH) and therefore signalling, redox regulation and defence58,59. Our study contributes to the understanding of how the redox chemistry of superoxide, pH and 1O2 formation are linked. We noted that in non-aqueous media, superoxide in contact with CO2 forms 1O2. Given the ubiquity of CO2 in organelles containing superoxide, further investigations into the aqueous chemistry of CO2 and superoxide are warranted.
For energy applications, further relevance and future research directions emerge: (1) For suppressing 1O2, generally, the driving force should be decreased, and the reorganization energy for the superoxide oxidation reaction should be increased. The classical equation by Marcus54, which connects the reorganization energy with the effective dielectric properties of the electrolyte and the separation of the redox centres, applies well to aqueous anionic redox couples56. For non-aqueous, low dielectric constant media, in which strong ion pairing occurs, particularly so with superoxide, the change in reorganization energy among different cations and electrolytes may not be predicted straightforwardly and would merit further investigation. (2) Oxygen evolution catalysis from water: metal-superoxo species have been identified as preceding the O2 evolution on, for example, the extensively studied Ni(Fe)OOH or CoOOH catalysts10. The metal Mn–1/n redox couple is considered to oxidize the superoxo moiety to O2 (refs. 10,60). Some of the most active Mn–1/n metal redox couples are typically a few 100 mV above the 1O2/superoxide potential shown in Fig. 4a and provide, in principle, enough driving force for 1O2 evolution. For example, at pH 14 \({E}_{{\genfrac{}{}{0ex}{}{1}{}{\rm{O}}}_{2}/{{{\rm{O}}}_{2}}^{-}}^{\circ }=1.32\,{\rm{V}}\), \({E}_{{{\rm{Co}}}^{{\rm{II}}/{\rm{III}}}}^{\circ }\approx 1.25\,{\rm{V}}\), \({E}_{{{\rm{Co}}}^{{\rm{III}}/{\rm{IV}}}}^{\circ }\approx 1.5\,{\rm{V}}\), \({E}_{{{\rm{Ni}}}^{{\rm{II}}/{\rm{III}}}}^{\circ }\approx 1.52\,{\rm{V}}\) and \({E}_{{{\rm{Ni}}}^{{\rm{III}}/{\rm{IV}}}}^{\circ }\approx 1.6\,{\rm{V}}\) on the RHE scale (refs. 10,60). Further investigations specifically on 1O2 evolution in oxygen evolution catalysis are therefore warranted. (3) Both Li-stoichiometric6 and Li-rich transition metal (TM, for example, Ni, Mn and Co) oxide2,7,61,62 intercalation materials used for positive electrodes in Li- or Na-ion batteries are known to undergo parasitic lattice oxygen loss at high states of charge. Both the intercalation material and the electrolyte degrade, hampering long-term cyclability. However, non-matching patterns of O2 and CO2/CO release from electrolyte decomposition (all containing lattice O as shown by isotopic labelling7,62) and enhanced lattice O loss with surface carbonates present7 remain unexplained and highlight the need for a deeper understanding of the prevailing ROS and decomposition pathways. For LiNiO2, 1O2 evolution has been suggested to result from the disproportionation of oxide radicals6. More generally, 1O2 may evolve from superoxo species (at the lattice surface, in (su)peroxocarbonates4,30) at the available driving forces. The oxidants could be a combination of (su)peroxides (for example, coordinated by TMs or carbonates) that get stabilized by further reduction and TM redox, such as CoIII/IV or NiIII/IV. Further investigations into the involvement of 1O2 evolution in TM oxide outgassing and surface reactions are therefore warranted.
The results expand the current knowledge on the electrogeneration of excited species more generally and pose open questions about the origin. Specifically, the process is more effective than typically assumed, given that \({\Delta G}_{1\leftarrow 3}^{\circ } < {\Delta H}_{1\leftarrow 3}^{\circ }\) and that Zel,1 ≫ Zel,3. Electrogeneration of excited ROS has significance ranging from biological systems to energy storage. Reactive excited species in life are very broadly associated with pathogenic events1,63. Recombination reactions in redox flow batteries are recognized to cause self-discharge, but this has so far not been recognized to potentially form extremely energetic excited species. Soluble parasitic oxidized and reduced species at the cathode and anode of Li- and Na-ion batteries may recombine to form energetic excited species.
Data availability
The data that support the plots in this paper and other findings of this study are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Sies, H., Mailloux, R. J. & Jakob, U. Fundamentals of redox regulation in biology. Nat. Rev. Mol. Cell Biol. 25, 701–719 (2024).
House, R. A. et al. Superstructure control of first-cycle voltage hysteresis in oxygen-redox cathodes. Nature 577, 502–508 (2020).
Liu, T. et al. Current challenges and routes forward for nonaqueous lithium–air batteries. Chem. Rev. 120, 6558–6625 (2020).
Lim, H.-K. et al. Toward a lithium–“air” battery: the effect of CO2 on the chemistry of a lithium–oxygen cell. J. Am. Chem. Soc. 135, 9733–9742 (2013).
Winterbourn, C. C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 4, 278–286 (2008).
Genreith-Schriever, A. R. et al. Oxygen hole formation controls stability in LiNiO2 cathodes. Joule 7, 1623–1640 (2023).
Kaufman, L. A. & McCloskey, B. D. Surface lithium carbonate influences electrolyte degradation via reactive oxygen attack in lithium-excess cathode materials. Chem. Mater. 33, 4170–4176 (2021).
Khan, A. U. Singlet molecular oxygen from superoxide anion and sensitized fluorescence of organic molecules. Science 168, 476–477 (1970).
Lopez, N. et al. Reversible reduction of oxygen to peroxide facilitated by molecular recognition. Science 335, 450–453 (2012).
Xiao, H., Shin, H. & Goddard, W. A. Synergy between Fe and Ni in the optimal performance of (Ni,Fe)OOH catalysts for the oxygen evolution reaction. Proc. Natl Acad. Sci. USA 115, 5872–5877 (2018).
Clayden, J., Greeves, N., Warren, S. & Wothers, P. Organic Chemistry (Oxford Univ. Press, 2001).
Schweitzer, C. & Schmidt, R. Physical mechanisms of generation and deactivation of singlet oxygen. Chem. Rev. 103, 1685–1758 (2003).
Wang, Y., Lu, Y.-R., Dong, C.-L. & Lu, Y.-C. Critical factors controlling superoxide reactions in lithium–oxygen batteries. ACS Energy Lett. 5, 1355–1363 (2020).
MacManus-Spencer, L. A. & McNeill, K. Quantification of singlet oxygen production in the reaction of superoxide with hydrogen peroxide using a selective chemiluminescent probe. J. Am. Chem. Soc. 127, 8954–8955 (2005).
Armstrong, D. A. et al. Standard electrode potentials involving radicals in aqueous solution: inorganic radicals. BioInorg. React. Mech. 9, 59–61 (2013).
Mourad, E. et al. Singlet oxygen from cation driven superoxide disproportionation and consequences for aprotic metal–O2 batteries. Energy Environ. Sci. 12, 2559–2568 (2019).
Pierini, A., Brutti, S. & Bodo, E. Superoxide anions disproportionation induced by Li+ and H+: pathways to 1O2 release in Li–O2 batteries. ChemPhysChem 21, 2060–2067 (2020).
Petit, Y. K. et al. Mechanism of mediated alkali peroxide oxidation and triplet versus singlet oxygen formation. Nat. Chem. 13, 465–471 (2021).
Ahn, S. et al. Why charging Li–air batteries with current low-voltage mediators is slow and singlet oxygen does not explain degradation. Nat. Chem. 15, 1022–1029 (2023).
Houchins, G., Pande, V. & Viswanathan, V. Mechanism for singlet oxygen production in Li-ion and metal–air batteries. ACS Energy Lett. 5, 1893–1899 (2020).
Wigner, E. & Witmer, E. E. Über die Struktur der zweiatomigen Molekelspektren nach der Quantenmechanik. Z. Physik 51, 859–886 (1928).
Marcus, R. A. On the theory of chemiluminescent electron‐transfer reactions. J. Chem. Phys. 43, 2654–2657 (1965).
Miao, W. Electrogenerated chemiluminescence and its biorelated applications. Chem. Rev. 108, 2506–2553 (2008).
McCloskey, B. D. et al. Limitations in rechargeability of Li–O2 batteries and possible origins. J. Phys. Chem. Lett. 3, 3043–3047 (2012).
Mahne, N. et al. Singlet oxygen generation as a major cause for parasitic reactions during cycling of aprotic lithium–oxygen batteries. Nat. Energy 2, 17036 (2017).
Hassoun, J., Croce, F., Armand, M. & Scrosati, B. Investigation of the O2 electrochemistry in a polymer electrolyte solid-state cell. Angew. Chem. Int. Ed. 50, 2999–3002 (2011).
Marcus, R. A. & Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 811, 265–322 (1985).
Bard, A. J., Faulkner, L. R. & White, H. S. Electrochemical Methods: Fundamentals and Applications 3rd edn (Wiley, 2022).
Closs, G. L. & Miller, J. R. Intramolecular long-distance electron transfer in organic molecules. Science 240, 440–447 (1988).
Mondal, S., Jethwa, R. B., Pant, B., Hauschild, R. & Freunberger, S. A. Singlet oxygen formation in non-aqueous oxygen redox chemistry: direct spectroscopic evidence for formation pathways and reliability of chemical probes. Faraday Discuss. 248, 175–189 (2024).
Prehal, C. et al. In situ small-angle X-ray scattering reveals solution phase discharge of Li–O2 batteries with weakly solvating electrolytes. Proc. Natl Acad. Sci. USA 118, e2021893118 (2021).
Johnson, L. et al. The role of LiO2 solubility in O2 reduction in aprotic solvents and its consequences for Li–O2 batteries. Nat. Chem. 6, 1091–1099 (2014).
Kwabi, D. G. et al. Experimental and computational analysis of the solvent-dependent O2/Li+-O2− redox couple: standard potentials, coupling strength, and implications for lithium–oxygen batteries. Angew. Chem. Int. Ed. 55, 3129–3134 (2016).
Das, U., Lau, K. C., Redfern, P. C. & Curtiss, L. A. Structure and stability of lithium superoxide clusters and relevance to Li–O2 batteries. J. Phys. Chem. Lett. 5, 813–819 (2014).
Casey, J. R., Grinstein, S. & Orlowski, J. Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 11, 50–61 (2010).
Kwak, W.-J. et al. Deactivation of redox mediators in lithium-oxygen batteries by singlet oxygen. Nat. Commun. 10, 1380 (2019).
Wilkinson, F., Helman, W. P. & Ross, A. B. Rate constants for the decay and reactions of the lowest electronically excited singlet state of molecular oxygen in solution. An expanded and revised compilation. J. Phys. Chem. Ref. Data 24, 663–677 (1995).
Hurvois, J. P. & Moinet, C. Reactivity of ferrocenium cations with molecular oxygen in polar organic solvents: decomposition, redox reactions and stabilization. J. Organomet. Chem. 690, 1829–1839 (2005).
Gao, X., Chen, Y., Johnson, L. & Bruce, P. G. Promoting solution phase discharge in Li–O2 batteries containing weakly solvating electrolyte solutions. Nat. Mater. 15, 882–888 (2016).
Evans, D. F. & Upton, M. W. Studies on singlet oxygen in aqueous solution. Part 3. The decomposition of peroxy-acids. J. Chem. Soc., Dalton Trans. 1985, 1151–1153 (1985).
Zhang, S. et al. On the incompatibility of lithium-O2 battery technology with CO2. Chem. Sci. 8, 6117–6122 (2017).
Roberts, J. L. Jr, Calderwood, T. S. & Sawyer, D. T. Nucleophilic oxygenation of carbon dioxide by superoxide ion in aprotic media to form the peroxydicarbonate(2–) ion species. J. Am. Chem. Soc. 106, 4667–4670 (1984).
Albery, W. J. et al. Pulsed titration sensors. 1: a breath-by-breath CO2 sensor. J. Electroanalyt. Chem. 340, 99–110 (1992).
Wadhawan, J. D. et al. Microelectrode studies of the reaction of superoxide with carbon dioxide in dimethyl sulfoxide. J. Phys. Chem. B 105, 10659–10668 (2001).
MacManus-Spencer, L. A., Edhlund, B. L. & McNeill, K. Singlet oxygen production in the reaction of superoxide with organic peroxides. J. Org. Chem. 71, 796–799 (2006).
Khan, A. U. & Kasha, M. Singlet molecular oxygen in the Haber-Weiss reaction. Proc. Natl Acad. Sci. USA 91, 12365–12367 (1994).
Donkers, R. L., Maran, F., Wayner, D. D. M. & Workentin, M. S. Kinetics of the reduction of dialkyl peroxides. New insights into the dynamics of dissociative electron transfer. J. Am. Chem. Soc. 121, 7239–7248 (1999).
Uhl, A. et al. in Ullmann’s Encyclopedia of Industrial Chemistry 1−45 (Wiley, 2018).
Mayeda, E. A. & Bard, A. J. Production of singlet oxygen in electrogenerated radical ion electron transfer reactions. J. Am. Chem. Soc. 95, 6223–6226 (1973).
Cheng, L. et al. Computational studies of solubilities of LiO2 and Li2O2 in aprotic solvents. J. Electrochem. Soc. 164, E3696–E3701 (2017).
Kim, J. et al. Dissolution and ionization of sodium superoxide in sodium–oxygen batteries. Nat. Commun. 7, 10670 (2016).
Arcelus, O., Li, C., Rojo, T. & Carrasco, J. Electronic structure of sodium superoxide bulk, (100) surface, and clusters using hybrid density functional: relevance for Na–O2 batteries. J. Phys. Chem. Lett. 6, 2027–2031 (2015).
Pierini, A., Brutti, S. & Bodo, E. Reactions in non-aqueous alkali and alkaline-earth metal–oxygen batteries: a thermodynamic study. Phys. Chem. Chem. Phys. 23, 24487–24496 (2021).
Marcus, R. A. Electron transfer reactions in chemistry. Theory and experiment. Rev. Mod. Phys. 65, 599–610 (1993).
Marcus, R. A. On the theory of electron-transfer reactions. VI. Unified treatment for homogeneous and electrode reactions. J. Chem. Phys. 43, 679–701 (1965).
Huang, B. et al. Cation-dependent interfacial structures and kinetics for outer-sphere electron-transfer reactions. J. Phys. Chem. C 125, 4397–4411 (2021).
Barthel, J. & Feuerlein, F. Dielectric properties of propylene carbonate-1,2-dimethoxyethane mixtures and their electrolyte solutions of NaClO4 and Bu4NClO4. Z. Phys. Chem. 148, 157–170 (1986).
Sies, H. & Jones, D. P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 21, 363–383 (2020).
Swietach, P., Boedtkjer, E. & Pedersen, S. F. How protons pave the way to aggressive cancers. Nat. Rev. Cancer 23, 825–841 (2023).
Moysiadou, A., Lee, S., Hsu, C.-S., Chen, H. M. & Hu, X. Mechanism of oxygen evolution catalyzed by cobalt oxyhydroxide: cobalt superoxide species as a key intermediate and dioxygen release as a rate-determining step. J. Am. Chem. Soc. 142, 11901–11914 (2020).
Zhang, M. et al. Pushing the limit of 3d transition metal-based layered oxides that use both cation and anion redox for energy storage. Nat. Rev. Mater. 7, 522–540 (2022).
Luo, K. et al. Charge-compensation in 3d-transition-metal-oxide intercalation cathodes through the generation of localized electron holes on oxygen. Nat. Chem. 8, 684–691 (2016).
Brash, D. E. & Goncalves, L. C. P. Chemiexcitation: mammalian photochemistry in the dark. Photochem. Photobiol. 99, 251–276 (2023).
Schafzahl, B. et al. Quantifying total superoxide, peroxide, and carbonaceous compounds in metal–O2 batteries and the solid electrolyte interphase. ACS Energy Lett. 3, 170–176 (2017).
Acknowledgements
S.A.F. thanks the Institute of Science and Technology Austria (ISTA) for the support. The Scientific Service Units of ISTA supported this research through resources provided by the Imaging and Optics Facility, the Lab Support Facility, the Miba Machine Shop and Scientific Computing. This research was partly funded by the Austrian Science Fund (FWF) (10.55776/P37169 and 10.55776/COE5). For open access purposes, the author has applied for a CC BY public copyright licence to any author-accepted manuscript version arising from this submission. R.H. acknowledges funding through CZI grant DAF2020-225401 (10.37921/120055ratwvi) from the Chan Zuckerberg Initiative DAF, an advised fund of Silicon Valley Community Foundation (10.13039/100014989). H.T.K.N. acknowledges funding by the European Commission Erasmus Mundus Joint Masters programme. We thank M. Sixt and M. Chinon for the discussions about O-redox in life and R. Jethwa for proofreading. Open access funding was provided by ISTA.
Funding
Open access funding provided by Institute of Science and Technology (IST Austria).
Author information
Authors and Affiliations
Contributions
S.A.F. and S.M. conceptualized the project and developed the methodology for all experiments, developed the model and code for data evaluation and performed simulations. S.M. and H.T.K.N. carried out electrochemistry and kinetics measurements. R.H., S.M. and S.A.F. developed the NIR setup, and S.M. carried out the chemiluminescence and time decay study. S.M. carried out the mass spectrometry. S.A.F. conceived and supervised the project, secured funding and wrote the paper. S.A.F. and S.M. evaluated and interpreted the results and revised the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Enrico Bodo, Kai Exner and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Kinetics and 1O2 yields during mediated superoxide oxidation in TEGDME electrolyte.
a, We measured the kinetic constants k for mediated KO2 oxidation with mediators using UV-Vis spectroscopy up to moderate mediator redox potentials. Higher potential mediators could not be used in the tetraglyme electrolyte, and errors were already significant at the upper end of driving forces. Plot of ln(k) versus mediator potential (\({E}_{{\text{RM}}^{\text{ox}/\text{red}}}^{\circ }\), top axis) and driving force (\(-\Delta G^\circ \), bottom axis). \(-\Delta G^\circ =({E}_{{\text{RM}}^{\text{ox}/\text{red}}}^{\circ }-{E}_{{{}^{3}{\rm{O}}}_{2}/{\text{KO}}_{2}}^{\circ })F\), where \({E}_{{{}^{3}{\rm{O}}}_{2}/{\text{KO}}_{2}}^{\circ }=2.48\,{\rm{V}}\) vs K/K+. The blue curve is a fit to Marcus kinetics in Eq. (1), which gives Zel = 6.4 × 10–3 cm s–1 and \(\lambda \) = 0.61 eV. The red dashed line indicates the driving force at \({E}_{{{}^{1}{\rm{O}}}_{2}/{\text{KO}}_{2}}^{\circ }={E}_{{{}^{3}{\rm{O}}}_{2}/{\text{KO}}_{2}}^{\circ }+0.97{\rm{V}}\), the commonly expected “threshold” for 1O2 formation. b, The formed 1O2 per mole of RMox as measured using 9,10-dimethylanthracene (DMA) conversion to DMA-O2 using HPLC. Data are presented as mean ± s.d. (n ≥ 3). Panel a and b adapted from ref. 18, Springer Nature Limited.
Extended Data Fig. 2 Oxidation stability of different solvents and potentials of the redox mediators used here.
a, Forward scan of cyclic voltammetry of dimethyl sulfoxide (DMSO), N,N-dimethylacetamide (DMAc), N,N-dimethylformamide (DMF), and acetonitrile (MeCN) containing 10 mM KClO4 The scan rate was 20 mVs–1 and the working electrode was a glassy carbon disc. b, The selected RMs, their abbreviations and measured redox potentials on the K/K+ scale as well as E° of the O2/KO2 couple.
Extended Data Fig. 3 From 1O2 formation rates to measurable NIR emission intensities.
a, Example for one mediator of the decay of the 1O2 phosphorescence at 1270 nm in MeCN after pulsed irradiation. The pulse frequency was 1 kHz and the signal was accumulated for 2 min. b, Example for one RM of the decay constant of 1O2 versus RMred concentration to get kQ(RM). Every value at any concentration is the mean of n ≥ 3 decay measurements. Error bars are too small to be visible. c, Measured 1O2 quenching rate constants kQ(RM) of the used mediators in their reduced form RMred as a function of their redox potential. The trend line with a slope of 103.5/V has been added to guide the eye. Each marker represents one set of measurements as shown in b. d, Segmented optical microscopy image of dispersed KO2 powder. e, Extracted particle size distribution from d, from which a surface area of A = 0.23 ± 0.04 m2g–1 (mean ± s.d., n = 3) was determined. f, Schematic Sankey plots of relative rates of the involved processes for the examples of Fc, MPT and ThA. The widths of the arrows are proportional to the rates. Relative rates at the left end are those given by the Marcus parabola for superoxide oxidation to 3O2 and 1O2 in Fig. 2a. Any 1O2 then undergoes various decay pathways. First, a combination of physical and reactive quenching by the solvent, 3O2 and superoxide, as denoted as kd. Second, strongly potential-dependent charge transfer quenching by the RM denoted as kq(RM). The fraction emitting NIR radiation is strongly exacerbated as it would otherwise be invisible relative to the others. Data are presented in b and c are from single experiment.
Extended Data Fig. 4 Ranges of driving forces for various systems, where superoxide oxidation occurs.
a, Comparison of individual kinetic parabolas as determined for mediated superoxide oxidation. Curves for MeCN are the Marcus fits in Fig. 2. For TEGDME the 3O2 parabola is the Marcus fit to data in Extended Data Fig. 1. The parabola for 1O2 evolution could not be measured in TEGDME, and is hence constructed using \({\triangle G}_{1\leftarrow 3}^{\circ }\) = 0.84 eV and Zel,1 = 6.3 × Zel,3 as determined with MeCN (Fig. 2). b, Ranges of driving forces for cation-induced O2– disproportionation (DISP) and for the indicated redox couples as defined in c and d. c, Commonly suggested peroxocarbonate species that form from O2– in contact with CO2 (refs. 41,42,43,44). For example, the superoxo species \({{\rm{CO}}}_{4}^{{\rm{\bullet }}-}\) or \({{\rm{C}}}_{2}{{\rm{O}}}_{6}^{{\rm{\bullet }}-}\) were suggested to be reduced by \({{\rm{O}}}_{2}^{-}\), liberating O2. Kang’s group4 suggested using DFT further peroxocarbonate redox couples and their redox potentials relative to \({{\rm{O}}}_{2}^{-}\)/3O2 (shown in b). d, Reactions of acyl and alkyl peroxides in contact with \({{\rm{O}}}_{2}^{-}\) which were found to form or not to form 1O2 (refs. 45,46) as explicable by their reduction potentials (shown in b).
Extended Data Fig. 5 Redox potentials in mixed Li+/TBA+ and Na+/TBA+ electrolytes.
a, Standard potentials of the O2/MO2(s) and O2/M2O2(s) redox couples with M = Li, Na, K as calculated from tabulated formation energies. The common scale is based on their \({E}_{{\rm{M}}/{{\rm{M}}}^{+}}^{\circ }\). b, Cyclic voltammograms (scan rate 100 mVs–1) in O2-saturated tetraglyme containing 100 mM of either TBAClO4, LiClO4, or NaClO4. c, Detailed thermodynamics of the 3O2/superoxide couple as a function of the fractions of Li+ and TBA+ salt. d, Thermodynamics of relevant redox couples for Na+-induced \({{\rm{O}}}_{2}^{-}\) disproportionation as a function of the fractions of Na+ and TBA+ salt. e, Normalised 1270 nm NIR emission signal as a function of Na+ mole fraction, which determines the driving force. Each bar represents a single measurement. Tetraglyme served as the electrolyte solvent.
Extended Data Fig. 6 NIR emission to measure 1O2 yields.
a, 1O2 NIR emission spectrum as reported in the literature12 and transmission of the used bandpass filter. b, Representative output signal upon injecting RMox solution onto KO2 powder. The integral of the recorded signal was used to determine the 1O2 yields. The example shown is with MPT. c, Representative output signal of the NIR detector upon injecting Li+ electrolyte solution onto KO2 powder. The setup was similar to the one shown in Extended Data Fig. 4, except that a cuvette replaced the glass vial and the NIR detector was placed next to it. The example shown is with a Li+ mole fraction of 0.166. d, Representative NIR signal upon injecting H+ solution containing buffer onto KO2 powder. The example shown is with BR buffer at pH 2.2. e, Output signal of the NIR detector upon injecting H2SO4 (pH 1.58) solution without any buffer onto KO2 powder.
Extended Data Fig. 7 Measuring KO2 oxidation kinetics using UV-Vis absorbance.
a, Exemplary evolution UV-Vis spectra of DPA and ThA. Concentration versus time for the indicated RMox in contact with KO2. b,c,e–m, RMox concentration versus time. For F4BQ (d), concentration versus time for RMred.
Extended Data Fig. 8 Measuring 3O2 yields and disproportionation kinetics.
a, Schematic set-up of the sample vial used for mass spectrometry and sequence of the solution addition during the 3O2 yield measurement. b, O2 evolution versus time upon immersing KO2 powder in RMox solution. The example shown is with DMPZ. c, Schematic of the set-up to measure disproportionation kinetics by pressure changes. d, Pressure evolution from three different cases where Li+/(Li++TBA+) mole fractions are 1, 0.33 and 0.166. e, Evolved moles versus time for superoxide disproportionation as measured by the pressure p. Fitting is done with p(t) = p0 + Δp(1–exp(–kt)). The example is shown for the case with the mole fraction Li+/(Li++TBA+) = 0.166. Panel a adapted with permission from ref. 64, American Chemical Society.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mondal, S., Nguyen, H.T.K., Hauschild, R. et al. Marcus kinetics control singlet and triplet oxygen evolving from superoxide. Nature (2025). https://doi.org/10.1038/s41586-025-09587-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41586-025-09587-7
