
Abstract
Embryonic genetic programs are reactivated in response to various types of tissue damage, providing cell plasticity for tissue regeneration or disease progression. In acute conditions, these programs remedy the damage and then halt to allow a return to homeostasis. In chronic situations, including inflammatory diseases, fibrosis and cancer, prolonged activation of embryonic programs leads to disease progression and tissue deterioration. Induction of progenitor identity and cell plasticity, for example, epithelial–mesenchymal plasticity, are critical outcomes of reactivated embryonic programs. In this Review, we describe molecular players governing reactivated embryonic genetic programs, their role during disease progression, their similarities and differences and lineage reversion in pathology and discuss associated therapeutics and drug-resistance mechanisms across many organs. We also discuss the diversity of reactivated programs in different disease contexts. A comprehensive overview of commonalities between development and disease will provide better understanding of the biology and therapeutic strategies.
Similar content being viewed by others


Main
In the early nineteenth century, Virchow noted embryonic-like characteristics, such as primitive cellular morphology, in cancerous tissue1. This observation was seminal for the concept of reactivation of embryonic genetic programs in pathological conditions. Today, we have evidence for common embryonic genetic programs underpinning cellular inflammatory responses upon tissue damage in regeneration and disease2. During regeneration, these programs are activated acutely and then suppressed to restore tissue homeostasis. In pathologies, inflammatory responses transition into chronic states, perpetuating tissue degeneration and contributing to disease progression. We are only starting to have a clear picture of these programs in detail.
‘Genetic programs’ comprise complex networks of DNA codes that include genes, regulatory sequences and instructions for controlling gene expression. These instructions are orchestrated by a multitude of inputs originating from macroenvironments and microenvironments, such as signaling pathways, extracellular components, cell–cell communication and DNA-level nucleotide changes. Regulation occurs at multiple levels, including chromatin conformation, cellular composition and transcriptional and post-translational modifications3. The molecular basis for the idea that embryonic genetic programs are reactivated is the reappearance of master embryonic transcription factors (TFs) that create interconnected feedback loops as instructors of major cellular plasticity and programming4,5,6. This reprogramming also involves changes in chromatin accessibility, leading to global transcriptional profile alterations and changes in cellular state7.
It remains to be determined whether the reactivations encompass entire original programs or only specific parts of certain pathways. Although regenerative processes may superficially resemble embryonic processes due to expression of similar genes, they can be mechanistically distinct, given the differences in cellular history and microenvironment between adult tissue and embryos. Tissue generation during embryogenesis and repair in adult tissue are fundamentally different processes, and only some of the underpinning mechanisms driving tissue repair may partially represent reactivated embryonic programs. Nevertheless, evidence from high-throughput omic techniques with single-cell resolution suggests that substantial portions of embryonic gene expression profiles are similarly expressed in disease contexts but potentially to different extents8,9.
Activated embryonic programs have also been linked to drug resistance in cancer treatment. Targeting distinct developmental factors may enable precise and efficient therapies10,11,12,13. Importantly, oncogenic mutations alone cannot induce many cancer types; tumorigenesis depends on additional, non-mutational mechanisms14,15,16,17. Such mechanisms may be inflammatory or regenerative programs activated in the tumor itself or in the surrounding tissue to induce phenotypic plasticity, which in turn drives disease progression and drug resistance in cancer16,17,18. Studying the function of these factors in the embryo and cancer, side by side, can point us toward essential and disease-specific targets.
In this Review, we explore the cellular and molecular hallmarks associated with reactivation of embryonic genetic programs in the adult context (Fig. 1), influencing both regeneration and disease progression in multiple organs. These programs primarily rely on cell plasticity, including epithelial–mesenchymal plasticity (EMP), and induction of stemness. We highlight similarities and differences of these programs between the original setting in the embryo and the reactivated setting in the adult. We also discuss the phenomenon of lineage reversion in disease pathology, particularly in cancer. We argue that diverse embryonic programs are reactivated in different regenerative and pathological contexts depending on the extent of tissue damage. Lastly, we explore therapeutic options and drug-resistance mechanisms linked to the reactivation of embryonic programs.

In adult tissue regeneration or in chronic diseases, reactivated embryonic genetic programs coordinate cellular and molecular programs that are involved in tissue repair or disease progression, respectively. These programs can induce cell plasticity (for example, EMP and dedifferentiation), stemness (for example, self-renewal, cell proliferation and survival), lineage reversion (inducing potent regenerative capacity and survival), drug resistance and immune evasion. These hallmarks may appear individually or together depending on the context. As indicated by bridging lines, hallmarks are linked to one another. The width of the lines is based on the availability of evidence.
Regenerative cell plasticity
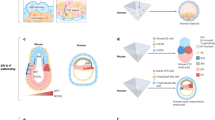
Cell plasticity (Box 1), denoting the dynamic reprogramming of cells that results in changing cellular behaviors and functions (cell state) without genetic mutation, plays a vital role in embryonic development but becomes limited during adult homeostasis (Fig. 2a). In healthy adult tissues, the main cell populations that display plasticity are resident adult stem cells. In tissues with a high turnover rate, such as the intestine, stem cells constantly replenish the cells that are lost (in this case, shed into the lumen)19. In the skin, dedifferentiation (Box 1) of transient amplifying cells to melanocyte stem cells is an integral process for homeostasis20. In other tissues, such as the liver, stem cells are quiescent until they are ‘activated’ to help regenerate damaged tissues. However, dedicated stem cells have hitherto not been identified in all adult tissues. The apparent absence of stem cells might be due to technical difficulties in detecting the cells21. However, if stem cells are indeed absent from some tissues in homeostasis, the key question is how do those tissues regenerate when damaged? Another important question is whether adult stem cells are also accountable for the regeneration process in tissues such as the intestine or skin. A potential mechanism has been suggested by recent work showing that regenerative tissues, for example, the intestine, do not rely solely on adult stem cells but that other cells in the damaged areas can dedifferentiate to a regenerative plastic state that resembles progenitor cells in fetal or embryonic stages8,19.
Tissue repair in mammals, particularly humans, is a highly complex process. Tissues normally consist of many cell types with intricate interactions. Regenerating extensive damage is beyond the capacity of resident adult stem cells and requires a more primitive and potent rescue plan8,22. Recent findings have suggested that this might be provided by embryonic genetic programs, mediated by master TFs and signaling pathways, which are reactivated to restore tissue homeostasis8,23,24,25 (Fig. 2b). These reactivated embryonic programs induce the higher degree of plasticity that is needed to repair injuries in complex tissues.

a, Cell plasticity is higher in early developmental stages and becomes more limited in later stages. Cell proliferation and cell movement are major cellular processes that are tightly regulated during embryonic development. Cell proliferation is regulated by a variety of different transcriptional programs such as those regulated by transcription factors (TFs; for example, MYC and YAP), and EMP (epithelial–mesenchymal plasticity) controls cell movement. EMP is regulated by a plethora of factors including TGFβ and WNT signaling pathways as well as EMT (epithelial–mesenchymal transition) TFs such as Snail family members. b, Genetic programs that regulate processes during embryonic development are reactivated in regeneration upon acute tissue damage to overcome cell loss and restore tissue architecture. EMT activation is mostly observed to a partial extent (partial EMT). c, Prolonged exposure to these programs due to chronic damage can cause inflammatory disease and fibrosis. Excess progenitor programs can cause tissue degeneration. d, Regenerative programs can either directly or indirectly, through inflammatory disease and/or fibrosis, give rise to cancer. Coincidence of embryonic progenitor and inflammatory programs with genetic mutation can lead to cancer progression. Cancer cells rely on stem–progenitor programs for excessive cell proliferation and self-renewal as well as invasive programs mostly activated by partial EMT to disseminate and generate metastatic tumors. Circular arrows represent reactivation of embryonic programs.
However, deregulated regenerative plasticity may also lead to disease. For example, chronic inflammatory responses can result in prolonged activation of embryonic programs, promoting excessive proliferation and cell migration (Fig. 2c,d). Interestingly, recent evidence suggests that, in cancer, the majority of gene expression heterogeneity arises from cellular plasticity rather than genetic mutations14,26. Remarkably, embryonic genetic programs can endow dedifferentiated cancer cells with enhanced survival and with resistance to the immune system and therapies as well as with metastatic potential10,11,27. Unbiased gene expression analyses demonstrate that metastatic tumors are more dedifferentiated than primary tumors and more benign conditions4,15,27, suggesting progressive plasticity in more advanced tissue damage (Fig. 2d). These plasticity programs encompass a diverse array of cellular and molecular attributes. These include the induction of ‘stemness’ (essential traits of stem cells, such as self-renewal and the ability to generate diverse cell types) (Box 1), along with cell movement. These programs are orchestrated by activating signaling pathways and master TFs that were originally active in the embryo, prompting drastic cellular changes, ranging from modification of cell–cell contacts down to altering chromatin conformation and accessibility.
Plasticity and chromatin accessibility
Cell plasticity requires major changes in gene expression, which arise from substantial chromatin reconfiguration28. One of the fundamental stages in cell plasticity involves activation and inhibition of genes regulating new (cell’s upcoming identity) and previous cell fates, achieved through the opening and closing of chromatin regions housing gene promoters and regulatory sequences7. These conformational rearrangements are directed by a variety of mechanisms, such as the recruitment of histone modifiers by master TFs6 (see below).
Genetic mutations in oncogenes as well as genes that encode chromatin remodelers can confer changes in chromatin accessibility and subsequent massive alterations in gene expression in cancer29. Notably, mutations in chromatin remodeler genes are found in around 10% of patients with pancreatic and lung cancers30,31. Furthermore, analysis of chromatin organization can be a clinical predictor. In colorectal cancer (CRC), for instance, more heterogeneous organization correlates with worse prognosis32. During cancer progression, particularly in early steps, chromatin accessibility is expanded at regulatory regions of regenerative or progenitor genes. As an important example in the mouse, Kras-mutated pancreatic cells in premalignant states contain open chromatin in the vicinity of genes encoding ligand and receptors that are essential for cell–cell communication and microenvironmental regulation. These cells also manifest progenitor-like identity from early stages throughout cancer progression14. Moreover, in Apc-mutated cells in early colonic neoplastic lesions in the mouse, SOX9 (a master TF highly expressed during embryonic development) regulates the chromatin-opening state at regenerative gene loci such as Tacstd2, Ly6a and Ly6e7. In melanoma, chromatin hyperaccessibility is shown to occur during dedifferentiation to a primitive neural crest state that confers a malignant phenotype and resistance to therapies33. This plasticity in chromatin accessibility is fundamental to induce disease progression by reactivation of embryonic programs that regulate stemness and invasion, for instance, by inducing epithelial–mesenchymal transition (EMT).
EMP
The best-studied type of plasticity that is common in development and disease is EMP, which refers to the capacity of cells to undergo EMT as well as the reverse process, mesenchymal–epithelial transition (MET)34,35. EMT is a fundamental developmental process enabling immotile epithelial cells to become motile mesenchymal counterparts36 (Box 2). EMT has also been observed in the context of tissue regeneration (Box 3) in the adult as well as pathologies, including inflammatory diseases, fibrosis and cancer34,36,37. Comparative and parallel studies have shown that, although common EMP regulators, particularly EMT TFs, are expressed in embryonic development and in adult contexts38,39, reactivation of EMP programs in regeneration and disease usually results in a spectrum of ‘partial EMT’ rather than ‘full EMT’ states34,36,40.
EMP in regeneration and fibrosis
In the adult, upon formation of a lesion in an epithelial tissue, the inflamed microenvironment can activate EMT in neighboring epithelial cells, allowing them to migrate and participate in wound closure. In skin injuries, for instance, expression of the SNAI2 TF in keratinocytes at the border of the lesion induces ‘partial EMT’ that permits collective cell migration. After the cells have closed the wound, they revert to an epithelial phenotype, undergoing MET36,41.
In chronic tissue damage (such as during fibrosis), resident and recruited fibroblasts differentiate into myofibroblasts that can generate collagen networks, creating fibrotic tissue. EMT is believed to be a key process for initiating this type of disease35. For instance, in kidney fibrosis, transforming growth factor-β (TGFβ) signaling reactivates expression of SNAI1 and TWIST1 EMT TFs in renal epithelial cells, leading to partial EMT in which renal tubules deteriorate concomitant with parenchymal damage, but cells do not disseminate from their primary sites. Epithelial cells in turn secrete cytokines including TGFβ that further induce fibrogenesis13,42. In Crohn’s disease, an inflammatory disease of the colon, Snail family members are reactivated in fibrotic lesions, resulting in a spectrum of EMT states43,44. Similar observations have been reported in different types of pulmonary fibrosis45. A key player in coordinating EMT and fibrogenesis is the RAS-responsive RREB1 protein. RREB1 regulates chromatin accessibility and genetic programs that include expression of SNAI1 in a context-dependent manner. During embryonic gastrulation, RREB1 collaborates with Nodal, a member of the TGFβ superfamily, to induce SNAI1 expression. In fibrosis, TGFβ–SMAD4 and RREB1 drive SNAI1 expression concomitant with fibrogenic factors stimulating myofibroblasts46. In addition to fibrosis and inflammatory diseases, EMP also has important roles in progression of other chronic diseases such as cancer.
EMP in cancer
Analyses of human and mouse tumors have provided extensive evidence of EMT during cancer progression. In most cases, EMT is induced partially39,47,48. Carcinomas, similar to embryos, exhibit specific combinations of EMT TFs and epithelial–mesenchymal effector proteins such as adhesion molecules: a concept called the ‘EMT code’ (ref. 49). In squamous cell carcinoma and lung cancer, for example, deleting protocadherin-encoding FAT1 induces ZEB1 and SOX2 expression, resulting in partial EMT and metastasis50. ZEB1 upregulation is associated with progression of many aggressive human tumors36,37. In particular, ZEB1 interacts with YAP1 (Hippo pathway effector; see below), leading to increased risk of metastasis in breast cancer51. Additionally, ZEB2 expression can be used as a prognostic marker to stratify patients with CRC and high risk of recurrence52.
In melanoma, gene expression programs resemble those expressed by early migratory neural crest cells and melanoblasts53. Subpopulations within the tumor activate EMT programs that resemble those in premigratory neural crest cells in the embryo54, including expression of NOTCH2 and nestin in addition to that of the pluripotency marker SOX2. Notably, PRRX1, which is also expressed in embryonic neural crest cells38, fuels melanoma metastasis by inducing a population of migratory metastasis-initiating cells55.
Additional evidence identifying the role of EMP in cancer metastasis arises from analyses of circulating tumor cells (CTCs). In several carcinomas, for example, breast and pancreatic cancers, CTCs often manifest features of EMT, a finding that correlates with poor clinical outcomes47,56. Molecular and cellular characterization of CTCs in patients as well as experimental models shows that CTCs are mostly found in partial EMT states. Intriguingly, CTCs that travel in clusters but not as single cells have higher metastatic potential57. Generation of these clusters requires some degree of epithelial cell–cell adhesion, which is compatible with partial EMT. In a mouse breast cancer model, it was shown that CTC clusters originate from hypoxic, highly epithelial regions of the primary tumor, while many captured CTCs coexpressed epithelial and mesenchymal genes, for example, Vim, Fn1, Zeb1 (mesenchymal) and Epcam (epithelial)58. Another report also showed expression of Snail family members in CTCs of mouse breast cancer models59. Similar observations were reported in pancreatic and prostate cancer mouse models60,61. Thus, partial (but not full) EMT is most likely the culprit driving metastasis in several carcinomas.
EMP and stemness in cancer
In addition to regulating cell movement and invasion, EMP also regulates ‘stemness’62 (Box 1). However, whether stemness is induced or inhibited depends on the specific EMT code and context. For instance, Snail and TWIST TFs induce stemness by regulating stem-inducing factors36,38,62,63. By contrast, cells expressing high levels of PRRX1 manifest low stemness features and poor plasticity. In experimental models of prostate and bladder carcinomas, extreme mesenchymal states correlate with suppression of embryonic stem cell gene expression such as SOX2 and MYC, while a more epithelial, likely partial EMT state coincides with more stem phenotype and tumor-initiating capacity64. Accordingly, in metastatic breast cancer models, cells with high PRRX1 expression levels are unable to cause metastases and suppression of PRRX1 expression can induce MET and metastatic colonization65. Considering that, during EMT induction, SNAI1 is rapidly upregulated and PRRX1 is only expressed later, it is plausible that, during the transition of cells from a fully epithelial to a fully mesenchymal state, cells are plastic only in intermediate, partial EMT states38,40,66. These findings depict the importance and complexity of regulation of several cellular processes, particularly stemness and EMT.
Regulating embryonic genetic programs, such as EMP, entails navigating a landscape of complexity that encompasses not only transcriptional regulatory mechanisms, including direct control by TFs (for example, EMT TFs; see below), but also intricate post-transcriptional regulation driven by instruments such as microRNA (miRNA).

miRNA
Reactivated embryonic programs include expression of miRNA species that act pleiotropically, influencing multiple downstream processes and cellular functions simultaneously. miRNA is a short noncoding regulatory RNA that post-transcriptionally attenuates gene expression by targeting mRNA67. Well-studied examples of miRNA species that control EMP are the miR-34 and the miR-200 families that target SNAI1 and ZEB1, respectively. These miRNA species, therefore, regulate the balance of the protection of the epithelial state and mesenchymal induction68. In addition, as part of a conserved gene regulatory network, PRRX1 regulates the expression of miR-15 family members to indirectly repress SNAI1, forming a negative reciprocal feedback loop. This mechanism ensures complementary expression and function of PRRX1 and SNAI1 EMT TFs during development and disease38. PRRX1 itself is targeted by several miRNA families including miR-17-92 and miR-34 (ref. 69). Other miRNA species known to regulate EMT include miR-30, miR-29, miR-10, miR-1 and miR-183-182, which can either promote or repress the EMT process by targeting different EMT factors70.
Additionally, a systematic comparison revealed that miRNA species expressed in the developing human colon were also expressed in colon cancer but not in a healthy adult colon. An example is miR-17-92, which functionally regulates cell proliferation by targeting the TF E2F1 (ref. 71). In pancreatic cancer, miR-181cd indirectly induces expression of the developmental factor HMGB3, leading to metastatic progression72.
Beyond the well-studied type of plasticity, numerous recent studies have identified additional factors that shape reactivated embryonic progenitor identities, involving signaling pathways and maser TFs.
Embryonic progenitor identity
During embryogenesis, developing tissues are populated by progenitor cells with high proliferative and stem capacities. A number of signaling pathways are critical regulators of embryonic progenitor identity during development including TGFβ–bone morphogenic protein (BMP) (reviewed in ref. 73), WNT (reviewed in ref. 74) and Hippo. These signaling pathways are among the main drivers of embryonic development, and, in the adult, they are essential modulators of regenerative progenitor plasticity in different damaged tissue contexts.
Hippo signaling
The Hippo pathway is frequently reactivated upon tissue damage or in cancer. In the intestine, for instance, regeneration is mediated by expansion of a population of stem-like progenitor cells that do not express adult stem cell markers such as LGR5 (refs. 8,22,24). These cells express Hippo pathway components, particularly the YAP1 transcriptional co-regulator. Other regenerative and embryonic progenitor genes include Ly6a (encoding SCA-1), Lrg1, Areg, Il33, S100a6 and Tacstd2 (refs. 24,75). In CRC, similar reactivation of genes, such as Yap1, Tacstd2 and S100a6, is associated with metastatic disease7,25,76. Particularly, YAP1 activation is important in early-stage Apc-mutant colonic cells with tumor-initiating capacity by inducing a regenerative program that includes EGF signaling75. This YAP1 activation is also observed in epithelial cells neighboring colon cancer cells77.
YAP1 activation is also important for metastatic colonization. Acting as a mechanotransduction effector, YAP1 can coordinate the spread of disseminated cancer cells along the capillaries mediated by expression of L1CAM78. Notably, expression of L1CAM is linked to metastasis-initiating capacity in human CRC and several other cancer types79,80, which points to the importance of this mechanism. Interestingly, this mechanism is also linked to regeneration, as expression of L1CAM is also activated in the damaged colon epithelium and is essential for tissue repair79.
YAP1 upregulation can induce progenitor-like plasticity and malignancy. In the pancreas, Kras mutation alone is not sufficient to induce adenocarcinoma and requires activation of YAP1 (ref. 81). In fact, early Kras-mutated cells can be indistinguishable from normal cells14. In liver cancer, YAP1 induces tumor progression through epigenetic reprogramming of cells via the DNA demethylase TET1 (ref. 82). Therefore, induction of progenitor-like plasticity programs, which, in these examples, are driven by YAP1, might be indispensable events in carcinogenesis.
Nonetheless, the role of YAP1 in reprogramming cells to an embryonic-like state has been challenged83. Contrary to previous models, blocking Hippo signaling during development did not affect liver size in mice; however, the ability of liver cells to differentiate was impaired. Additionally, gene expression profiling in YAP1-overexpression experiments showed discrete genes that did not fully recapitulate embryonic signatures. These findings suggested that Hippo pathway hyperactivation leads to ectopic overgrowth that does not necessarily resemble developmental programs83. Although YAP1 is expressed both in development and in cancer, perhaps due to its ectopic high expression, the downstream outcomes do not necessarily correspond to those triggered in developmental programs. Other reasons for the discrepancy could be differences in the cohort of signaling pathways active in disease versus development and microenvironmental differences (Fig. 3). However, YAP1 is not the only master TF that is deregulated in disease.

a, Similar but not identical signals, such as growth factors and microenvironmental components, can induce embryonic TFs. b, Due to the differences between embryonic signals and those activated in adult disease, the network of activated TFs, although including many of the same factors, can be substantially distinct. These differences can include the abundance of a TF and ectopic interactions with other factors that are not present in the embryo. Importantly, the dynamic regulation that exists in the embryo can be lost in the reactivated adult context. c, In many cases, embryonic TFs can induce similar downstream genes in the adult (for example, no. 3). However, variabilities can also occur, for instance, if these factors may differentially induce some genes due to alterations in chromatin accessibility (for example, nos. 4 and 6). In addition, varied levels of TFs can induce excess gene expression (for example, no. 2). Altered levels of post-translational modification can also occur in reactivated factors (for example, no. 5). Mutations in the disease can also lead to production of a mutated product (missense, for example, no. 1) or even absence of gene expression (for example, if deleted like in no. 7). d, Similarities and differences between original embryonic genetic programs and those reactivated in the adult can induce similar functional outcomes such as cell proliferation and cell movement, while the differences can make these outcomes uncontrolled and deleterious, for example, during tumor growth and invasion.
Master plasticity TFs
The cellular plasticity that underpins regenerative programs in tissue damage or chronic diseases is regulated by master TFs. Many of these are conductors orchestrating embryonic morphogenetic programs. Master TFs accomplish this by establishing superenhancer domains at genes that control the pluripotent state6. Importantly, some superenhancers are found to operate in both development and cancer progression. These regulate genes essential to promote plasticity and confer enhanced responsiveness to oncogenic signaling cues, thus inducing tumorigenesis5.
Pluripotency factors
OCT4, NANOG, MYC and SOX2 are prime examples of master TFs; together, they activate the pluripotency gene expression program in embryonic stem cells. They induce and maintain the precommitted epiblast state as well as proper deployment of commitment programs, including regulation of Hox cluster gene expression84,85. SOX2 expression levels are tightly regulated and, depending on the level, control different modes of whole-genome TCF–β-catenin axis occupancy in epiblast progenitors86 (Box 3). NANOG, OCT4, SOX2 and MYC are upregulated in poorly differentiated human tumors, for instance, in human patients with aggressive cancer types including breast cancer, bladder carcinomas and glioblastoma4,87. Extensive whole-epigenome analysis of patient samples shows SOX2–OCT4 TF-binding motifs as one of the most highly enriched regions in many cancer types4. Another player in regulating pluripotency is SALL4, which is reactivated in aggressive liver cancer88.
In cancer, these pluripotency TFs are reactivated and confer progenitor-like stem plasticity that fuels disease progression and poor clinical outcome89. For instance, distinct early embryonic pluripotency programs are active in some CTC clusters in breast cancers of human and mouse origin. Binding motifs for the TFs OCT4, NANOG, SOX2 and SOX18 as well as the SIN3A transcription cofactor were hypomethylated across the genome, granting tumor-initiating capacity features and metastatic potential90. In liver cancer, NANOG expression drives self-renewal features downstream of STAT3 signaling91. In skin squamous cell carcinoma and lung cancers, loss of the protocadherin FAT1 induces SOX2 expression. This also coincides with YAP1 activation and EMP50.
MYC
MYC is one of the most frequently upregulated TFs in cancer, and its expression correlates with stemness signatures in many human tumors4. The oncogene is overactivated by gene amplification or downstream of aberrant signaling pathways including WNT and TGFβ. MYC was originally an essential embryonic transcriptional regulator in the early pluripotent state92. It is known as a ‘grand orchestrator’ of tumor progression because of the many biological processes it regulates (reviewed in ref. 93). Expression of MYC together with pluripotency TFs correlates with poor differentiation and worse clinical outcome in many tumor types87. MYC regulates phenotypic plasticity of CRC, melanoma and pancreatic carcinoma9,26,94.
MYC synergizes with mutant RAS to induce inflammatory reprogramming95. Notably, in the mouse, Kras oncogenic mutation cannot induce tumorigenesis without MYC96. In this context, inflammatory or regenerative reprogramming of cells with MYC activation can be stored as a transcriptional and epigenetic memory in epithelial cells even after resolving the damage. Consequently, the previously damaged tissue is more prone to tumorigenesis23. The memory includes genes that stay upregulated after recovery, for instance, Sox9 (ref. 23). Therefore, it can be inferred that oncogenic mutations need embryonic progenitor-like programs to cause cancer. This notion is in line with finding oncogenic mutations in healthy tissues17,97.
SOX families
Many SOX TFs have crucial functions in development and disease. SOX2 is a prominent example (Pluripotency factors). Another example is SOX9, which is expressed across several different tissues during embryogenesis, including mesodermal progenitors98. Expression of SOX9 is expanded and upregulated during colonic and liver inflammation8,99, during skin wound repair100 and during esophageal metaplasia101, regulating cell fate and plasticity. In CRC, overactivation of SOX9 is essential for reactivation of regenerative progenitor programs and the transition to adenoma7. In breast cancer, SOX9 potentiates the acquisition of tumor-initiating capacity and metastatic features by SNAI2-expressing cells102 and upregulates microenvironmental factors essential for stemness maintenance (for example, tenascin C (TNC) and POSTN)63,103. In lung adenocarcinoma, a continuum of progenitor-like gene expression consisting of sequential expression of SOX2 and SOX9 has been shown to control metastasis progression27. SOX4 is another SOX family member that exerts master regulatory functions in different contexts. In different cancer types, such as breast, colon and prostate, SOX4 expression promotes survival, invasion and EMP104. Moreover, SOX10 and SOX11, which are essential for nervous system development, are upregulated in breast cancer and associated with embryonic progenitor properties and EMP as well as disease progression105.
Pluripotency TFs, MYC and SOX family members are typically triggered in response to crucial signaling cues during early development. Many of these cues originate from the microenvironment. However, in inflamed microenvironments characteristic of chronic diseases like cancer, these signaling cues are markedly overactive, potentially leading to aberrant activation of these master TFs. This dysregulation can disrupt the precise developmental control mechanisms observed during development. Therefore, the microenvironment plays a crucial role in regulating master TFs and, hence, the genetic programs that regulate cell plasticity. But how similar is the inflamed microenvironment in disease compared to in embryonic development?
Plasticity and the microenvironment
The microenvironment varies in different contexts: development, regeneration and disease. The resulting differences are reflected in the details of the triggered genetic programs. For example, inflamed tissues may receive similar signals as embryos but with distinct combinations and levels. Consequently, cells near the damaged area activate embryonic programs, yet the output is influenced by altered inputs. This concept remains relatively underexplored, necessitating systematic analyses across various systems to comprehensively grasp differences and similarities.
Immune responses, such as inflammation, both drive and are driven by changes in the microenvironment, for example, by reprogramming progenitor-like cells in the damaged area. In inflammatory skin diseases, such as atopic dermatitis and psoriasis, vascular endothelial cells and macrophages are reprogrammed by reactivating prenatal programs106. In cancer, MYC expression can also modify the microenvironment by suppressing the host immune response93. For example, in hepatocellular carcinoma, embryonic reprogramming of the microenvironment leads to immune suppression. This includes reappearance of embryonic endothelial cell and macrophage programs107.
Specific components and interactions of the extracellular matrix can play important roles in regulating cell plasticity. For instance, collagen I is critical during development108,109. In the adult intestinal extracellular matrix, collagen I is substituted with laminin, depicting a transition from embryonic development to adulthood109. In colonic inflammation, collagen is enriched again and induces YAP–TAZ-dependent reprogramming of epithelial cells into an embryonic-like state22. In pancreatic cancer, collagen I homotrimers produced by cancer cells affect the tumor microenvironment and microbiome by binding to integrin α3β1. This follows integrin α3β1-dependent modification of the immune response and state of hypoxia in the tumor microenvironment110. In breast cancer, dense collagen deposition generates matrix stiffness that in turn induces EMT by upregulation of TWIST1 (ref. 111).
Stromal cells are important signaling hubs during tissue damage. Stromal fibroblasts secrete ligands and cytokines that are vital for homeostatic and regenerative plasticity8,112,113. This is subverted in disease. For example, in the colon, WNT-secreting fibroblasts, which are needed for homeostasis, are expanded upon inflammation113. In acute prostatitis, the inflamed microenvironment is crucial for cell plasticity and lineage conversion, leading to induction of prostate cancer114. In prostate cancer, cancer-associated fibroblasts trigger SOX9 expression, which promotes disease progression and leads to poor clinical outcome115. In several cancer types, PRRX1 acts as a master regulator of the cancer-associated myofibroblast lineage. Although its activation in cancer cells can have favorable outcomes38,65, PRRX1 upregulation in cancer-associated fibroblasts mediated by TGFβ signaling correlates with unfavorable outcome in patients116. PRRX1 expression in melanoma triggers cross-talk involving the microenvironment, particularly endothelial cells, which impacts tumorigenic competence and metastasis55. Moreover, SNAI1 expression in melanoma tumor stromal cells, particularly fibroblasts, promotes immunosuppression, leading to tumor growth and metastasis117. Thus, reprogramming of microenvironmental components to embryonic progenitor states directly regulates disease progression.
Lineage reversion in pathology
Despite the storm of microenvironmental signaling pathways and regulatory elements in diseases causing tissue damage, reprogrammed cells may still retain their lineage identity. This is plausible due to the presence of lineage-specific TFs, which are master regulators that define a cellular lineage by controlling gene expression patterns via binding to superenhancer sequences and promoters of lineage-identifying genes5. In cancer, selective programs specific to tissue lineage are reported to even be essential for tumor progression, often triggered by aberrant regulation of TFs normally associated with developmental or committed tissue lineage specification (Table 1). Some of these TFs are expressed at low levels in homeostasis, but their expression is expanded and upregulated, whereas some others are only expressed in late developmental stages and reactivated upon damage. While, in tissue damage, these TFs activate regenerative programs, co-occurrence with oncogenic mutations can lead to cancer progression. Co-opting with activation of progenitor programs, the presence of lineage TFs that are expressed during development in progenitor cells is a sign of ‘lineage reversion’. These lineage TFs induce ‘cancer dependency’ whereby losing expression of these TFs will generally result in apoptosis118.
For example, progenitor lineage programs that are inactive after maturation reappear during breast cancer metastasis119. These include p63, distinct SOX family members and FOXA1120,121. p63 is a lineage marker that is expressed since the early multipotent state in the mammary gland122. Ectopic expression of p63 in differentiated luminal cells induces their reprogramming into basal-like cells, and p63 overactivation induces WNT-dependent development of basal-like breast cancer122,123. Additionally, SOX4, which is expressed in early progenitors of luminal mammary cells120, is an activator of EMT and a driver of breast cancer metastasis124,125. SOX4 directly controls expression of EZH2, which epigenetically regulates the EMT state126. By contrast, SOX11 is only expressed in early multipotent embryonic progenitors and then reactivated in breast cancer, inducing invasive features and poor prognosis127,128. Furthermore, expression of FOXA1, which is a fetal factor, creates a core program that regulates a cohort of genes associated with high risk of endocrine-resistant breast cancer129. FOXA1 is a key determinant of the cellular response to estrogen receptor α by regulating the interaction between the receptor and chromatin130. Mechanistically, upregulation of FOXA1, as a pioneer TF, leads to wide reprogramming composed of activation of hypoxia-inducible factor (HIF1–HIF2, SOX family members and other metastasis-promoting programs131.
As another example, in gastrointestinal tissues, SOX and caudal-type homeobox (CDX) factors are expressed in lineage progenitors. The early-stage developing stomach (foregut) and colon (hindgut) express the lineage TFs SOX2 and CDX2, respectively132. SOX2 (together with SOX9) and CDX2 also promote programs leading to gastric cancer and CRC, respectively133,134.
Similar to diseases of breast and gastrointestinal organs, lineage reversion also occurs in many other diseases across different organs such as skin, nervous system, bone, lung, prostate, etc. (summarized in Table 1). These instances of lineage reversion and reactivation of embryonic programs that were discussed earlier depict a diverse temporal pattern. It seems that there are important stages during development that serve as docking points for adult cells in the damaged tissue to revert back to2.
Diversity of reactivated embryonic programs
Embryonic genetic programs are diversely reactivated in different regenerative and pathological contexts. Depending on the extent of tissue damage being acute or chronic, adult cells revert back to different embryonic states (discussed in ref. 2) (Fig. 4). In an acute process such as colonic inflammation, TFs and cellular states resemble the molecular and cellular makeup of late embryogenesis, for instance, the reappearance of Apoa1-positive embryonic enterocyte progenitors8 or other fetal progenitor markers such as Tacstd2 (ref. 24). EMT TFs primarily active during mid-stage embryogenesis, such as SNAI1, are activated in tissue fibrosis, in which more prolonged and expanded damage occurs36. And, interestingly, in many cancer types, in addition to lineage TFs that are typically active during late embryogenesis, very early programs are also reactivated, controlled, for example, by pluripotency factors. In this case, a mix of ectopic progenitor programs can potentially increase tumor heterogeneity and malignancy2.

a–c, Different embryonic genetic programs are active during different stages of development. d–g, Tissue damage in the adult leads to loss of homeostatic programs and activates regenerative programs, which, depending on the extent of the damage being acute or chronic, can include activation of late, intermediate or early embryonic programs, respectively. Identifying common programs in development and disease will provide new specific targets. Studying these targets in development can reveal their importance. In the disease context, these reactivated embryonic program components can serve as specific drug targets for therapy. IBD, inflammatory bowel disease.
The importance of embryonic program reactivation in cancer has been realized, and different terms, such as ‘onco-fetal’ or ‘carcino-embryonic’, have been coined135,136, but neither of the terms are satisfactory. Considering the mixture of reactivated programs in cancer, from early to late developmental stages, the term onco-fetal as a general identifier for this phenomenon is not comprehensive. This will limit the programs to only a ‘fetal’ period (after 8 weeks in humans, which is equal to about 14 days in mice2), therefore neglecting the programs in earlier (embryonic) stages. In turn, using the term ‘carcino’ in ‘carcino-embryonic’ is ambiguous. On the one hand, it can be misleading if understood as short for ‘carcinoma’, therefore being limited to cancers arising from epithelial tissue; on the other hand, it can be accurate if assumed to mean cancer in general. As a more encompassing term, we propose ‘onco-embryonic’ to describe reactivation of embryonic programs in cancer, as ‘embryonic’ is often used to address all stages of embryogenesis.
Therapeutics and drug resistance
Understanding the key features of reactivated embryonic programs in diseases, particularly cancer (onco-embryology), offers potential for new therapies. For example, inhibiting specific drivers only active in the embryo and disease states could minimize side effects (Fig. 4). Moreover, targeting embryonic factors critical for tumor cell plasticity and potency may prove most efficient.
Cell plasticity and cancer therapy
The plasticity and stemness of cancer cells make them resistant to treatments. Lineage plasticity, for example, can allow tumor cells to escape therapy33. An example is prostate cancer: tumors resistant to antiandrogen treatment (enzalutamide) undergo a phenotypic switch from androgen receptor-dependent cells to an independent luminal state. In another example, melanoma cells in patients undergo dedifferentiation to a neural crest state, a process that induces resistance to targeted therapies and immunotherapies33. CTCs from patients with breast cancer who express EMT markers are associated with disease progression and resistance to therapy47. In a proof-of-principle study, reverting mesenchymal cells to a more epithelial counterpart (by inducing MET) resulted in tumors with no tumor-initiating capacity and blocked disease progression137. Remarkably, blocking EMT has been shown to be beneficial in mouse models of skin squamous cell carcinoma and endometrial cancer as well as in patients with endometrial cancer. Targeting netrin 1 (a laminin-related secreted protein) in these contexts can block transition of cancer cells to full EMT and reduce metastasis. Upon netrin 1 blockade, cancer cells were more susceptible to therapy138,139. Importantly, netrin blockade does not affect transition of cells to partial EMT. This may be essential to block EMT-driven metastasis in other contexts (see above; EMP). The outcome of blocking netrin 1 should be assessed in other cancer types.
In another study, invasive breast cancer cells that underwent EMT were reprogrammed to adipocytes, making them less invasive. The treatment comprised a combination of MEK inhibitors and the anti-diabetic agent rosiglitazone and was tested in several experimental models140. Although these attempts have proven effective in the preclinical phase, their efficacy in a clinical setting still needs to be evaluated. One drawback of targeting mesenchymal cells (generated from EMT) is their similarity to tissue mesenchymal cells such as fibroblasts. Similarly, these methods may also target tissue-resident mesenchymal cells. The consequence of the effect on resident fibroblasts is not clear.
EMT can also induce immune escape as well as resistance to immunotherapy. EMT induces cell plasticity and heterogeneity, in particular, shaping the tumor microenvironment to control antitumor immunity141. In a melanoma mouse model, targeting SNAI1 expression in the tumor microenvironment decreased melanoma growth and lung metastasis117. Additionally, breast cancer cells in a partial EMT state create an immunosuppressive microenvironment that impairs immune checkpoint blockade therapy. Interestingly, disrupting this protective state by blocking key factors that are derived from partial EMT cells, including CD73, CSF1 and SPP1, has been shown to sensitize tumors to the treatment142.
Embryonic latent cell states and cancer therapy
Cancer cells hijack embryonic programs to enter a transient ‘drug-tolerant persister’ state. Persister cells are quiescent, resistant to chemotherapy and, long after apparently successful therapy, can be reawakened to form metastases. Quiescent tumor cells express embryonic TFs, including SOX2 (ref. 143). The persister state shares transcriptional programs with embryonic diapause, which is a reversible state during which embryonic development is suspended due to environmental stress10,11. Intriguingly, in this state, cancer cells suppress MYC. Artificially repressing MYC can also induce the diapause state11. MYC is an attractive target as several cancers have been suggested to be addicted to its expression93. On the flip side, if MYC reduction induces a persister state, then this treatment may have unwanted long-term consequences.
Another developmental process that has crucial roles during regeneration and cancer progression is senescence. Senescence instructs patterning and growth of the embryo, through nonproliferative states, driven by mediators such as p21 and senescence-associated secretory components, such as pro-inflammatory cytokines144,145. In adults, these factors drive cell plasticity and stem cell functions via genome-wide epigenetic remodeling as a stress response, for instance, induced by upregulation of Ras oncogene or as a side effect of anticancer therapies. WNT signaling is an essential driver for this process145. Persistently residing senescent cells are capable of resuming proliferation and consequently hold an important capacity for relapse. Therefore, targeting senescence in cancer is considered a treatment strategy that is unlikely to affect healthy cells145.
Therapeutic targeting of embryonic TFs
TFs are attractive but challenging targets for therapy. For example, SOX2 is crucial for inducing progenitor and stem phenotypes and therapy resistance in several cancer types, but directly targeting it in patients has proven difficult146. Nevertheless, targeting SOX11 in mantle cell lymphoma has shown promise. Small-molecule inhibitors blocking the DNA-binding ability of SOX11 can disrupt its functional outputs, and downstream targets can be abrogated12. Notably, CRISPR interference with SOX11-binding (and HNRNPH1-binding) motifs in superenhancers regulating MYC expression effectively reduced MYC levels147. In another example of successful targeting of TFs, SNAI1 knockdown using interfering RNA in mice blocked and reverted kidney fibrosis13. Similarly, blocking TGFβ or SNAI1 has been suggested as a therapeutic option in fibrosis of the liver and lung36,45.
On the other hand, inducing embryonic programs in tissues lacking regenerative capacity could improve repair potency. For example, overexpressing pluripotency factors in adult mouse cardiomyocytes improves heart regeneration after myocardial infarction by inducing embryonic gene expression and less mature cardiomyocytes148. This approach could be extended to tissues such as the nervous system with limited regeneration capability. However, caution is necessary, as overexpression of TFs might lead to deleterious outcomes due to random mutations or unspecific delivery to various cell types in vivo.
Understanding the regulatory networks of reactivated embryonic programs in disease could be an important step to overcome the inherent challenge of targeting TFs. Identifying the molecular structure, regulation and cellular functions of TFs is essential before using them as targets. In addition, targeting several types of malignant cancers, similar to other more complex diseases, could require targeting several programs encompassing early, intermediate and late developmental stages.
Conclusions and perspectives
There is overwhelming evidence that embryonic programs are reactivated upon tissue damage or cellular stress and have essential roles in promoting adult diseases by inducing cellular plasticity, stemness and cell movement. By systematically comparing embryonic programs and their respective corrupted versions active in diseases, we can uncover disease-specific targets for therapies. Using embryonic development as a navigation platform to understand disease will open new ways to better understand the biology of adult pathology.
Importantly, in disease states, reactivated embryonic programs will not be exact copies of those found in the embryo, as the history of cells, the cellular context and/or the concomitant signals they receive can impact cellular response. Even differences in expression levels of the responsible master TFs may result in major differences in regulating downstream targets. The effect of disparate SOX2 or YAP1 expression levels are two prominent examples83,86. Moreover, we need to define ‘reactivation of an embryonic or developmental program’ more precisely: it is not one single process. Depending on the identity of the reactivated programs, for example, from early versus late embryonic stages, different effects will be exerted in terms of self-renewal versus cell proliferation and cell migration.
In sum, a holistic view of commonalities between development and disease will provide better understanding of the underlying process and should spotlight therapeutic targets.
References
Virchow, R. L. K. Die Cellularpathologie in ihrer Begründung auf physiologische und pathologische Gewebelehre (A. Hirschwald, 1859).
Fazilaty, H. Restoration of embryonic gene expression patterns in tissue regeneration and disease. Nat. Rev. Mol. Cell Biol. 24, 375–376 (2023).
Peluffo, A. E. The ‘genetic program’: behind the genesis of an influential metaphor. Genetics 200, 685–696 (2015).
Malta, T. M. et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 173, 338–354 (2018).
Hnisz, D. et al. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 58, 362–370 (2015).
Whyte, W. A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Bala, P. et al. Aberrant cell state plasticity mediated by developmental reprogramming precedes colorectal cancer initiation. Sci. Adv. 9, eadf0927 (2023).
Fazilaty, H. et al. Tracing colonic embryonic transcriptional profiles and their reactivation upon intestinal damage. Cell Rep. 36, 109484 (2021).
Varum, S. et al. Yin yang 1 orchestrates a metabolic program required for both neural crest development and melanoma formation. Cell Stem Cell 24, 637–653 (2019).
Rehman, S. K. et al. Colorectal cancer cells enter a diapause-like DTP state to survive chemotherapy. Cell 184, 226–242 (2021).
Dhimolea, E. et al. An embryonic diapause-like adaptation with suppressed Myc activity enables tumor treatment persistence. Cancer Cell 39, 240–256 (2021).
Jatiani, S. S. et al. SOX11 inhibitors are cytotoxic in mantle cell lymphoma. Clin. Cancer Res. 27, 4652–4663 (2021).
Grande, M. T. et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 21, 989–997 (2015).
Burdziak, C. et al. Epigenetic plasticity cooperates with cell–cell interactions to direct pancreatic tumorigenesis. Science 380, eadd5327 (2023).
Moorman, A. R. et al. Progressive plasticity during colorectal cancer metastasis. Preprint at bioRxiv https://doi.org/10.1101/2023.08.18.553925 (2023).
Li, Y. et al. Mutant Kras co-opts a proto-oncogenic enhancer network in inflammation-induced metaplastic progenitor cells to initiate pancreatic cancer. Nat. Cancer 2, 49–65 (2021).
Hill, W. et al. Lung adenocarcinoma promotion by air pollutants. Nature 616, 159–167 (2023).
Salgia, R. & Kulkarni, P. The genetic/non-genetic duality of drug ‘resistance’ in cancer. Trends Cancer 4, 110–118 (2018).
Shivdasani, R. A., Clevers, H. & de Sauvage, F. J. Tissue regeneration: reserve or reverse? Science 371, 784–786 (2021).
Sun, Q. et al. Dedifferentiation maintains melanocyte stem cells in a dynamic niche. Nature 616, 774–782 (2023).
Bhartiya, D. Adult tissue-resident stem cells—fact or fiction? Stem Cell Res. Ther. 12, 73 (2021).
Yui, S. et al. YAP/TAZ-dependent reprogramming of colonic epithelium links ECM remodeling to tissue regeneration. Cell Stem Cell 22, 35–49 (2018).
Del Poggetto, E. et al. Epithelial memory of inflammation limits tissue damage while promoting pancreatic tumorigenesis. Science 373, eabj0486 (2021).
Nusse, Y. M. et al. Parasitic helminths induce fetal-like reversion in the intestinal stem cell niche. Nature 559, 109–113 (2018).
Mustata, R. C. et al. Identification of Lgr5-independent spheroid-generating progenitors of the mouse fetal intestinal epithelium. Cell Rep. 5, 421–432 (2013).
Househam, J. et al. Phenotypic plasticity and genetic control in colorectal cancer evolution. Nature 611, 744–753 (2022).
Laughney, A. M. et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med. 26, 259–269 (2020).
Perino, M. & Veenstra, G. J. C. Chromatin control of developmental dynamics and plasticity. Dev. Cell 38, 610–620 (2016).
Hanahan, D. Hallmarks of cancer: new dimensions. Cancer Discov. 12, 31–46 (2022).
Hayashi, A., Hong, J. & Iacobuzio-Donahue, C. A. The pancreatic cancer genome revisited. Nat. Rev. Gastroenterol. Hepatol. 18, 469–481 (2021).
Dagogo-Jack, I. et al. Clinicopathologic characteristics of BRG1-deficient NSCLC. J. Thorac. Oncol. 15, 766–776 (2020).
Kleppe, A. et al. Chromatin organisation and cancer prognosis: a pan-cancer study. Lancet Oncol. 19, 356–369 (2018).
Kim, Y. J. et al. Melanoma dedifferentiation induced by IFN-γ epigenetic remodeling in response to anti-PD-1 therapy. J. Clin. Invest. 131, e145859 (2021).
Yang, J. et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 21, 341–352 (2020).
Thiery, J. P., Acloque, H., Huang, R. Y. J. & Nieto, M. A. Epithelial–mesenchymal transitions in development and disease. Cell 139, 871–890 (2009).
Nieto, M. A., Huang, R. Y.-J., Jackson, R. A. & Thiery, J. P. EMT: 2016. Cell 166, 21–45 (2016).
Brabletz, T., Kalluri, R., Nieto, M. A. & Weinberg, R. A. EMT in cancer. Nat. Rev. Cancer 18, 128–134 (2018).
Fazilaty, H. et al. A gene regulatory network to control EMT programs in development and disease. Nat. Commun. 10, 5115 (2019).
Zhang, Y. et al. Genome-wide CRISPR screen identifies PRC2 and KMT2D-COMPASS as regulators of distinct EMT trajectories that contribute differentially to metastasis. Nat. Cell Biol. 24, 554–564 (2022).
Lambert, A. W. & Weinberg, R. A. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat. Rev. Cancer 21, 325–338 (2021).
Shaw, T. J. & Martin, P. Wound repair: a showcase for cell plasticity and migration. Curr. Opin. Cell Biol. 42, 29–37 (2016).
Frangogiannis, N. G. Transforming growth factor-β in tissue fibrosis. J. Exp. Med. 217, e20190103 (2020).
Scharl, M. et al. Hallmarks of epithelial to mesenchymal transition are detectable in Crohn’s disease associated intestinal fibrosis. Clin. Transl. Med. 4, 1 (2015).
Scharl, M. & Rogler, G. Pathophysiology of fistula formation in Crohn’s disease. World J. Gastrointest. Pathophysiol. 5, 205–212 (2014).
Rout-Pitt, N., Farrow, N., Parsons, D. & Donnelley, M. Epithelial mesenchymal transition (EMT): a universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir. Res. 19, 136 (2018).
Su, J. et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature 577, 566–571 (2020).
Yu, M. et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584 (2013).
Lüönd, F. et al. Distinct contributions of partial and full EMT to breast cancer malignancy. Dev. Cell 56, 3203–3221 (2021).
Nieto, M. A. Context-specific roles of EMT programmes in cancer cell dissemination. Nat. Cell Biol. 19, 416–418 (2017).
Pastushenko, I. et al. Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature 589, 448–455 (2021).
Lehmann, W. et al. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat. Commun. 7, 10498 (2016).
Sreekumar, R. et al. Assessment of nuclear ZEB2 as a biomarker for colorectal cancer outcome and TNM risk stratification. JAMA Netw. Open 1, e183115 (2018).
Marie, K. L. et al. Melanoblast transcriptome analysis reveals pathways promoting melanoma metastasis. Nat. Commun. 11, 333 (2020).
Soldatov, R. et al. Spatiotemporal structure of cell fate decisions in murine neural crest. Science 364, eaas9536 (2019).
Karras, P. et al. A cellular hierarchy in melanoma uncouples growth and metastasis. Nature 610, 190–198 (2022).
Semaan, A. et al. Characterisation of circulating tumour cell phenotypes identifies a partial-EMT sub-population for clinical stratification of pancreatic cancer. Br. J. Cancer 124, 1970–1977 (2021).
Aceto, N. et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122 (2014).
Donato, C. et al. Hypoxia triggers the intravasation of clustered circulating tumor cells. Cell Rep. 32, 108105 (2020).
Bonnomet, A. et al. A dynamic in vivo model of epithelial-to-mesenchymal transitions in circulating tumor cells and metastases of breast cancer. Oncogene 31, 3741–3753 (2012).
Ting, D. T. et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep. 8, 1905–1918 (2014).
Ruscetti, M., Quach, B., Dadashian, E. L., Mulholland, D. J. & Wu, H. Tracking and functional characterization of epithelial–mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Cancer Res. 75, 2749–2759 (2015).
Mani, S. A. et al. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (2008).
Fazilaty, H., Gardaneh, M., Akbari, P., Zekri, A. & Behnam, B. SLUG and SOX9 cooperatively regulate tumor initiating niche factors in breast cancer. Cancer Microenviron. 9, 71–74 (2016).
Celià-Terrassa, T. et al. Epithelial–mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J. Clin. Invest. 122, 1849–1868 (2012).
Ocaña, O. H. et al. Metastatic colonization requires the repression of the epithelial–mesenchymal transition inducer Prrx1. Cancer Cell 22, 709–724 (2012).
Pastushenko, I. et al. Identification of the tumour transition states occurring during EMT. Nature 556, 463–468 (2018).
Bartel, D. P. Metazoan microRNAs. Cell 173, 20–51 (2018).
Cano, A. & Nieto, M. A. Non-coding RNAs take centre stage in epithelial-to-mesenchymal transition. Trends Cell Biol. 18, 357–359 (2008).
Rago, L. et al. MicroRNAs establish the right-handed dominance of the heart laterality pathway in vertebrates. Dev. Cell 51, 446–459 (2019).
Díaz-López, A., Moreno-Bueno, G. & Cano, A. Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives. Cancer Manag. Res. 6, 205–216 (2014).
Monzo, M. et al. Overlapping expression of microRNAs in human embryonic colon and colorectal cancer. Cell Res. 18, 823–833 (2008).
Saghafinia, S. et al. Cancer cells retrace a stepwise differentiation program during malignant progression. Cancer Discov. 11, 2638–2657 (2021).
Lee, J. H. & Massagué, J. TGF-β in developmental and fibrogenic EMTs. Semin. Cancer Biol. 86, 136–145 (2022).
Rim, E. Y., Clevers, H. & Nusse, R. The Wnt pathway: from signaling mechanisms to synthetic modulators. Annu. Rev. Biochem. 91, 571–598 (2022).
Gregorieff, A., Liu, Y., Inanlou, M. R., Khomchuk, Y. & Wrana, J. L. Yap-dependent reprogramming of Lgr5+ stem cells drives intestinal regeneration and cancer. Nature 526, 715–718 (2015).
Solé, L. et al. p53 wild-type colorectal cancer cells that express a fetal gene signature are associated with metastasis and poor prognosis. Nat. Commun. 13, 2866 (2022).
Jacquemin, G. et al. Paracrine signalling between intestinal epithelial and tumour cells induces a regenerative programme. eLife 11, e76541 (2022).
Er, E. E. et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat. Cell Biol. 20, 966–978 (2018).
Ganesh, K. et al. L1CAM defines the regenerative origin of metastasis-initiating cells in colorectal cancer. Nat. Cancer 1, 28–45 (2020).
Altevogt, P. et al. Recent insights into the role of L1CAM in cancer initiation and progression. Int. J. Cancer 147, 3292–3296 (2020).
Zhang, W. et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci. Signal. 7, ra42 (2014).
Wu, B.-K., Mei, S.-C., Chen, E. H., Zheng, Y. & Pan, D. YAP induces an oncogenic transcriptional program through TET1-mediated epigenetic remodeling in liver growth and tumorigenesis. Nat. Genet. 54, 1202–1213 (2022).
Kowalczyk, W. et al. Hippo signaling instructs ectopic but not normal organ growth. Science 378, eabg3679 (2022).
Tiana, M. et al. Pluripotency factors regulate the onset of Hox cluster activation in the early embryo. Sci. Adv. 8, eabo3583 (2022).
Li, M. & Belmonte, J. C. I. Ground rules of the pluripotency gene regulatory network. Nat. Rev. Genet. 18, 180–191 (2017).
Blassberg, R. et al. Sox2 levels regulate the chromatin occupancy of WNT mediators in epiblast progenitors responsible for vertebrate body formation. Nat. Cell Biol. 24, 633–644 (2022).
Ben-Porath, I. et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 40, 499–507 (2008).
Yong, K. J. et al. Oncofetal gene SALL4 in aggressive hepatocellular carcinoma. N. Engl. J. Med. 368, 2266–2276 (2013).
Hepburn, A. C. et al. The induction of core pluripotency master regulators in cancers defines poor clinical outcomes and treatment resistance. Oncogene 38, 4412–4424 (2019).
Gkountela, S. et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell 176, 98–112 (2019).
Lee, T. K. W. et al. CD24+ liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 9, 50–63 (2011).
Kim, J. et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 143, 313–324 (2010).
Dhanasekaran, R. et al. The MYC oncogene — the grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 19, 23–36 (2022).
Sodir, N. M. et al. MYC instructs and maintains pancreatic adenocarcinoma phenotype. Cancer Discov. 10, 588–607 (2020).
Kortlever, R. M. et al. Myc cooperates with Ras by programming inflammation and immune suppression. Cell 171, 1301–1315 (2017).
Sodir, N. M. et al. Reversible Myc hypomorphism identifies a key Myc-dependency in early cancer evolution. Nat. Commun. 13, 6782 (2022).
Lee-Six, H. et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 574, 532–537 (2019).
Jo, A. et al. The versatile functions of Sox9 in development, stem cells, and human diseases. Genes Dis. 1, 149–161 (2014).
Campana, L., Esser, H., Huch, M. & Forbes, S. Liver regeneration and inflammation: from fundamental science to clinical applications. Nat. Rev. Mol. Cell Biol. 22, 608–624 (2021).
Ge, Y. et al. Stem cell lineage infidelity drives wound repair and cancer. Cell 169, 636–650 (2017).
Drubbel, A. V. et al. Reactivation of the Hedgehog pathway in esophageal progenitors turns on an embryonic-like program to initiate columnar metaplasia. Cell Stem Cell 28, 1411–1427 (2021).
Guo, W. et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 148, 1015–1028 (2012).
Fazilaty, H., Gardaneh, M., Bahrami, T., Salmaninejad, A. & Behnam, B. Crosstalk between breast cancer stem cells and metastatic niche: emerging molecular metastasis pathway? Tumour Biol. 34, 2019–2030 (2013).
Hanieh, H., Ahmed, E. A., Vishnubalaji, R. & Alajez, N. M. SOX4: epigenetic regulation and role in tumorigenesis. Semin. Cancer Biol. 67, 91–104 (2020).
Dravis, C. et al. Epigenetic and transcriptomic profiling of mammary gland development and tumor models disclose regulators of cell state plasticity. Cancer Cell 34, 466–482 (2018).
Reynolds, G. et al. Developmental cell programs are co-opted in inflammatory skin disease. Science 371, eaba6500 (2021).
Sharma, A. et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell 183, 377–394 (2020).
Chen, Y. et al. Type-I collagen produced by distinct fibroblast lineages reveals specific function during embryogenesis and osteogenesis imperfecta. Nat. Commun. 12, 7199 (2021).
Ramadan, R. et al. The extracellular matrix controls stem cell specification and crypt morphology in the developing and adult mouse gut. Biology Open 11, bio059544 (2022).
Chen, Y. et al. Oncogenic collagen I homotrimers from cancer cells bind to α3β1 integrin and impact tumor microbiome and immunity to promote pancreatic cancer. Cancer Cell 40, 818–834 (2022).
Wei, S. C. et al. Matrix stiffness drives epithelial–mesenchymal transition and tumour metastasis through a TWIST1–G3BP2 mechanotransduction pathway. Nat. Cell Biol. 17, 678–688 (2015).
Brügger, M. D., Valenta, T., Fazilaty, H., Hausmann, G. & Basler, K. Distinct populations of crypt-associated fibroblasts act as signaling hubs to control colon homeostasis. PLoS Biol. 18, e3001032 (2020).
Degirmenci, B., Valenta, T., Dimitrieva, S., Hausmann, G. & Basler, K. GLI1-expressing mesenchymal cells form the essential Wnt-secreting niche for colon stem cells. Nature 558, 449–453 (2018).
Kwon, O.-J., Zhang, L., Ittmann, M. M. & Xin, L. Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. Proc. Natl Acad. Sci. USA 111, E592–E600 (2014).
Qin, H. et al. SOX9 in prostate cancer is upregulated by cancer-associated fibroblasts to promote tumor progression through HGF/c-Met–FRA1 signaling. FEBS J. 288, 5406–5429 (2021).
Lee, K.-W. et al. PRRX1 is a master transcription factor of stromal fibroblasts for myofibroblastic lineage progression. Nat. Commun. 13, 2793 (2022).
Arumi-Planas, M. et al. Microenvironmental Snail1-induced immunosuppression promotes melanoma growth. Oncogene 42, 2659–2672 (2023).
Tsherniak, A. et al. Defining a cancer dependency map. Cell 170, 564–576 (2017).
Giraddi, R. R. et al. Single-cell transcriptomes distinguish stem cell state changes and lineage specification programs in early mammary gland development. Cell Rep. 24, 1653–1666 (2018).
Pal, B. et al. Construction of developmental lineage relationships in the mouse mammary gland by single-cell RNA profiling. Nat. Commun. 8, 1627 (2017).
Liu, Y. & Guo, W. SOX factors as cell-state regulators in the mammary gland and breast cancer. Semin. Cell Dev. Biol. 114, 126–133 (2021).
Wuidart, A. et al. Early lineage segregation of multipotent embryonic mammary gland progenitors. Nat. Cell Biol. 20, 666–676 (2018).
Chakrabarti, R. et al. ΔNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat. Cell Biol. 16, 1004–1015 (2014).
Bagati, A. et al. Integrin αvβ6–TGFβ–SOX4 pathway drives immune evasion in triple-negative breast cancer. Cancer Cell 39, 54–67 (2021).
Roukens, M. G. et al. Regulation of a progenitor gene program by SOX4 is essential for mammary tumor proliferation. Oncogene 40, 6343–6353 (2021).
Tiwari, N. et al. Sox4 is a master regulator of epithelial–mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell 23, 768–783 (2013).
Oliemuller, E. et al. SOX11 promotes invasive growth and ductal carcinoma in situ progression: SOX11 promotes invasion and DCIS progression. J. Pathol. 243, 193–207 (2017).
Zvelebil, M. et al. Embryonic mammary signature subsets are activated in Brca1−/− and basal-like breast cancers. Breast Cancer Res. 15, R25 (2013).
Wen, W. et al. Genetic variations of DNA bindings of FOXA1 and co-factors in breast cancer susceptibility. Nat. Commun. 12, 5318 (2021).
Hurtado, A., Holmes, K. A., Ross-Innes, C. S., Schmidt, D. & Carroll, J. S. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat. Genet. 43, 27–33 (2011).
Fu, X. et al. FOXA1 upregulation promotes enhancer and transcriptional reprogramming in endocrine-resistant breast cancer. Proc. Natl Acad. Sci. USA 116, 26823–26834 (2019).
Sherwood, R. I., Chen, T.-Y. A. & Melton, D. A. Transcriptional dynamics of endodermal organ formation. Dev. Dyn. 238, 29–42 (2009).
Francis, R. et al. Gastrointestinal transcription factors drive lineage-specific developmental programs in organ specification and cancer. Sci. Adv. 5, eaax8898 (2019).
Salari, K. et al. CDX2 is an amplified lineage-survival oncogene in colorectal cancer. Proc. Natl Acad. Sci. USA 109, E3196–E3205 (2012).
Gold, P. & Freedman, S. O. Specific carcinoembryonic antigens of the human digestive system. J. Exp. Med. 122, 467–481 (1965).
Banwo, O., Versey, J. & Hobbs, J. R. New oncofetal antigen for human pancreas. Lancet 303, 643–645 (1974).
Pattabiraman, D. R. et al. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 351, aad3680 (2016).
Cassier, P. A. et al. Netrin-1 blockade inhibits tumour growth and EMT features in endometrial cancer. Nature 620, 409–416 (2023).
Lengrand, J. et al. Pharmacological targeting of netrin-1 inhibits EMT in cancer. Nature 620, 402–408 (2023).
Ishay-Ronen, D. et al. Gain fat—lose metastasis: converting invasive breast cancer cells into adipocytes inhibits cancer metastasis. Cancer Cell 35, 17–32 (2019).
Terry, S. et al. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 11, 824–846 (2017).
Dongre, A. et al. Direct and indirect regulators of epithelial–mesenchymal transition-mediated immunosuppression in breast carcinomas. Cancer Discov. 11, 1286–1305 (2021).
Hu, J. et al. STING inhibits the reactivation of dormant metastasis in lung adenocarcinoma. Nature 616, 806–813 (2023).
Storer, M. et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155, 1119–1130 (2013).
Milanovic, M., Yu, Y. & Schmitt, C. A. The senescence–stemness alliance — a cancer-hijacked regeneration principle. Trends Cell Biol. 28, 1049–1061 (2018).
Hüser, L., Novak, D., Umansky, V., Altevogt, P. & Utikal, J. Targeting SOX2 in anticancer therapy. Expert Opin. Ther. Targets 22, 983–991 (2018).
Luo, Z. et al. Human fetal cerebellar cell atlas informs medulloblastoma origin and oncogenesis. Nature 612, 787–794 (2022).
Chen, Y. et al. Reversible reprogramming of cardiomyocytes to a fetal state drives heart regeneration in mice. Science 373, 1537–1540 (2021).
Chen, J., Ju, H. L., Yuan, X. Y., Wang, T. J. & Lai, B. Q. SOX4 is a potential prognostic factor in human cancers: a systematic review and meta-analysis. Clin. Transl. Oncol. 18, 65–72 (2016).
Pomerantz, M. M. et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat. Genet. 52, 790–799 (2020).
Ma, F. et al. SOX9 drives WNT pathway activation in prostate cancer. J. Clin. Invest. 126, 1745–1758 (2016).
Mu, P. et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355, 84–88 (2017).
Rudin, C. M. et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 44, 1111–1116 (2012).
Tanaka, H. et al. Lineage-specific dependency of lung adenocarcinomas on the lung development regulator TTF-1. Cancer Res. 67, 6007–6011 (2007).
Jing, X., Wang, T., Huang, S., Glorioso, J. C. & Albers, K. M. The transcription factor Sox11 promotes nerve regeneration through activation of the regeneration-associated gene Sprr1a. Exp. Neurol. 233, 221–232 (2012).
Hamon, A., Roger, J. E., Yang, X.-J. & Perron, M. Müller glial cell-dependent regeneration of the neural retina: an overview across vertebrate model systems. Dev. Dyn. 245, 727–738 (2016).
Decaesteker, B. et al. SOX11 regulates SWI/SNF complex components as member of the adrenergic neuroblastoma core regulatory circuitry. Nat. Commun. 14, 1267 (2023).
Angelozzi, M., da Silva, R. P., Gonzalez, M. V. & Lefebvre, V. Single-cell atlas of craniogenesis uncovers SOXC-dependent, highly proliferative, and myofibroblast-like osteodermal progenitors. Cell Rep. 40, 111045 (2022).
Storz, P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 14, 296–304 (2017).
Dassaye, R., Naidoo, S. & Cerf, M. E. Transcription factor regulation of pancreatic organogenesis, differentiation and maturation. Islets 8, 13–34 (2015).
Patel, S. A. et al. The renal lineage factor PAX8 controls oncogenic signalling in kidney cancer. Nature 606, 999–1006 (2022).
Imgrund, M. et al. Re-expression of the developmental gene Pax-2 during experimental acute tubular necrosis in mice. Kidney Int. 56, 1423–1431 (1999).
Kimura, S. Thyroid-specific transcription factors and their roles in thyroid cancer. J. Thyroid Res. 2011, e710213 (2011).
Vu-Phan, D. et al. The thyroid cancer PAX8–PPARG fusion protein activates Wnt/TCF-responsive cells that have a transformed phenotype. Endocr. Relat. Cancer 20, 725–739 (2013).
Shakhova, O. et al. Sox10 promotes the formation and maintenance of giant congenital naevi and melanoma. Nat. Cell Biol. 14, 882–890 (2012).
Garraway, L. A. et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436, 117–122 (2005).
Miao, Q. et al. SOX11 and SOX4 drive the reactivation of an embryonic gene program during murine wound repair. Nat. Commun. 10, 4042 (2019).
Limaye, P. B. et al. Expression of specific hepatocyte and cholangiocyte transcription factors in human liver disease and embryonic development. Lab. Invest. 88, 865–872 (2008).
Sun, R. et al. SOX4 contributes to the progression of cervical cancer and the resistance to the chemotherapeutic drug through ABCG2. Cell Death Dis. 6, e1990 (2015).
Mills, J. C., Stanger, B. Z. & Sander, M. Nomenclature for cellular plasticity: are the terms as plastic as the cells themselves? EMBO J. 38, e103148 (2019).
Acknowledgements
We thank E. Brunner and T. Dalessi for discussions as well as other members of the Basler laboratory including M. D. Brügger, G. Hausmann, T. Valenta, D. King, G. Moro, A. F. Guthörl and L. F. Rago for critical input on the manuscript. Considering the broad topic of this Review and limited space, we were not able to include all essential studies and therefore apologize to authors whose work we did not cite. H.F. was supported by the Forschungskredit of the University of Zürich (grant FK-21-119) and a grant from the University and Medical Faculty of Zürich and the Comprehensive Cancer Center Zürich. K.B. is supported by Swiss National Science Foundation (grants 192475 and 207594) project funding in biology and medicine.
Author information
Authors and Affiliations
Contributions
H.F. conceived and wrote the paper. K.B. provided support and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Joan Massague, Joseph Taube and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fazilaty, H., Basler, K. Reactivation of embryonic genetic programs in tissue regeneration and disease. Nat Genet 55, 1792–1806 (2023). https://doi.org/10.1038/s41588-023-01526-4
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41588-023-01526-4
This article is cited by
-
Epigenomic profiling of immune cell subtypes reveals H3K27ac-marked stress signatures after long-duration spaceflight
Scientific Reports (2025)
-
Circulating cell-free DNA methylation patterns indicate cellular sources of allograft injury after liver transplant
Nature Communications (2025)
-
A patient-derived organoid model captures fetal-like plasticity in colorectal cancer
Cell Research (2025)
-
Molecular mechanisms altering cell identity in cancer
Oncogene (2025)
-
CUT&Tag applied to zebrafish adult tail fins reveals a return of embryonic H3K4me3 patterns during regeneration
Epigenetics & Chromatin (2024)
