
Abstract
The major spliceosome includes five small nuclear RNA (snRNAs), U1, U2, U4, U5 and U6, each of which is encoded by multiple genes. We recently showed that mutations in RNU4-2, the gene that encodes the U4-2 snRNA, cause one of the most prevalent monogenic neurodevelopmental disorders. Here, we report that recurrent germline mutations in RNU2-2 (previously known as pseudogene RNU2-2P), a 191-bp gene that encodes the U2-2 snRNA, are responsible for a related disorder. By genetic association, we identified recurrent de novo single-nucleotide mutations at nucleotide positions 4 and 35 of RNU2-2 in nine cases. We replicated this finding in 16 additional cases, bringing the total to 25. We estimate that RNU2-2 syndrome has a prevalence of ~20% that of RNU4-2 syndrome. The disorder is characterized by intellectual disability, autistic behavior, microcephaly, hypotonia, epilepsy and hyperventilation. All cases display a severe and complex seizure phenotype. We found that U2-2 and canonical U2-1 were similarly expressed in blood. Despite mutant U2-2 being expressed in patient blood samples, we found no evidence of missplicing. Our findings cement the role of major spliceosomal snRNAs in the etiologies of neurodevelopmental disorders.
Similar content being viewed by others

Main
More than 4,000 genes have been established as etiological for a rare disease, of which only 69 are noncoding1. Three of these noncoding genes—RNU4ATAC, RNU12 and RNU4-2—encode snRNAs that have crucial roles in pre-messenger RNA (mRNA) splicing. Variants in RNU4ATAC are responsible for microcephalic osteodysplastic primordial dwarfism type I (refs. 2,3), Roifman syndrome4 and Lowry–Wood syndrome5, whereas variants in RNU12 cause early-onset cerebellar ataxia6 and CDAGS syndrome7. These pathologies are inherited in an autosomal-recessive manner. Both RNU4ATAC and RNU12 encode components of the minor spliceosome, a molecular complex that catalyzes splicing for fewer than 1% of all introns in humans8. However, more than 99% of introns are spliced by the major spliceosome. Recently, we reported that de novo mutations in RNU4-2, which is transcribed into the U4-2 snRNA component of the major spliceosome, cause one of the most prevalent monogenic neurodevelopmental disorders (NDDs)9. The discovery was published independently by a separate group10.
To explore whether other noncoding genes might also be causal for NDDs, we performed a refined statistical analysis of the 100,000 Genomes Project (100KGP) data in the National Genomic Research Library (NGRL)11. Following a previously described approach9,12, we used the BeviMed genetic association method13 to compare rare variant genotypes in the 41,132 canonical transcript entries in Ensembl v.104 with a biotype other than ‘protein_coding’ (Supplementary Data), which included 14,307 entries annotated as pseudogene transcripts, between 7,452 unrelated, unexplained cases annotated with the ‘Neurodevelopmental abnormality’ (NDA) Human Phenotype Ontology (HPO) term and 43,727 unrelated participants without the NDA term. Notably, whereas our previous analyses filtered out single-nucleotide variants with combined annotation-dependent depletion (CADD)14 score < 10, our present analysis removed this threshold to expand the variant search space.
Our analysis yielded only two genes with a posterior probability of association (PPA) with NDA > 0.5. RNU4-2, which we have reported previously9, had a PPA of ~1, and RNU2-2P (now called RNU2-2) had a PPA of 0.97. The association with RNU2-2 depended on inclusion of variants with CADD scores ≤ 10 (Extended Data Fig. 1). Conditional on the association, two variants, at nucleotide positions 4 and 35, had a BeviMed posterior probability of pathogenicity (PPP) > 0.5 (Fig. 1a). The nine NDA cases with either of the variants had a significantly greater phenotypic homogeneity based on HPO terms than expected under random selection of nine NDA cases from unexplained and unrelated NDA study participants in the 100KGP (P = 1.33 × 10−3, Fig. 1b), supporting causality for a distinct NDD. RNU2-2 has a 191-bp sequence that is identical to that of the canonical gene RNU2-1, except for eight single-nucleotide substitutions (all within n.108–191). Unlike RNU2-1, which has a variable copy number within a region on chromosome 17, RNU2-2 has a unique sequence occurring in only one location on chromosome 11. Although at the time of analysis, RNU2-2 was known as RNU2-2P and annotated as one of many U2 pseudogenes in bioinformatics databases15, it has recently been shown to be expressed in cell lines, and its transcripts, U2-2P (now U2-2), have been shown to have the greatest abundance and stability of all noncanonical U2 snRNAs16. After aggregation over the 11 copies of RNU2-1 in the GRCh38 build of the reference genome, RNU2-1 and RNU2-2 show comparable levels of expression in whole blood and in blood cells (Fig. 1c). RNU2-2 resides in a 5′ untranslated exon of WDR74 that had previously been identified as being enriched for hotspot mutations in cancer, although the existence of RNU2-2 at that locus was not known at the time17. A recent study showed that both RNU2-1 and RNU2-2 carry recurrent somatic mutations (n.28C>T) that drive B cell-derived tumors, prostate cancers and pancreatic cancers18. The same study showed that RNU2-2 is a functional gene that is transcribed independently of WDR74—a finding that we recapitulated in blood and blood cells (Extended Data Fig. 2)—and that both the canonical U2-1 and noncanonical U2-2 snRNAs are incorporated into the spliceosome18.

a, BeviMed PPAs between each of RNU4-2 and RNU2-2 (previously known as RNU2-2P) and NDA. All other noncoding genes and pseudogenes had PPA < 0.5. Only two RNU2-2 variants had conditional PPP > 0.5: n.4G>A and n.35A>G. Prob., probability. b, Distribution of phenotypic homogeneity scores for 100,000 randomly selected sets of nine participants chosen from 9,112 unrelated NDA-coded participants. The score corresponding to the nine identified cases with one of the two RNU2-2 variants with PPP > 0.5 is indicated with a red line. c, Scatter plot of log10 expression of RNU2-1 against that of RNU2-2 in whole-blood samples from a random subset of 500 participants in the NGRL and in four blood cell types from 204 NBR participants. TPM, transcripts per million. d, Top, numbers of participants with a rare allele at each of the 191 bases of RNU2-2, stratified by affection status and inheritance information of the carried allele. The two variants with PPP > 0.5 are indicated with green arrows. The color-coded track shows the aggregated (over distinct alleles at a position) minor allele count (aMAC) in gnomAD v.4.1.0 (gn.) at each position, and the black bars show the numbers of distinct alternate alleles in gnomAD at each position (multiple insertions and multiple deletions at a given position each count as one). Variants failing quality control (QC) in gnomAD are not shown in this subpanel. Bottom, data corresponding to nucleotide positions 1 to 41 in greater detail, including gnomAD-QC-failing variant n.35A>T. Above and below the RNU2-2 cDNA sequence (Seq.), the alternate alleles in 100KGP participants and the distinct alleles in gnomAD are shown, respectively; ‘+’ indicates insertions, and the variant that failed QC in gnomAD is indicated. e, Pedigrees for participants with a rare alternate allele n.4 or n.35 in RNU2-2. Pedigrees used for discovery have a ‘G’ prefix and are labeled in black. Pedigrees used for replication in the IMPaCT-GENóMICA, URDCat and ENoD-CIBERER aggregate collection; the 100KGP; the NBR; Erasmus MC UMC; the GMS; Radboud UMC; deCODE or the ZOEMBA study have an ‘I’, ‘M’, ‘N’, ‘R’, ‘S’, ‘W’, ‘Y’ or ‘Z’ prefix, respectively, and are labeled in blue. Hom., homozygous; ref., reference.
The two germline variants with a high PPP, n.4G>A and n.35A>G, are located in a genomic locus spanning a region of approximately 40 nucleotides at the 5′ end of the 191-bp RNU2-2 gene. The locus has a markedly reduced density of population genetic variation in gnomAD19, consistent with the effects of negative selection (Fig. 1d). Published secondary structure data of the U2 snRNA show that r.4 is located within the helix II U2–U6 interaction domain, whereas r.35 is part of the highly conserved recognition domain GUAGUA that binds the branch sites of introns20,21,22 (Extended Data Fig. 3). Trio sequencing of four of the five cases with n.4G>A and three of the four cases with n.35A>G showed that the variants were de novo in each case. A variant with a different alternate allele at nucleotide 35, n.35A>T, was called in eight unaffected participants; it was also present in gnomAD but failed quality control (QC) (Fig. 1d). Analysis of whole-genome sequencing (WGS) and Sanger sequencing data suggested that n.35A>G is a germline variant, but n.35A>T is a recurring somatic mosaic variant. This somatic variant is observed only in individuals over the age of 40 years, consistent with clonal hematopoiesis (Extended Data Fig. 4).
To replicate our findings in the nine NDD cases, we examined eight additional rare disease collections: a component of the 100KGP not included in the discovery dataset (10,373 participants, of whom 1,736 have an NDA); the NIHR BioResource-Rare Diseases (NBR) data23 (7,388 participants, of whom 731 have an NDA); the UK Genomic Medicine Service (GMS) data (32,030 participants, of whom 6,469 have an NDA); data from the Erasmus MC UMC (1,527 participants, of whom approximately 400 have an NDA); an aggregate of the IMPaCT-GENóMICA, URDCat and ENoD-CIBERER programs for undiagnosed rare diseases24 (1,707 probands with NDDs and WGS data); clinical data from Radboud UMC Nijmegen (1,037 probands with an NDA); WGS data from deCODE genetics (73,821 participants, of whom 4,416 have an NDA) and data from the ZOEMBA study (127 participants, of whom 71 have an NDA). We identified a further 16 cases in these replication collections (Fig. 1e), all but two of whom were confirmed to have a de novo variant. There were no unaffected carriers of either variant. Eight replication cases had n.4G>A, seven replication cases had n.35A>G, and one replication case had a different alternate allele at nucleotide 35, n.35A>C. Although this case represented the only individual harboring n.35A>C, modeling of the interactions between U2-2 snRNA and canonical branch site sequences suggested that n.35A>C has a destabilizing effect on binding that is greater than that of the n.35A>G variant and in many cases similar in magnitude to that of the n.4G>A variant with respect to its cognate partner U6 (Extended Data Fig. 5). All these variants were called confidently by WGS (Extended Data Fig. 6). In the 100KGP, RNU2-2 was a more prevalent etiological gene than all but 29 of the ~1,400 known etiological genes for intellectual disability, explaining about one-fifth the number of cases as RNU4-2, the etiological gene for RNU4-2 syndrome, also known as ReNU syndrome (Fig. 2). This relative prevalence was consistent with observations in the IMPaCT-GENóMICA, URDCat and ENoD-CIBERER aggregate collection, which identified 27 cases with RNU4-2 syndrome and six cases (that is, 4.5 times fewer) with RNU2-2 syndrome.

Of the 9,112 unrelated NDA-coded cases in the 100KGP, the numbers solved through pathogenic or likely pathogenic variants in a gene are shown, provided at least nine cases were diagnosed. For RNU2-2, the number of NDA-coded cases in the 100KGP with one of the recurring de novo variants is shown.
Analysis of HPO terms for the nine uniformly phenotyped 100KGP cases revealed that 100% were assigned ‘Intellectual disability’ and ‘Global developmental delay’, 89% were assigned ‘Delayed speech and language development’, 78% were assigned ‘Motor delay’ and 56% were assigned ‘Autistic behavior’, in line with frequencies among NDA cases generally (Fig. 3). However, certain terms were enriched in RNU2-2 cases: ‘Seizure’ was annotated in 89% of RNU2-2 cases (versus 27% in other NDA cases, Bonferroni-adjusted (BA) P = 2.44 × 10−3) but later confirmed to be present in 100%, ‘Microcephaly’ in 78% of cases (versus 18%, BA P = 1.62 × 10−3), ‘Generalized hypotonia’ in 56% of cases (versus 13%, BA P = 3.56 × 10−2), ‘Severe global developmental delay’ in 44% (versus 2.7%, BA P = 8.89 × 10−4) and ‘Hyperventilation’ in 33% of cases (versus 0.16%, BA P = 7.56 × 10−6). No HPO terms were significantly underrepresented in the RNU2-2 cases. Of the terms that were enriched among cases of RNU4-2 syndrome, ‘Seizure’, ‘Microcephaly’ and ‘Generalized hypotonia’ were also enriched in RNU2-2 cases. However, ‘Severe global developmental delay’ and ‘Hyperventilation’ were only enriched in RNU2-2 cases, suggesting that these may be differentiating phenotypic features. Strikingly, three RNU2-2 cases were coded with the seldom-used ‘Hyperventilation’ term by three independent clinicians.

Graph showing the ‘is-a’ relationships among HPO terms present in at least three of the nine NDA-coded RNU2-2 cases in the discovery collection or significantly enriched among them relative to 9,112 unrelated NDA-coded participants of the 100KGP. The significantly overrepresented terms are highlighted. For each term, the number of cases with the term and the proportion that number represents out of nine is shown. For each overrepresented term, the proportion of NDA-coded participants with the term and the proportion of NDA-coded RNU2-2 cases with the term are represented as the horizontal coordinate of the base and the head of an arrow, respectively. *, Only eight of the nine (89%) of the cases had the ‘Seizure’ HPO term in the NGRL, but epilepsy was confirmed in the case without the HPO term by inspecting the individual’s electronic health record and the numbers attached to ‘Seizure’ were updated accordingly.
Detailed clinical vignettes for the 15 cases in pedigrees G1–2, G4, I1–6, M2, R1, S3, W1, Y1 and Z1 are provided in Supplementary Note and Supplementary Table 1. These indicate that the neurodevelopmental phenotype caused by the RNU2-2 variants typically manifests from 3 to 6 months of age but is progressive, frequently severe and accompanied by characteristic dysmorphic features (Fig. 4). All the cases displayed prominent epilepsy, usually from the first few months of life, and seizures were severe and pharmacoresistant. Seizures were characteristically complex and included spasms, tonic, tonic clonic, myoclonic and absence types, classified in some probands as Lennox–Gastaut syndrome. These features distinguish the RNU2-2 cases from previously reported cases of RNU4-2 syndrome, in which the developmental phenotype was reported as less severe, some of the dysmorphic features were different, and epilepsy was typically later in onset, less severe and more commonly focal9,10,25. Extraordinarily, case M2 also harbored a de novo truncating variant in SPEN predicted to cause Radio–Tartaglia syndrome26. However, the individual in this case had short stature (<−2.65 s.d.) and microcephaly (<−2.65 s.d.), which are not characteristic of Radio–Tartaglia syndrome, as well as having a craniofacial morphology that more closely resembled that of other RNU2-2 patients than Radio–Tartaglia syndrome patients (Supplementary Note). This atypical presentation is consistent with a dual rare genetic diagnosis.

Clinical photographs of individuals from pedigrees G1, G4, S3, R1 and I1–6. The individuals in these cases show common features of long palpebral fissures with slight eversion of the lateral lower lids, long eyelashes, broad nasal root, large low set ears, wide mouth and wide spaced teeth. The approximate ages of the individuals when the photographs were taken are shown. Photographs of individual M2, who has Radio–Tartaglia syndrome in addition to RNU2-2 syndrome, are included in the Supplementary Note. We have obtained specific consent from the families to publish these clinical photographs. m, months; yr, years.
Using trio WGS data, which were available for 17 families, we were able to determine the parental origin of the de novo mutations for ten of those families. Echoing observations in cases with RNU4-2 syndrome, the pathogenic RNU2-2 mutations were ubiquitously of maternal origin, suggesting that they may affect spermatogenesis. Analysis of uniquely aligned reads at heterozygous sites in whole-blood RNA sequencing (RNA-seq) data revealed that both alleles of RNU2-2 were expressed robustly in cases (Extended Data Fig. 7). However, a genome-wide comparison of the RNA-seq alignments between five cases and 495 unrelated unexplained NDA-coded participants did not reveal differential gene expression, differential splice junction usage or any pattern of aberrant splicing in the cases (Extended Data Fig. 8), suggesting that transcriptomic analysis of other tissue types will be required to uncover the underlying molecular mediators of disease.
U2 is involved in all stages of pre-mRNA splicing and contains distinct domains that interact with the catalytic U6, intronic branch sites and scaffolding of several protein assemblies27. Notably, the U6 binding domain and the branch site recognition domain of U2-2 are transcribed from a region in RNU2-2 exhibiting markedly reduced population genetic variation (Fig. 1d). Studies in the 1990s of yeast U2 snRNA showed that variants in branch site recognition sequence GUAGUA inhibit splicing and even generate a dominant lethal phenotype when the recognition sequence is changed entirely28,29. Position r.35 in the human U2 sequence corresponds to r.36 in the yeast U2 sequence, where n.36A>G and n.36A>T result in 0–10% and 10–20% splicing activity, respectively, compared with the wild-type sequence29. Although the U2–U6 recognition sequences are not conserved between yeast and human, a similar organization is retained. The U2–U6 interaction in yeast is not very sensitive to variation in U2 snRNA29, but genetic suppression experiments that changed multiple residues within U2 or U6 snRNAs, including position r.4 in U2 snRNA, have demonstrated that the U2–U6 helix II plays a part in the regulation of splicing in mammalian cells30,31. Mice with variants in a direct ortholog of RNU2-2 do not exist; however, mice with a homozygous 5-bp deletion in U2 ortholog Rnu2-8 present with ataxia and neurodegeneration32. Transcriptomic analysis of the mutant cerebellum detected aberrant splicing, particularly increased retention of short introns. Although it remains unclear how this splicing defect might cause neuronal death, it has been hypothesized that premature translation termination codons within the retained introns could trigger the nonsense-mediated decay (NMD) pathway. We and others have shown that the recessive human disorders caused by variants in RNU4ATAC and RNU12 result in minor intron retention in blood cells and fibroblasts2,4,6,33,34. By contrast, we have been unable to detect any significant and reproducible large-scale splicing defect in the blood cells of patients with dominant germline variants in the major spliceosome gene RNU2-2. Although a recent study described systematic disruption of 5′ splice site usage in the whole blood of some patients with de novo RNU4-2 variants10, RNA-seq of fibroblasts in a separate case study could not detect any defect in splicing25. Moreover, transcriptomic analysis of primary hematological tumors and cell lines transfected with vectors expressing the n.28C>T RNU2-2 mutation did not reveal any significant differences in splicing18. Therefore, further studies are required to understand how RNU4-2 and RNU2-2 mutations affect splicing. It might be that, in contrast to recessive splicing disorders, it is challenging to detect widespread splicing defects in these newly discovered dominant disorders because wild-type transcripts are expressed in combination with misspliced transcripts from the same gene that are subjected to NMD. In certain cell types, the effects of NMD might be overcome such that the overall expression levels of mRNAs remain unchanged, owing to rapid mRNA turnover and dosage compensation35. However, certain cell types, such as stem cells, which we have not yet been able to study, might be more sensitive to high NMD dosage than terminally differentiated cells. Neuronal stem cells and mouse models of RNU4-2 and RNU2-2 pathologies may be needed to resolve these mechanistic questions.
Methods
Ethics
Participants in the 100KGP, the 100KGP Pilot Project and the GMS were enrolled to the NGRL under a protocol approved by the East of England–Cambridge Central Research Ethics Committee (ref: 20/EE/0035). We obtained written informed consent to publish additional clinical data from a subset of the affected cases in the NGRL following local best practices. NBR participants were enrolled under a protocol approved by the East of England–Cambridge South Research Ethics Committee (ref. 13/EE/0325). The investigations at Erasmus MC UMC were approved by the center’s institutional review board (MEC-2012-387). Informed consent at that institution was obtained for all diagnostics, and written informed consent was obtained from the parents of participants for publication of medical data including photographs, in line with the Declaration of Helsinki. Participants in the IMPaCT-GENóMICA, URDCat and ENoD-CIBERER programs were enrolled through clinical services under a protocol approved by the Instituto de Salud Carlos III Research Ethics Committee (CEI-PI01_2022) and endorsed by the institutional review boards of the participating hospitals. The ZOEMBA study was approved by the institutional review board of Amsterdam UMC (registration number NL67721.018.19). Written informed consent to publish clinical data and photographs of the affected individuals were obtained following local best practices.
Enrollment
The enrollment criteria for participants in the NGRL are available from the Genomics England website36. The available enrollment criteria for replication cohorts are given in refs. 23,24.
Genetic association analysis
The genetic association analysis was conducted as described previously9,12, except that variants were not thresholded on CADD score. Cases comprised all the 9,112 unrelated cases in the 100KGP included in the merged variant call format file provided by the 100KGP that were annotated with the NDA HPO term, whereas the controls comprised all the 40,937 unrelated participants in the merged variant call format file who were not assigned the NDA HPO term. Of the 9,112 cases, 7,452 had been previously solved through pathogenic or likely pathogenic variants. Cases explained by variants in a given gene were reassigned to the control group in the genetic association analyses for genes other than that gene.
Phenotypic homogeneity analysis
To assess the phenotypic homogeneity of the nine participants in the discovery collection with n.4G>A or n.35A>G in RNU2-2, we computed a phenotype homogeneity score for that group with respect to unexplained and unrelated NDA study participants. We calculated this score using the get_sim_grid and get_sim_p functions from the ontologySimilarity R package37, as previously described9. We then obtained a Monte Carlo P value as the proportion of random sets of nine unexplained unrelated NDA cases with a homogeneity score greater than or equal to the homogeneity score of the group carrying either of the RNU2-2 variants.
Analysis of HPO terms
To identify enriched or depleted HPO terms among the nine NDA-annotated cases with n.4G>A or n.35A>G in RNU2-2 in the discovery collection, compared with unrelated NDA-coded participants without either of these two variants, we computed P values of association using Fisher’s two-sided exact test. We only tested enrichment for terms that were attached to at least three of the nine cases and belonged to the set of nonredundant terms at each level of frequency among the cases. To account for multiple comparisons, we adjusted the P values by multiplying them by the number of tests. An adjusted P < 0.05 was deemed to indicate statistical significance. To visualize both common and distinctive HPO terms for RNU2-2 cases, we selected terms that were either statistically significant or present in at least 50% of the cases, removed redundant terms at each level of frequency among the nine cases, and arranged the terms along with a nonredundant set of ancestral terms as a directed acyclic graph of ‘is-a’ relations. These analyses were conducted using the ontologyX R packages37.
Analysis of expression levels of U2-1 and U2-2
The NBR Molecular Phenotyping Study is a multicenter multiomics study of approximately 1,000 patients. It consists of RNA-seq and proteomics data for platelets, neutrophils, monocytes and CD4+ T cells. Approximately 5,000 study participants in the NGRL also underwent whole-blood RNA-seq. We aligned the NBR blood cell RNA-seq data to the GRCh38 reference genome using STAR to assess coverage in the RNU2-2 locus. We did the same for NGRL participants using RNA-seq reads aligned by DRAGEN to the GRCh38 reference genome. Both the NBR and the NGRL data were generated following a ribosomal RNA depletion and fragment size selection protocol that enables sequencing of short RNAs. To quantify expression of U2-1 and U2-2 in the NBR and the NGRL participants, we used the kallisto v.0.51.1 pseudoaligner to map reads against a GRCh38 reference transcriptome composed of all transcript sequences in Ensembl v.104 after removing duplicate sequences using the rmdup function from seqkit v.2.9.0. As only one of the 11 copies of the RNU2-1 sequence was included in the reference transcriptome, this approach ensured that quantification of U2-1 expression was not diluted over repeated entries of the RNU2-1 sequence.
Mosaicism analysis
To compute the proportions of WGS reads supporting alternate alleles, we extracted the sequencing depth and the number of reads supporting each alternate allele at n.4 and n.35 of RNU2-2 from BAM files using ‘samtools mpileup’ with default settings.
Sanger sequencing
We used the following primers to amplify genomic DNA containing the RNU2-2 gene before Sanger sequencing: forward primer, 5′-CCAATCCCAGGATCCTAAAAA-3′; reverse primer, 5′-GAAGACCACATGGAGATACTACG-3′. The amplified fragments corresponded to chr. 11:62841419–62842071 in version GRCh38 of the human reference genome.
Modeling free energies of association
We calculated the free energy of duplex formation ΔG38 of duplex formation with U6-1 and with branch site sequences for wild-type and mutant U2-2 using the RNA.fold_compound.eval_structure function in the ViennaRNA (v.2.6.4) Python package. This enabled us to calculate the difference in stability change on mutation, ΔΔG.
Parental origin of de novo mutations
For each proband for which trio WGS data were available, we selected read pairs overlapping the position of the de novo variant in question. For each inherited variant called in the mother but not in the father that was supported by such read pairs, we constructed a 2 × 2 contingency table indicating the number of read pairs supporting each allele across the inherited and the de novo variant. If across all of these maternally inherited variants, the number of reads supporting linkage between the reference allele for one variant and the alternate allele for the other variant was equal to zero, and if at least one read supported linkage between the de novo alternate allele and at least one maternally inherited alternate allele, then the origin was determined to be maternal. If across all of the paternally inherited variants, the number of reads supporting linkage between the two reference alleles was equal to zero and the number of reads supporting linkage between the two alternate alleles was equal to zero, and at least one read supported linkage between the reference allele at the de novo variant position and at least one paternally inherited alternate allele, then the origin was determined to be maternal. The same logic was applied to determine a paternal origin. If none of the above conditions was met, the origin was determined to be inconclusive.
Gene expression and splicing analysis
We performed QC on RNA-seq data derived from the whole blood of 5,546 participants in the NGRL as follows. Based on visual inspection of QC parameter distributions, we filtered out samples with a percentage of RNA fragments larger than 200 bases (as measured using an Agilent TapeStation 4200) of ≤65%, a total read count outside the range (108M, 592M), a genome mapping rate <0.85 or a high-quality read rate <0.9 (where reads were deemed to be of high quality if they aligned as proper pairs, had fewer than seven mismatches and had a mapping quality ≥60). After QC filtering, 5,165 samples remained for analysis, including five cases with implicated variants in RNU2-2. We assessed allele-specific expression in cases by counting genome-aligned RNA-seq reads overlapping heterozygous sites using ‘samtools mpileup’ with default settings. We selected 500 samples for differential gene expression and splice junction usage analysis by taking samples from the five cases and 495 samples selected at random from those passing the QC criteria and belonging to unrelated NDA-coded individuals presently unexplained. We used DESeq2 (ref. 39) to conduct differential gene expression analysis, taking the transcript quantifications generated by the Salmon software40 and aggregated by gene with the tximport BioConductor package41. For the differential splicing analysis, we used the 905,036 junctions observed (that is, supported by at least one spliced read) in at least five of the 500 samples. We obtained one-sided P values by permutation of case labels within the 500 NGRL samples for the lowness of the sum of ranks of normalized numbers of reads supporting groups of splice junctions ranked from high to low and low to high, assigning the maximum rank in the event of ties. We grouped the splice junctions by dinucleotide pairs at the splice sites, quantile of GC content in the region encompassed by the splice junction and quantile of splice junction length. The numbers of reads for each sample were normalized by dividing by the total number of uniquely aligned reads supporting splice junctions genome-wide. To identify differentially spliced individual junctions, we also computed the mean ranks from low to high (assigning the average rank in the event of ties) of normalized splice junction usage across the five cases among the 500 samples for all the 905,036 selected junctions. The mean rank for the splice junction with the lowest mean rank (among the 87,067 splice junctions observed in at least 495 of the 500 samples) and highest mean rank (among all 905,036 splice junctions) was recorded. These values were then compared with equivalents for 500 randomly selected sets of five samples from among all 500 samples to assess whether there was at least one splice junction with extreme usage among the five RNU2-2 cases.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Genetic and phenotypic data for participants in the 100KGP study, 100KGP Pilot study and the GMS are available through the Genomics England Research Environment via the application at https://www.genomicsengland.co.uk/join-a-gecip-domain. WGS data from the NGRL were obtained for 78,132 100KGP participants, 4,054 100KGP Pilot participants and 32,030 GMS participants (v.3). RNA-seq data from the NGRL and corresponding quality control metrics were obtained for 5,546 participants of the 100KGP from the ‘transcriptome_file_paths_and_types’ and ‘rnaseq_qc_metrics’ tables (Main Programme v.18). Access to blood cell RNA-seq data generated by the NIHR BioResource can be requested by contacting the NIHR BioResource Data Access Committee at dac@bioresource.nihr.ac.uk. HPO phenotype data in the NGRL were obtained from the ‘rare_diseases_participant_phenotype’ table (Main Programme v.14), ‘observation’ table (GMS v.3) and ‘hpo’ table (Rare Diseases Pilot v.3); specific disease class data from the ‘rare_diseases_participant_disease’ table (Main Programme v.13); ICD-10 codes from the ‘hes_apc’ table (Main Programme v.13); pedigree information from the ‘rare_diseases_pedigree_member’ table (Main Programme v.13), ‘referral_participant’ table (GMS v.3), and ‘pedigree’ table (Rare Diseases Pilot v.3); and explained and/or unexplained status of cases from the ‘gmc_exit_questionnaire’ tables (Main Programme v.18, GMS v.3). Ensembl v.104 (http://may2021.archive.ensembl.org/index.html), gnomAD v.3.0 (https://gnomad.broadinstitute.org/) and CADD v.1.6 (https://cadd.gs.washington.edu/) were used for transcript selection and variant annotation against reference genome GRCh38. A more recent version of gnomAD, v.4.1.0, was used to assign the variant allele frequencies in RNU2-2 shown in Fig. 1. Data presented in this paper were requested from the Genomics England Airlock on 13 August 2024 at 03:39 BST. The manuscript was submitted to the Genomics England Publication Committee on 21 August 2024 at 23:51 BST and approved for submission on 27 August 2024 at 15:52 BST.
Code availability
Software packages rsvr v.1.0, bcftools v.1.16, samtools v.1.9/1.16.1 and Perl v.5 were used to build the 100KGP Rareservoir. The Rareservoir software is available from https://github.com/turrogroup/rsvr. R v.3.6.2 and v.4.3.3 and all R packages that were used for data analysis and visualization (Matrix v.1.2-18, dplyr v.0.8.5, bit64 v.0.9-7, bit v.1.1-14, DBI v.1.1.0, RSQLite v.2.1.4, BeviMed v.5.7, ontologyIndex v.2.12, ontologySimilarity v.2.7, ontologyPlot v.1.7, ggplot2 v.3.5.0, tximport v.1.32.0 and DESeq2 v.1.44) are available via the Comprehensive R Archive Network site (https://cran.r-project.org/) or Bioconductor (https://bioconductor.org). The ViennaRNA v.2.6.4, salmon v.1.10.0, seqkit v.2.9.0 and kallisto v0.51.1 packages can be installed via the conda package manager, available from https://anaconda.org/anaconda/conda.
References
Martin, A. R. et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat. Genet. 51, 1560–1565 (2019).
He, H. et al. Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science 332, 238–240 (2011).
Edery, P. et al. Association of TALS developmental disorder with defect in minor splicing component U4atac snRNA. Science 332, 240–243 (2011).
Merico, D. et al. Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman syndrome by disrupting minor intron splicing. Nat. Commun. 6, 8718 (2015).
Farach, L. S. et al. The expanding phenotype of RNU4ATAC pathogenic variants to Lowry Wood syndrome. Am. J. Med. Genet. A 176, 465–469 (2018).
Elsaid, M. F. et al. Mutation in noncoding RNA RNU12 causes early onset cerebellar ataxia. Ann. Neurol. 81, 68–78 (2017).
Xing, C. et al. Biallelic variants in RNU12 cause CDAGS syndrome. Hum. Mutat. 42, 1042–1052 (2021).
Moyer, D. C. et al. Comprehensive database and evolutionary dynamics of U12-type introns. Nucleic Acids Res. 48, 7066–7078 (2020).
Greene, D. et al. Mutations in the U4 snRNA gene RNU4-2 cause one of the most prevalent monogenic neurodevelopmental disorders. Nat. Med. 30, 2165–2169 (2024).
Chen, Y. et al. De novo variants in the RNU4-2 snRNA cause a frequent neurodevelopmental syndrome. Nature 632, 832–840 (2024).
Caulfield, M. et al. National Genomics Research Library. figshare https://doi.org/10.6084/m9.figshare.4530893 (2020).
Greene, D. et al. Genetic association analysis of 77,539 genomes reveals rare disease etiologies. Nat. Med. 29, 679–688 (2023).
Greene, D., Richardson, S. & Turro, E. A fast association test for identifying pathogenic variants involved in rare diseases. Am. J. Hum. Genet. 101, 104–114 (2017).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Dyer, S. C. Ensembl 2025. Nucleic Acids Res. 53, D948–D957 (2025).
Mabin, J. W., Lewis, P. W., Brow, D. A. & Dvinge, H. Human spliceosomal snRNA sequence variants generate variant spliceosomes. RNA 27, 1186–1203 (2021).
Weinhold, N. et al. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 46, 1160–1165 (2014).
Bousquets-Muñoz, P. et al. PanCancer analysis of somatic mutations in repetitive regions reveals recurrent mutations in snRNA U2. NPJ Genom. Med. 7, 19 (2022).
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625, 92–100 (2024).
Xie, J., Wang, L. & Lin, R. J. Variations of intronic branchpoint motif: identification and functional implications in splicing and disease. Commun. Biol. 6, 1142 (2023).
Nez-Lumbreras, S., Morguet, C. & Sattler, M. Dynamic interactions drive early spliceosome assembly. Curr. Opin. Struct. Biol. 88, 102907 (2024).
Taggart, A. J. et al. Large-scale analysis of branchpoint usage across species and cell lines. Genome Res. 27, 639–649 (2017).
Turro, E. et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature 583, 96–102 (2020).
Luque, J. et al. CIBERER: Spanish national network for research on rare diseases: a highly productive collaborative initiative. Clin. Genet. 101, 481–493 (2022).
Schot, R. et al. Re-analysis of whole genome sequencing ends a diagnostic odyssey: case report of an RNU4-2 related neurodevelopmental disorder. Clin. Genet. 106, 512–517 (2024).
Radio, F. C. et al. SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females. Am. J. Hum. Genet. 108, 502–516 (2021).
Van der Feltz, C. & Hoskins, A. A. Structural and functional modularity of the U2 snRNP in pre-mRNA splicing. Crit. Rev. Biochem. Mol. Biol. 54, 443–465 (2019).
Miraglia, L., Seiwert, S., Igel, A. H. & Ares, M. Limited functional equivalence of phylogenetic variation in small nuclear RNA: yeast U2 RNA with altered branchpoint complementarity inhibits splicing and produces a dominant lethal phenotype. Proc. Natl Acad. Sci. USA 88, 7061–7065 (1991).
McPheeters, D. S. & Abelson, J. Mutational analysis of the yeast U2 snRNA suggests a structural similarity to the catalytic core of group I introns. Cell 71, 819–831 (1992).
Wu, J. A. & Manley, J. L. Base pairing between U2 and U6 snRNAs is necessary for splicing of a mammalian pre-mRNA. Nature 352, 818–821 (1991).
Datta, B. & Weiner, A. M. Genetic evidence for base pairing between U2 and U6 snRNA in mammalian mRNA splicing. Nature 352, 821–824 (1991).
Jia, Y., Mu, J. C. & Ackerman, S. L. Mutation of a U2 snRNA gene causes global disruption of alternative splicing and neurodegeneration. Cell 148, 296–308 (2012).
Heremans, J. et al. Abnormal differentiation of B cells and megakaryocytes in patients with Roifman syndrome. J. Allergy Clin. Immunol. 142, 630–646 (2018).
Cologne, A. et al. New insights into minor splicing—a transcriptomic analysis of cells derived from TALS patients. RNA 25, 1130–1149 (2019).
Lindeboom, R. G. H., Supek, F. & Lehner, B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 48, 1112–1118 (2016).
Devereau, A., Scott, R. & Thomas, E. Rare Disease Eligibility Criteria: 100,000 Genomes Project (Genomics England, 2018); https://files.genomicsengland.co.uk/forms/Rare-Disease-Eligibility-Criteria.pdf
Greene, D., Richardson, S. & Turro, E. ontologyX: a suite of R packages for working with ontological data. Bioinformatics 33, 1104–1106 (2017).
Tinoco, I., Uhlenbeck, O. C. & Levine, M. D. Estimation of secondary structure in ribonucleic acids. Nature 230, 362–367 (1971).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Patro, R. et al. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521 (2015).
Choi, S., Cho, N. & Kim, K. K. The implications of alternative pre-mRNA splicing in cell signal transduction. Exp. Mol. Med. 55, 755–766 (2023).
Rhode, B. M., Hartmuth, K., Westhof, E. & Hrmann, R. Proximity of conserved U6 and U2 snRNA elements to the 5′ splice site region in activated spliceosomes. EMBO J. 25, 2475–2486 (2006).
Wilkinson, M. E., Charenton, C. & Nagai, K. RNA splicing by the spliceosome. Annu. Rev. Biochem. 89, 359–388 (2020).
Zhang, Z. et al. Cryo-EM analyses of dimerized spliceosomes provide new insights into the functions of B complex proteins. EMBO J. 43, 1065–1088 (2024).
Boesler, C. et al. A spliceosome intermediate with loosely associated tri-snRNP accumulates in the absence of Prp28 ATPase activity. Nat. Commun. 7, 11997 (2016).
Acknowledgements
This research was made possible through access to data in the NGRL, which is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The NGRL holds data provided by patients and collected by the NHS as part of their care and data collected as part of their participation in research. The NGRL is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK and the Medical Research Council have also funded research infrastructure. We thank NIHR BioResource volunteers for their participation, and gratefully acknowledge NIHR BioResource centers, NHS Trusts and staff for their contribution. We thank the National Institute for Health and Care Research, NHS Blood and Transplant, and Health Data Research UK as part of the Digital Innovation Hub Programme. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. The Barakat laboratory was supported by the Netherlands Organisation for Scientific Research (ZonMw Vidi, grant 09150172110002) and acknowledges support from EpilepsieNL and CURE Epilepsy. These funding bodies had no influence over the study design, results, data interpretation or final manuscript. We thank all participants and families involved in the programs ‘Infraestructura de Medicina de Precisión asociada a la Ciencia y la Tecnología en Medicina Genómica (IMPaCT-GENóMICA)’ and ‘Programes de Malalties Rares no Diagnosticades de Catalunya i CIBERER (URDCat/ENoD-CIBERER)’. IMPaCT-GENóMICA was supported by Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación and the European Union European Regional Development Fund (IMP/00009) (principal investigator: Á.C.). URDCat was supported by the Department of Health of Catalonia (grant SLT002/16/00174) (principal investigator: L.A.P.-J.). The ENoD-CIBERER program was funded by the Biomedical Network Research Center for Rare Diseases-CIBER-ER-ISCIII (principal investigator: L.A.P.-J.). The ZOEMBA study was funded by Metakids and the United for Metabolic Diseases consortium, who thank M. Oud for bioinformatic support. K.F. was supported by Katholieke Universiteit (KU) Leuven Special Research Fund (BOF) (C14/23/121), Research Foundation – Flanders (G072921N) and NIH award R01HL161365. K.D.W. was supported by the Belgian American Education Foundation and NIH award R01HL161365. A.D.M. was supported by NIH award R01HL161365. D.G. and E.T. were supported by NIH awards R01HL161365 and R03HD111492, and E.T. was further supported by the Lowy Foundation USA.
Author information
Authors and Affiliations
Contributions
D.G. conducted statistical and bioinformatic analyses and cowrote the paper. K.D.W. analyzed RNA-seq data, generated expression scatterplots and made the illustration showing molecular interactions. J.L. modeled free energies of association. A.K. processed NBR RNA-seq data. S.P. performed PCR and Sanger sequencing. E.H. oversaw recruitment to the NBR RNA-seq project. M.C.-S., I.V. and E.F.T. designed primers, selected cases for sequencing and provided early access to detailed phenotype data on RNU4-2 cases for comparative analysis. I.V. summarized the vignettes in a table. R.S. and F.S. coordinated WGS of Erasmus MC cases. R.P. and P.R. provided data for the family that gave consent at Radboud UMC Nijmegen. R.W., C.V.K. and M.E. recruited and provided data for the ZOEMBA study participants. B.O.J., P.S. and K. Stefansson provided data for the deCODE study participant. G.A., T.S.B., D.D., N.F., J.J., S.G.K., S.M., M.O’D., M.S. and P.V. obtained consent and provided detailed phenotype information. I.A.C., D.C.-A., A.D.R., B.F.G., D.G.-N.N., E.G.A., I.M.B., A.F.M.-M., N.V.O.C., L.R.-R.B., A.S.J. and A.I.V.P. obtained consent from and provided clinical information on individuals recruited to the IMPaCT-GENóMICA, URDCat and ENoD-CIBERER programs. E.N.M. and M.S.P. were responsible for implementing the IMPaCT-GENóMICA, URDCat and ENoD-CIBERER programs under the supervision of Á.C., P.L., B.M. and L.A.P.-J. T.S.B. and M.O’D. also provided expert clinical interpretation. K. Stirrups and N.P.M. oversaw the NBR RNA-seq study. K.F. provided biological interpretation and cowrote the paper. A.D.M. coordinated clinical contacts, provided clinical and biological interpretation, and cowrote the paper. E.T. oversaw the study and cowrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors affiliated with deCODE genetics/Amgen Inc. (B.O.J., K. Stefansson and P.S.) are employed by the company. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Effect on PPAs of relaxing the CADD score threshold.
Histograms of the posterior probability of association (PPA) between the 41,132 canonical Ensembl transcripts not annotated as being protein-coding and neurodevelopmental abnormality (NDA), with and without filtering out variants with a CADD v1.6 score <10. The CADD v1.6 scores for n.4 G > A, n.35 A > G and n.35 A > C were 7.7, 9.4 and 9.1, respectively. The more recent CADD v1.7 gives scores >10 for these variants.
Extended Data Fig. 2 RNA-seq coverage in the RNU2-2 locus.
Coverage of uniquely aligned RNA-seq reads from the whole blood of five RNU2-2 cases in the NGRL and in four blood cell types of an exemplar participant in the NBR demonstrating that RNU2-2 (previously annotated as the pseudogene RNU2-2P) is expressed abundantly in blood cells.
Extended Data Fig. 3 Location of the pathogenic variants in U2-2 snRNA within the major spliceosome.
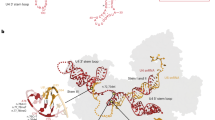
Assembly of the spliceosome A complex is initiated by binding of the intronic 5′ splice site (5′SS) to the U1 snRNA and the intronic branch site sequence to the U2-2 snRNA through Watson-Crick pairing of cognate ribonucleotides. The branch site sequence is depicted as the human YNYUNAY consensus motif (Y means C or T; N means any ribonucleotide), which interacts with the GUAGUA sequence at positions 33 to 38 in the U2-2 snRNA (depicted in red)20. The spliceosome pre-B complex is formed by incorporation of the U4/U6.U5 tri-small nuclear ribonucleoprotein (snRNP) complex that contains the U4, U5 and U6 snRNAs. This requires interactions between U5 snRNA and the 5′ and 3′ exons42 and further interactions between nucleotides near the 3′ end of the U6 snRNA and a cognate CGCUUCUCG sequence (nucleotides 3–11) close to the 5′ end of the U2-2 snRNA (depicted in blue)43. Tethering of U4/U6.U5 tri-snRNP to U2-2 within the spliceosome pre-B complex enables displacement of U1 to enable a new interaction between U6 snRNA with the 5′SS and reconfiguration of U4/U6.U5 tri-snRNP to form the catalytically active spliceosome B complex, which is a prerequisite for the splicing reaction44. The critical U6 snRNA region that interacts with the intronic 5′SS45 is maintained in correct orientation by conserved regions in the adjacent U4 snRNA (depicted in orange), which are the sites of destabilizing variants responsible for the recently described RNU4-2 syndrome9. The variants responsible for RNU2-2 syndrome occur at critical interaction sites between U2-2 snRNA near r.4 and U6 snRNA and between U2-2 snRNA near r.35 and intronic branch sites. These interactions are necessary for intron recognition and the correct assembly of the catalytically active spliceosome B complex46.
Extended Data Fig. 4 Mosaicism analysis.
a, For each of the three rare variants at positions n.4 and n.35 of RNU2-2 called in the discovery collection, truncated bar charts showing the distribution of the proportions of reads supporting the alternate allele over participants, partitioned into 0% and all left-open intervals of size 4% up to 100%. In contrast to n.4 G > A and n.35 A > G, the reads in the eight participants with the n.35 A > T heterozygous call exhibit a strong skew in favor of the reference allele. Furthermore, seven participants with a homozygous reference call at n.35 have at least 8% of aligned reads at that position supporting the ‘T’ allele, suggesting that n.35 A > T is not a germline variant, but rather a low-frequency somatic mosaic variant. b, Histogram of age at enrollment of participants in the discovery collection. The purple points show the age at enrollment of study participants with at least 8% of aligned reads supporting the ‘T’ allele at n.35. These participants are significantly older than expected by chance (P = 1.3 × 10−3, Kolmogorov-Smirnoff test). To comply with Genomics England’s rules on identifiability, all ages of at least 95 years are included in the same x = 95 bin. c, Sanger sequencing traces from an NDA case (in pedigree N1) with the n.4 G > A call, an unaffected participant with the n.35 A > T call, and a control with neither call, showing that n.4 G > A is a germline variant while n.35 A > T is a likely somatic mosaic variant.
Extended Data Fig. 5 Predicted effects of the mutants on duplex binding stability.
a, Differential binding stability (ΔΔG) values between U2-2 and U6-1 for the A4 mutant allele compared to the reference G4 allele and between U2-2 and each of 16 branch site sequences consistent with the human YUNAY motif. Each of the substitutions reduces the predicted free energies of association relative to the corresponding reference allele. b, For each of the alleles observed at r.4 of RNU2-2 (the reference G4 and the mutant A4), a graphical representation of Watson-Crick interactions between the U6-interacting region in U2-2 (encompassing UCGCU at r.2–6) and the corresponding U6-1 snRNA region. Hydrogen bonding between cognate nucleotides is depicted with dotted lines. c, For each of the germline alleles observed at r.35 (the reference A35 and the mutant G35 and C35 alleles), a graphical representation of Watson-Crick interactions between the branch site recognition region in U2-2 (GUAG at n.33–36) and an example branch site sequence (CUUAU). Hydrogen bonding between cognate nucleotides is depicted with dotted lines.
Extended Data Fig. 6 Read pileups in the replication collections.
Sequencing read pileups for cases identified in the replication collections. The reads supporting the reference allele are in blue and those supporting the variant allele are in red.
Extended Data Fig. 7 Allele-specific expression of RNU2-2 in cases.
Coverage of RNA-seq reads from whole blood aligned to the genome near RNU2-2 in five cases. The coverage levels of reads containing alternate alleles at heterozygous sites are shown in red. The locations of the mutant alleles at n.4 and n.35 are indicated with green arrows. The aligned reads overlapping heterozygous sites show that both alleles are expressed robustly in the cases in pedigrees G6, M1 and S3. The cases in pedigrees G1 and G5 were heterozygous only at n.4, where coverage was too low to assess allele-specific expression.
Extended Data Fig. 8 Aberrant splicing analysis.
a, Histogram of the number of differentially expressed genes controlling FDR at 0.05 with the Benjamini-Hochberg procedure for randomly selected sets of five from 500 RNA-seq samples (five cases with implicated variants in RNU2-2 and 495 unexplained unrelated NDD cases). The number of such genes for the five cases is shown with a red line. b, Histogram of the proportion of unique RNA-seq alignments that contain a splice junction in the 500 RNA-seq samples. The proportions corresponding to the samples from the five cases with implicated variants in RNU2-2 are shown with red bars. c, Histogram of the mean (over randomly selected sets of five samples) rank of normalized splice junction (SJ) usage of the splice junction with the lowest (left) and highest (right) mean rank. The red lines correspond to the lowest and highest mean ranks for the five RNU2-2 cases. d, One-sided P values obtained by permutation of case labels within the 500 NGRL samples for the lowness of the sum of ranks of normalized numbers of reads supporting groups of splice junctions ranked from high to low (the upward facing blue triangles) and low to high (the downward facing red triangles), assigning the maximum rank in the event of ties. The splice junctions were grouped by: dinucleotide pairs at the splice sites (for N ≥ 5), quantile of GC content in the region encompassed by the splice junction, and quantile of splice junction length. The dashed line at y = 0.05/102 indicates the P value significance threshold to control the family-wise error rate at 0.05.
Supplementary information
Supplementary Information
Supplementary Note.
Supplementary Data 1
GRCh38 coordinates for the 41,132 canonical transcript entries in Ensembl v.104 with a biotype other than ‘protein_coding’.
Supplementary Table 1
Table summarizing the contents of the 15 clinical vignettes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Greene, D., De Wispelaere, K., Lees, J. et al. Mutations in the small nuclear RNA gene RNU2-2 cause a severe neurodevelopmental disorder with prominent epilepsy. Nat Genet 57, 1367–1373 (2025). https://doi.org/10.1038/s41588-025-02159-5
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41588-025-02159-5
