
Abstract
Alfalfa (Medicago sativa L.), a globally important forage crop, is valued for its high nutritional quality and nitrogen-fixing capacity. Here, we present a high-quality pan-genome constructed from 24 diverse alfalfa accessions, encompassing a wide range of genetic backgrounds. This comprehensive analysis identified 433,765 structural variations and characterized 54,002 pan-gene families, highlighting the pivotal role of genomic diversity in alfalfa domestication and adaptation. Key structural variations associated with salt tolerance and quality traits were discovered, with functional analysis implicating genes such as MsMAP65 and MsGA3ox1. Notably, overexpression of MsGA3ox1 led to a reduced stem–leaf ratio and enhanced forage quality. The integration of genomic selection and marker-assisted breeding strategies improved genomic estimated breeding values across multiple traits, offering valuable genomic resources for advancing alfalfa breeding. These findings provide insights into the genetic basis of important agronomic traits and establish a solid foundation for future crop improvement.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

Data availability
The sequencing raw data have been deposited in the NCBI database under accession code BioProject PRJNA1197171. The haploid reference genome is derived from a previously published study5. The assembled data have been deposited in the NCBI database under the BioProject accession code PRJNA1220045. Additionally, the data are available via Zenodo at https://doi.org/10.5281/zenodo.14118213 (ref. 85) and via Figshare at https://figshare.com/articles/dataset/Alfalfa/28426967 (ref. 86). Resequencing data used in this study were obtained from Zhang’s research, and the relevant data have been provided in his published article45. The RNA sequence data from this study have been deposited in the NCBI database under accession code BioProject PRJNA1083622. The phenotypes used in GWAS and GS studies are available via Zenodo at https://doi.org/10.5281/zenodo.14869063 (ref. 87).
Code availability
All codes associated with this project are available via GitHub at https://github.com/hefei0609-afk/Alfalfa and via Zenodo at https://doi.org/10.5281/zenodo.14800545 (ref. 88).
References
Annicchiarico, P., Barrett, B., Brummer, E. C., Julier, B. & Marshall, A. H. Achievements and challenges in improving temperate perennial forage legumes. Crit. Rev. Plant Sci. 34, 327–380 (2015).
Shen, C. et al. The chromosome-level genome sequence of the autotetraploid alfalfa and resequencing of core germplasms provide genomic resources for alfalfa research. Mol. Plant 13, 1250–1261 (2020).
Li, X. & Brummer, E. C. Applied genetics and genomics in alfalfa breeding. Agronomy 2, 40–61 (2012).
Chen, H. et al. Allele-aware chromosome-level genome assembly and efficient transgene-free genome editing for the autotetraploid cultivated alfalfa. Nat. Commun. 11, 2494 (2020).
Long, R. et al. Genome assembly of alfalfa cultivar Zhongmu-4 and identification of SNPs associated with agronomic traits. Genomics Proteomics Bioinformatics 20, 14–28 (2022).
Jayakodi, M., Schreiber, M., Stein, N. & Mascher, M. Building pan-genome infrastructures for crop plants and their use in association genetics. DNA Res. 28, dsaa030 (2021).
Pang, A. W. et al. Towards a comprehensive structural variation map of an individual human genome. Genome Biol. 11, R52 (2010).
Zhang, Z. et al. Genome-wide mapping of structural variations reveals a copy number variant that determines reproductive morphology in cucumber. Plant Cell 27, 1595–1604 (2015).
Zhou, Y. et al. The population genetics of structural variants in grapevine domestication. Nat. Plants 5, 965–979 (2019).
Saxena, R. K., Edwards, D. & Varshney, R. K. Structural variations in plant genomes. Brief. Funct. Genomics 13, 296–307 (2014).
Gabur, I., Chawla, H. S., Snowdon, R. J. & Parkin, I. A. Connecting genome structural variation with complex traits in crop plants. Theor. Appl. Genet. 132, 733–750 (2019).
Chen, S. et al. Gene mining and genomics-assisted breeding empowered by the pangenome of tea plant Camellia sinensis. Nat. Plants 9, 1986–1999 (2023).
Gaut, B. S., Seymour, D. K., Liu, Q. & Zhou, Y. Demography and its effects on genomic variation in crop domestication. Nat. Plants 4, 512–520 (2018).
Wellenreuther, M., Mérot, C., Berdan, E. & Bernatchez, L. Going beyond SNPs: the role of structural genomic variants in adaptive evolution and species diversification. Mol. Ecol. 28, 1203–1209 (2019).
Huang, K. & Rieseberg, L. H. Frequency, origins, and evolutionary role of chromosomal inversions in plants. Front. Plant Sci. 11, 296 (2020).
Kirkpatrick, M. & Barton, N. Chromosome inversions, local adaptation and speciation. Genetics 173, 419–434 (2006).
Zhang, X. et al. Haplotype-resolved genome assembly provides insights into evolutionary history of the tea plant Camellia sinensis. Nat. Genet. 53, 1250–1259 (2021).
Simão, F. A., Waterhouse, R. M., Panagiotis, I., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Ou, S., Chen, J. & Jiang, N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 46, e126 (2018).
Alonge, M. et al. Automated assembly scaffolding using RagTag elevates a new tomato system for high-throughput genome editing. Genome Biol. 23, 258 (2022).
Li, A. et al. A chromosome-scale genome assembly of a diploid alfalfa, the progenitor of autotetraploid alfalfa. Hortic. Res. 7, 194 (2020).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 (2014).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Zhou, S., Chen, Q., Li, X. & Li, Y. MAP65-1 is required for the depolymerization and reorganization of cortical microtubules in the response to salt stress in Arabidopsis. Plant Sci. 264, 112–121 (2017).
Liang, M. et al. Comprehensive analyses of microtubule-associated protein MAP65 family genes in Cucurbitaceae and CsaMAP65s expression profiles in cucumber. J. Appl. Genet. 64, 393–408 (2023).
Dwiningsih, Y. & Al-Kahtani, J. Genome-wide association study of complex traits in maize detects genomic regions and genes for increasing grain yield and grain quality. Adv. Sustain. Sci. Eng. Technol. 4, 0220209 (2022).
Liu, R. et al. GWAS analysis and QTL identification of fiber quality traits and yield components in upland cotton using enriched high-density SNP markers. Front. Plant Sci. 13, 1067 (2018).
Kephart, K. D., Buxton, D. & Hill, R. Jr Digestibility and cell‐wall components of alfalfa following selection for divergent herbage lignin concentration. Crop Sci. 30, 207–212 (1990).
Han, R.-H., Lu, X.-S., Gao, G.-J. & Yang, X.-J. Analysis of the principal components and the subordinate function of alfalfa drought resistance. Acta Agrestia Sin. 14, 142 (2006).
Reinecke, D. M. et al. Gibberellin 3-oxidase gene expression patterns influence gibberellin biosynthesis, growth, and development in pea. Plant Physiol. 163, 929–945 (2013).
Wu, H., Bai, B., Lu, X. & Li, H. A gibberellin-deficient maize mutant exhibits altered plant height, stem strength and drought tolerance. Plant Cell Rep. 42, 1687–1699 (2023).
Ameur, A. Goodbye reference, hello genome graphs. Nat. Biotechnol. 37, 866–868 (2019).
Garrison, E. et al. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol. 36, 875–879 (2018).
Qin, P. et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 184, 3542–3558.e16 (2021).
Alonge, M. et al. Major impacts of widespread structural variation on gene expression and crop improvement in tomato. Cell 182, 145–161.e23 (2020).
He, Q. et al. A graph-based genome and pan-genome variation of the model plant Setaria. Nat. Genet. 55, 1232–1242 (2023).
Huang, Y. et al. Pangenome analysis provides insight into the evolution of the orange subfamily and a key gene for citric acid accumulation in citrus fruits. Nat. Genet. 55, 1964–1975 (2023).
Liu, Y. et al. Pan-genome of wild and cultivated soybeans. Cell 182, 162–176.e13 (2020).
Hu, J. et al. Potential sites of bioactive gibberellin production during reproductive growth in Arabidopsis. Plant Cell 20, 320–336 (2008).
Sun, H. et al. Gibberellins inhibit flavonoid biosynthesis and promote nitrogen metabolism in Medicago truncatula. Int. J. Mol. Sci. 22, 9291 (2021).
Dalmadi, Á. et al. Dwarf plants of diploid Medicago sativa carry a mutation in the gibberellin 3-β-hydroxylase gene. Plant Cell Rep. 27, 1271–1279 (2008).
Israelsson, M., Mellerowicz, E., Chono, M., Gullberg, J. & Moritz, T. Cloning and overproduction of gibberellin 3-oxidase in hybrid aspen trees. Effects on gibberellin homeostasis and development. Plant Physiol. 135, 221–230 (2004).
Zheng, L. et al. From model to alfalfa: gene editing to obtain semidwarf and prostrate growth habits. Crop J. 10, 932–941 (2022).
He, X. et al. Accuracy of genomic selection for alfalfa biomass yield in two full-sib populations. Front. Plant Sci. 13, 1037272 (2022).
Zhang, F. et al. Evolutionary genomics of climatic adaptation and resilience to climate change in alfalfa. Mol. Plant 17, 867–883 (2024).
Li, H. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Alexander, D. H. & Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinformatics 12, 246 (2011).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Zhang, J. et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 50, 1565–1573 (2018).
Wu, T. D. & Watanabe, C. K. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21, 1859 (2005).
Tang, H. et al. An improved genome release (Version Mt4.0) for the model legume Medicago truncatula. BMC Genomics 15, 312 (2014).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 36, 2896–2898 (2020).
Zhao, X. & Hao, W. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 4.10.11–14.10.14 (2004).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 3, lqaa108 (2021).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275 (2019).
Su, W., Gu, X. & Peterson, T. TIR-Learner, a new ensemble method for TIR transposable element annotation, provides evidence for abundant new transposable elements in the maize genome. Mol. Plant 12, 447–460 (2019).
Xiong, W. et al. HelitronScanner uncovers a large overlooked cache of Helitron transposons in many plant genomes. Proc. Natl Acad. Sci. USA 111, 10263–10268 (2014).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl Acad. Sci. USA 117, 9451–9457 (2020).
Lavigne, R., Seto, D., Mahadevan, P., Ackermann, H.-W. & Kropinski, A. M. Unifying classical and molecular taxonomic classification: analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 159, 406–414 (2008).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Wang, D., Zhang, Y., Zhang, Z., Zhu, J. & Yu, J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics 8, 77–80 (2010).
Wang, D.-P., Wan, H.-L., Zhang, S. & Yu, J. γ-MYN: a new algorithm for estimating Ka and Ks with consideration of variable substitution rates. Biol. Direct 4, 20 (2009).
Sedlazeck, F. J. et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468 (2018).
Heller, D. & Vingron, M. SVIM: structural variant identification using mapped long reads. Bioinformatics 35, 2907–2915 (2019).
Jiang, T. et al. Long-read-based human genomic structural variation detection with cuteSV. Genome Biol. 21, 189 (2020).
Jeffares, D. C. et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 8, 14061 (2017).
Marçais, G. et al. MUMmer4: a fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944 (2018).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Zadeh, L. A. Fuzzy sets as a basis for a theory of possibility. Fuzzy Sets Syst. 1, 3–28 (1978).
VanRaden, P. M. Efficient methods to compute genomic predictions. J. Dairy Sci. 91, 4414–4423 (2008).
Cortes, C. & Vapnik, V. Support-vector networks. Machine Leaning 20, 273–297 (1995).
Fu, C., Hernandez, T., Zhou, C. & Wang, Z.-Y. Alfalfa (Medicago sativa L.). Methods Mol. Biol. 1223, 213–221 (2015).
Abràmoff, M. D., Magalhães, P. J. & Ram, S. J. Image processing with ImageJ. Biophotonics Int. 11, 36–42 (2004).
He, F. Pan-genomic analysis highlights genes associated with agronomic traits and enhances genomics-assisted breeding in alfalfa. Zenodo https://doi.org/10.5281/zenodo.14118212 (2024).
He, F. Alfalfa. Figshare https://doi.org/10.6084/m9.figshare.28426967.v1 (2025).
Fei, H. Alfalfa. Zenodo https://doi.org/10.5281/zenodo.14869062 (2025).
Fei, H. Alfalfa pan-genome. Zenodo https://doi.org/10.5281/zenodo.14800544 (2025).
Acknowledgements
This work was supported by China Agriculture Research System of MOF and MARA (grant no. CARS-34 to Q.Y.), the Biological Breeding-National Science and Technology Major Project (grant no. 2022ZD04011 to R.L.), the Key Projects in Science and Technology of Inner Mongolia (grant no. 2021ZD0031 to R.L.) and Agricultural Science and Technology Innovation Program of CAAS (grant no. ASTIP-IAS14 to Q.Y.).
Author information
Authors and Affiliations
Contributions
Q.Y., R.L. and X.Z. designed this project and coordinated the research activities. F.Z., J.K., H.L., L.C., Xianyang Li, M.L., X.W., X.J., B.S., M.X. and Y.L. collected and provided plant materials. F.Z., R.L. and X.Z. participated in the genome sequencing and resequencing. S.C., S.Q. and K.C. assembled the genomes. S.C., W.K., Q.Z., K.C. and S.Q. performed the gene annotation. S.C. and F.H. analyzed RNA-seq data. F.H. constructed the sequence and gene-based pan-genome. F.Z., S.C. and F.H. contributed to population GWAS analysis. Y.Z. performed functional verification. X.H., Xiao Li and T.Z. conducted a whole-genome selection analysis. F.H., S.C., X.Z., R.L. and Q.Y. interpreted the data and contributed to the manuscript writing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Eric von Wettberg and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Population structure and Fixation Index of the global alfalfa diversity panel.
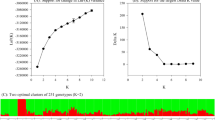
a. Population structure of the alfalfa panel was inferred by assuming three subpopulations (K). Each color represents a different subpopulation. b. Word cloud of the primary origin countries for alfalfa varieties in Group1, Group2, and Group3. Font size represents the relative proportion of varieties from each country. Group1 is predominantly from the United States, Group2 from China, and Group3 from Turkey, with contributions from other countries as well. c. The PCA scatter plot shows the distribution of PC1 and PC2, with different colors representing different groups (Group1, Group2, Group3). d. Fixation Index (FST) values among Group1, Group2, and Group3 alfalfa accessions.
Extended Data Fig. 2 The genome structure variations (SVs) between species of alfalfa.
a, Chromosome. b–h, means the distribution of repeat density, gene density, SNP/Indel density, deletions, insertion, duplication and inversion.
Extended Data Fig. 3 Genome-wide association study (GWAS) for monosaccharide content, In Vitro True Dry Matter Degradability at 24 h (IVTDMD24), and In Vitro True Dry Matter Degradability at 30 h (IVTDMD30).
a, c, e, present the Manhattan and QQ plots of the GWAS results for monosaccharide, IVTDMD24, and IVTDMD30, respectively, using structural variation (SV) markers. b, d, f, show the Manhattan and QQ plots for the same traits using single nucleotide polymorphism (SNP) markers. The red dashed line indicates the Bonferroni-corrected genome-wide significance threshold (α = 0.05/n, where 'n' is the total number of independent SNPs and effective SVs). g, i, k, depict scatter plots of the peak structural variations in chromosome 1 for the three traits, with the horizontal line marking the Bonferroni-corrected genome-wide significance threshold. h, j, l, display boxplots of the three traits across different accessions, categorized by the alleles they carry. The sample sizes for the REF and ALT groups are 171 and 5, respectively. In boxplots, the 25% and 75% quartiles are shown as lower and upper edges of boxes, respectively, and central lines denote the median. The whiskers extend to 1.5 times the inter-quartile range. P-values were computed from two-tailed Student’ s t-test.
Extended Data Fig. 4 Impact of MsGA3ox1 overexpression on alfalfa morphology traits.
a, Comparison between WT alfalfa plants and overexpression lines (OE3, OE7, and OE12). b-e, Quantitative measurements of MsGA3ox1 expression levels, plant height, SLR, and biomass. f, Photographs of leaves from WT, OE3, OE7, and OE12 lines at the 3rd, 4th, and 5th stem nodes. g-i, Comparative assessments of leaf area, leaf length, and leaf width between WT and MsGA3ox1 overexpression lines as shown in f. j, Comparison of WT and MsGA3ox1 overexpression lines in the number of trifoliolate leaves. The scale bar represents 5 cm. Asterisks denote statistical significance with ‘*’ ‘**’ and ‘***’ indicating P < 0.05, P < 0.01and P < 0.001, respectively. Data are presented as means ± SEM, with three independent experimental replicates for panel b, six independent experimental replicates for panels c, d, e, and j, and nine independent experimental replicates for panels g, h, and i. The control group (WT) is the Zhongmu No.1 variety of Medicago sativa L.
Extended Data Fig. 5 Phenotypic characterization of alfalfa quality traits in MsGA3ox overexpression lines.
The bar graphs depict a comparative analysis of crude protein (CP) (a), acid detergent fiber (ADF) (b), neutral detergent fiber (NDF) (c), lignin content (d), total digestible nutrients (TDN) (e), and net energy for gain (NEg) (f) between WT and overexpressed lines OE1 and OE3. Asterisks denote levels of statistical significance compared to WT (*P < 0.05, **P < 0.01, ***P < 0.001). Data are presented as means ± SEM, with four biological replicates per group. The control group (WT) is the Zhongmu No.1 variety of Medicago sativa L.
Supplementary information
Supplementary Information
Supplementary Figs. 1–7 and Tables 1–10.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
He, F., Chen, S., Zhang, Y. et al. Pan-genomic analysis highlights genes associated with agronomic traits and enhances genomics-assisted breeding in alfalfa. Nat Genet 57, 1262–1273 (2025). https://doi.org/10.1038/s41588-025-02164-8
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41588-025-02164-8
