
Abstract
Genetic variants associated with autoimmune diseases are highly enriched within putative cis-regulatory regions of CD4+ T cells, suggesting that they could alter disease risk through changes in gene regulation. However, very few genetic variants have been shown to affect T cell gene expression or function. Here we tested >18,000 autoimmune disease-associated variants for allele-specific effects on expression using massively parallel reporter assays in primary human CD4+ T cells. We find 545 variants that modulate expression in an allele-specific manner (emVars). Primary T cell emVars greatly enrich for likely causal variants, are mediated by common upstream pathways and their putative target genes are highly enriched within a lymphocyte activation network. Using bulk and single-cell CRISPR-interference screens, we confirm that emVar-containing T cell cis-regulatory elements modulate both known and previously unappreciated target genes that regulate T cell proliferation, providing plausible mechanisms by which these variants alter autoimmune disease risk.
Similar content being viewed by others

Main
CD4+ T cells have an essential role in immunity and autoimmune disease1,2,3,4, but how common genetic variants affect human CD4+ T cell function and disease pathology remain mostly unknown5,6. Genome-wide association studies (GWAS) have identified tens of thousands of genetic variants associated with autoimmune diseases, but the vast majority are in non-coding regions and in tight linkage disequilibrium with many other variants5,7,8. Hence, >99% of causal variants that drive complex traits such as autoimmune diseases have yet to be determined9. Identifying causal variants and the cells in which they operate would aid in defining disease-relevant target genes, pathways and cell types, and inform the development of more effective treatments10.
Complex trait-associated variants are enriched within chromatin that is accessible and/or actively modulating gene expression (that is, within regions of H3K27ac-deposited chromatin), which we define here as putative cis-regulatory elements (CREs). CREs differ between cell types11, and autoimmune GWAS variants enrich most highly within the CREs of CD4+ T cells5,12,13,14. Recent studies have shown that massively parallel reporter assays (MPRAs) can identify variants that alter cis-regulatory activity by changes in reporter gene expression15,16,17. Combining MPRAs with readouts of T cell-accessible chromatin enriched for variants with high statistical fine-mapping posterior inclusion probabilities (PIP > 0.5) up to 58-fold, suggesting that many emVars are putatively disease-causal variants18. Once putative causal variants are identified, a further challenge is to determine how they affect gene expression networks and cellular function. Online databases of functional genomic data, such as Open Targets Genetics, have been helpful for prioritizing genes that are probable targets of variants, but these databases lack perturbational data that directly link variants and CREs to gene expression19. To address this problem, recent studies have used bulk and single-cell CRISPR-interference (CRISPRi) screens to link variant CREs to the genes that they regulate20,21,22.
Although the above experiments support the probable cis-regulatory role of GWAS variants in T cells and other blood cells, both MPRA and CRISPRi screens have rarely been applied in primary cells. Instead, they tend to be performed in immortalized cell lines such as those derived from T cell leukemia (Jurkat) or erythroleukemia (K562). These cell lines may not accurately recapitulate the transcriptional regulation of primary cells or their associated phenotypes23. Testing these assays in primary cells containing transcriptional signatures and phenotypes that more closely reflect cells that have a role in disease may aid in highlighting the most relevant functional consequences of risk variants.
Here, we tested >18,000 variants for their effects on CRE activity with MPRAs in primary human T cells. We identified primary T cell expression-modulating variants (or emVars) that are largely distinct from those found in Jurkat cells. Primary T cell emVars tend to alter the binding of inflammatory transcription factors (TFs), enrich highly for fine-mapped variants with PIP > 0.8 and are often within loci containing genes that control T cell activation, among other pathways related to transcription and translation. Using single-cell and proliferation-based CRISPRi screens in primary T cells, we found that emVar CREs modulate genes within the T cell networks that control lymphocyte activation and mRNA processing, thus linking risk variants to T cell expression and function.
Results
Primary human CD4+ T cell emVars enrich for causal variants
We assessed the regulatory effects of genetic variants associated with multiple sclerosis, type 1 diabetes, psoriasis, rheumatoid arthritis and inflammatory bowel disease (IBD)24,25,26,27,28 in activated primary human CD4+ T cells using MPRA (Fig. 1a). We used a library previously described for an experiment performed on Jurkat T cells totaling 578 indexed single nucleotide polymorphisms (SNPs) and 18,312 total variants in tight linkage disequilibrium (R2 > 0.8) in the European subset of the 1000 Genomes Project cohort18.

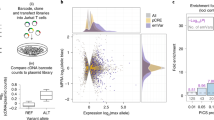
a, Primary T cell MPRA workflow. b, Volcano plot. The log2 expression of the highest-expressing allele is on the x axis, and the log2 of the activity of allele1/allele2 is on the y axis. emVars are labeled red, pCRE elements are dark gray and elements with no activity are light gray. c, Enrichment of pCRE and emVar elements for the accessible chromatin profiles of all ENCODE cell types. The −log10P from a two-sided Fisher’s exact test for the enrichment of pCRE and emVar elements is on the y axis, and the rank according to this P value is on the x axis. Cell lineages are depicted according to the colors in the legend. d, Bar plot showing the enrichment of DHS elements, emVars identified in primary T cells and emVars in DHS elements for PICS statistically fine-mapped variants using probability thresholds indicated on the x axis. Bar plot shade indicates −log10P enrichment. Numbers below bars indicate the number of emVars that are statistically fine-mapped at a given PICS probability. Enrichment was calculated as a risk ratio, with P values determined through a two-sided Fisher’s exact test. e, Scatterplot comparing element allelic skew between Jurkat and primary T cell MPRA libraries. Color indicates emVar positivity in the primary T cell MPRA, Jurkat MPRA, both or neither assay. The log2 allelic bias levels of MPRA elements tested in primary T cells are plotted on the x axis and in Jurkat on the y axis. f, Venn diagram depicting called emVars in only primary T cell MPRAs (red), Jurkat MPRAs (blue) or both (purple).
We observed high reproducibility of the primary T cell MPRA across independent donors (Pearson correlation > 0.93; Extended Data Fig. 1a–c), identifying 1,125 elements with putative CRE activity greater than baseline (Extended Data Figs. 1d and 2), 545 of which were emVars (Fig. 1b and Supplementary Table 1). We found primary T cell emVars in 39.2% of tested loci (Extended Data Fig. 3a), with only one emVar on the haplotype in 53.8% of tested loci (Extended Data Fig. 3b). Primary T cell emVars and putative CREs were both enriched within active chromatin marks and other readouts of cis-regulatory activity and were found preferentially in the accessible chromatin of primary T cells compared to other cell types (Fig. 1c, Extended Data Fig. 4, Supplementary Note and Supplementary Tables 2–4)18,29.
We next assessed emVar enrichment for both statistically fine-mapped variants using probabilistic identification of causal SNPs (PICS) PIPs for each trait locus5,18 and SuSiE fine-mapping data for UK Biobank traits (Fig. 1d, Extended Data Fig. 5 and Supplementary Tables 5 and 6)30. In all loci, emVars enriched twofold to fourfold for high posterior probability variants compared to all other tested variants (Extended Data Fig. 5a,b and Supplementary Note). Focusing on loci in which at least one emVar is observed, emVars were enriched for variants with PIP > 0.8 upwards of 71-fold (PICS) and 50-fold (SuSiE) (Fig. 1d, center, Extended Data Fig. 5c, center and Supplementary Tables 7 and 8). When considering only variants in T cell DNase hypersensitivity (DHS) sites in these loci, emVars enrich 122-fold (PICS) to 200-fold (SuSiE) for high posterior probability variants (Fig. 1d, right, Extended Data Fig. 5c, right and Supplementary Tables 7 and 8). Overall, we found that our primary T cell MPRAs had a sensitivity of 27% and a specificity of 88% for identifying variants in fine-mapped 95% credible sets in risk loci. Thus, primary T cell emVars in T cell-accessible chromatin enrich highly for fine-mapped variants, indicating that they enrich for causal variants.
TF usage varies by cell type and disease
Although both primary T cell and Jurkat emVars enrich highly for causal variants (Fig. 1d and Extended Data Fig. 5)18, only 45 emVars overlapped between datasets; however, this was significantly more than expected by chance (hypergeometric P = 5.4 × 10−29). Primary T cell and Jurkat MPRAs differed largely in activity and allelic bias in reporter expression (Fig. 1e,f and Supplementary Tables 1 and 9); therefore, we reasoned that differences in signaling and activation between cell types may alter the TF programs operating in each cell type23,31. To assess how each variant is predicted to change TF binding, we used known catalogs of TF motifs to assess variant-mediated TF motif disruption and compared this to the allelic effect on reporter expression within MPRA (Supplementary Note). We identified TF motifs that, when disrupted by variants, reduce expression in the MPRA, suggesting that these are transcriptional activators, such as ATF1 (Fig. 2a). Conversely, we also identified variant-disrupted motifs with a corresponding increase in expression, suggesting that these are transcriptional repressors, such as GFI1B, consistent with GFI1B’s known repressor role (Fig. 2a)32. Although many TF effects were shared between both T cells and Jurkat cells, transcription conferred by TFs associated with inflammation was more likely to be disrupted by variants in primary T cell MPRAs, including NFKB1 (n = 67 variants), STAT3 (n = 71 variants) and JUN and FOSB (n = 72 and 61 variants, respectively) (Fig. 2a, Extended Data Fig. 6a,b and Supplementary Tables 10–13). TF motifs whose perturbation by variants had concordant effects on MPRA expression in both cell types include ATF1, ETS factors ELK1 and ELK4 and ELF1 and ELF2, and GFI1B (Fig. 2a, Extended Data Fig. 6a,b and Supplementary Tables 10–13). To validate whether differences in TF programs within primary T cells versus Jurkat cells drove this difference, we performed SCENIC on single-cell RNA sequencing (scRNA-seq) data from each cell type to relate gene expression programs to TF activity33. In agreement with our TF motif disruption analysis, we found that NF-κB, JUNB, RUNX1, MYB and ZEB1 expression programs were highly operant in primary T cells but not within Jurkat cells, whereas Jurkat cell programs were driven more by TFs such as SOX4, ATF4 and ELF2 (Fig. 2b). Therefore, although both Jurkat and primary T cell MPRAs enriched for causal variants, differences in emVar identification in each setting are probably driven by alternative cellular programs and orchestrated by TFs.

a, Scatterplot comparing the effect of variants that are predicted to disrupt a TF motif and subsequent cumulative effect on MPRA expression between both primary T cell (red outline) and Jurkat (blue fill) experiments. The effect size is calculated using Cohen’s d for variant alleles predicted to disrupt a given TF motif, and P values are calculated using a two-sided t-test comparing the effect on expression of variants that disrupt a given motif versus all other variants. b, Scatterplot comparing cumulative effect of disruption of a given factor on MPRA expression (as in a) for Jurkat (blue) and primary T cell (red) results (x axis) and the AUCell activity score indicating TF regulon activity within a given cellular population based on single-cell RNA-seq data from Jurkat and primary T cells. The shade of each dot is the −log10P from a, calculated using a two-sided t-test comparing the effect on expression of variants that disrupt a given motif versus all other variants.
To determine whether we can identify TFs that drive risk in a disease-specific manner, we grouped variants by disease and repeated the analysis. We observed several TFs with disease-specific enrichment, including the ZNF563 motif, whose disruption is highly activating at IBD (Cohen’s d = 0.35, P = 0.0006) and rheumatoid arthritis loci (Cohen’s d = 0.45, P = 0.13), but repressive at psoriasis loci (Cohen’s d = −0.84, P = 0.005), and disruption of the GATA3 motif to be highly activating at multiple sclerosis loci (Cohen’s d = 0.30, P = 0.002) but with an average of no effect in loci associated with other diseases (Extended Data Fig. 6c). Therefore, we find MPRAs to be sensitive to TF usage in different cellular contexts and we define TFs that may be more important at specific disease loci.
emVars connect to T cell networks through multiple pathways
Motivated by the observation that the critical transcriptional regulators of T cell responses appear to mediate some primary T cell emVars, we sought to understand the pathways that emVars modulate to increase disease risk. To this end, we compared putative target genes of emVars in primary T cell DHS sites that were identified in primary T cell versus Jurkat MPRAs using the Open Targets Variant to Gene (V2G) dataset (Supplementary Tables 14 and 15 and Supplementary Note)19. We input these genes into STRING34 to define a primary T cell network of genes according to gene interaction experiments, co-expression and text mining. The resulting primary T cell network was more highly connected than expected when compared to a background of all 3,100 V2G genes linked to all MPRA-tested variants in T cell DHS sites (STRING protein–protein interaction enrichment, P < 1 × 10−16; Fig. 3a and Supplementary Data 1). Overall, the primary T cell network was enriched for T cell activation according to EnrichR35 even when compared to the refined background of the 3,100 V2G genes (false discovery rate, 0.026; Panther module; Supplementary Table 16). We then defined clusters within the network and observed that the largest clusters were involved in lymphocyte activation, translation, transcriptional regulation, antigen processing, mRNA processing and mRNA splicing (Fig. 3a,b and Supplementary Table 17).

a, STRING network showing V2G genes linked to 79 emVars in T cell DHS sites (nodes) and edges representing the strength of gene–gene interactions. Colors represent different network subclusters. P value is calculated using a two-sided hypergeometric test. b, The subclusters with the most genes from the larger network in a with gene nodes labeled. c–e, The lymphocyte activation (c), translation (d) and transcriptional regulation (e) clusters with each emVar on the x axis and target gene on the y axis. Fill color indicates that the gene is a V2G gene of the indicated emVar.
Within the lymphocyte activation cluster were known costimulatory genes expressed in T cells that encode proteins that regulate T cell activation, including CD28, CTLA4, ICOS, GITR, OX40 and SLAM family members (Fig. 3b, pink cluster). We also found that the transcriptional regulation cluster contained several genes encoding members of the NF-κB signaling family NFKB1, NFKB2, NFKBIA, TNFAIP3 and TNIP1 (Fig. 3b, yellow-green cluster). Both costimulatory genes and NF-κB signaling family members were absent from comparable clusters within a network built on V2G genes linked to Jurkat emVars in T cell DHS sites (Extended Data Fig. 7a,b, pink and yellow-green clusters and Supplementary Table 18). To connect genes within these networks to potential therapeutic targets, we used the Connectivity Map, finding that the top three primary T cell clusters were more significantly associated with NF-κB-driven gene programs compared to the Jurkat emVar clusters (Fig. 3b, Extended Data Fig. 7b,c and Supplementary Note). We created an emVar-by-gene matrix for each cluster for both primary T cell and Jurkat networks, which showed largely distinct gene targets in primary T cell emVar versus Jurkat emVar clusters (Fig. 3c–e, Extended Data Fig. 7d–g and Supplementary Note). Thus, the putative target genes of emVars found in primary T cell MPRAs are more involved in T cell costimulation and NF-κB signaling than those of Jurkat emVars.
Single-cell CRISPRi screens connect emVars to target genes
Although V2G data provide putative gene targets of variants, we sought to connect variants directly to the genes they regulate using a single-cell CRISPRi (scCRISPRi) approach in primary T cells. We used guide RNAs (gRNAs) and catalytically dead Cas9 (dCas9) tethered to a chromatin-repressing ZIM3–KRAB domain to target variant CREs and assessed local effects on gene expression (within 1 Mb) with scRNA-seq (Fig. 4a)36. We created two gRNA libraries to test 56 total emVar CREs. The first library targeted 20 emVars and three non-emVars in T cell-accessible chromatin, prioritizing variants more likely to be causal variants by PIP. The second library targeted 49 T cell emVars in T cell-accessible chromatin (emVar CREs) > 3,500 bp from transcription start sites (TSSs), to avoid silencing of promoter regions. A total of 13 emVar CREs overlapped between both libraries (Supplementary Tables 19 and 20). We used SCEPTRE37 to connect CREs to local genes (<1 Mb) based on differential gene expression in cells containing gRNAs targeting the CRE versus those containing non-target gRNAs.

a, Workflow for scCRISPRi screens. MOI, multiplicity of infection. b, Volcano plots depicting significantly differentially expressed genes when targeting a given emVar CRE, with the distance of emVar CRE to gene indicated by dot color; log2(fold change) is on the x axis and −log10P for differential gene expression is on the y axis. The dotted line indicates the empirical significance cutoff determined by SCEPTRE based on calibration with the non-target control gRNAs. c–e, Locus plots of the IL2RA (c), SESN3 (d) and PLEC loci (e). In d, inset scale for genome tracks is 0–3. pcHiC loops from primary human T cells are depicted below genes in the locus plot. Disease-associated variants (dots) are red if they are emVars in T cell DHS sites, blue if they are emVars not within DHS sites and gray if they are non-emVars. Accessible chromatin data from T cells are depicted as read pileups (peaks) on the locus track from various T cell types. The pink lines represent the location of emVars in DHS. Violin plots depict genes that are differentially expressed when targeting CRISPRi to the emVar using gRNAs compared to cells containing non-target gRNAs. f, Network of genes identified using scCRISPRi screens compared to all tested V2G genes for 56 emVar CREs. The red subcluster indicates the lymphocyte activation network. In b–e, two-sided SCEPTRE P values are false discovery rate-corrected using the Benjamini–Hochberg method. NT, non-target gRNA; FC, fold change.
We found 13 of the 56 tested emVar CREs to impact at least one gene in cis, with a total of 18 significant emVar CRE:gene interactions (Fig. 4b, Extended Data Fig. 8a,b and Supplementary Tables 21 and 22). Among them, we found that rs61839660 (type 1 diabetes and IBD PICS = 0.98), an IL2RA intronic variant (9 kb from TSS) previously associated with the timing of IL-2RA protein expression in murine T cells38, was associated with a downregulation of IL2RA but also an upregulation of several nearby genes, including IL15RA, a gene involved in homeostatic proliferation of memory T cells, and RBM17, which encodes a protein involved in non-sense mediated decay (Fig. 4b,c)39. We also identified rs887314 (psoriasis PICS = 0.13) within the promoter of BAD, which we found to not only regulate BAD, which encodes a protein involved in T cell development and apoptosis40, but also GPR137 and OTUB1 expression (Fig. 4b and Extended Data Fig. 8c). Other notable hits include rs56095240 (multiple sclerosis PICS = 0.18) in an intergenic region 456 kb from the SESN3 TSS, which regulates SESN3 expression (Fig. 4b,d), encoding a protein involved in negative regulation of reactive oxygen species signaling41 and T cell MAPK signaling42, and rs60600003 (multiple sclerosis PICS = 0.48) in intron 1 of ELMO1, 106 kb from the ELMO1 TSS, which had a substantial effect on ELMO1 expression, encoding a gene involved in lymphocyte motility (Fig. 4b, left and Extended Data Fig. 8d)43. In addition, two other emVars, rs1250567 (multiple sclerosis PICS = 0.03) and rs7441808 (rheumatoid arthritis PICS = 0.007 and eosinophil counts UK Biobank PIP = 0.18 (ref. 30)) were significantly associated with RBPJ and ZMIZ1, respectively, both encoding proteins involved in WNT signaling in T cells (Fig. 4b, right)44,45, and rs61907765 (psoriasis PICS = 0.43) was associated with ETS1 expression, which encodes a TF involved in survival and activation of T cells and the development of natural regulatory T cells (Fig. 4b, right)46. We also identified genes that could have a role in disease biology, but with limited or no previous evidence of contributing to T cell biology. For example, targeting three emVars in separate CREs in a large haplotype within PLEC with CRISPRi leads to an upregulation in GRINA expression, which encodes a glutamate receptor (Fig. 4e); neither PLEC nor GRINA has an established role in T cells. Furthermore, between both screens, we identified two emVars that regulate PPP5C expression, which encodes a phosphatase that acts on ERK signaling47 but, to our knowledge, has no established role in T cell biology (Fig. 4b).
Finally, to assess whether genes identified by our scCRISPRi screens were enriched in T cell-related networks, we relaxed our calling threshold to include variant CRE:gene interactions of marginal significance (P < 0.05). We were able to connect 37 of the 56 tested variants (66%) to 61 genes, of which 49 overlapped with V2G genes (Supplementary Tables 21 and 22). Interestingly, through creating a STRING network based on the hits versus all local genes that were tested in the single-cell screen, we again found that the T cell activation cluster was the most predominant cluster in the network (Fig. 4f, Supplementary Table 23 and Supplementary Note), further supporting the importance of T cell activation programs in genetic risk for autoimmunity.
Linking variant CREs to T cell proliferation
Although many primary T cell emVar target genes were found within or connected to T cell activation networks, whether emVar CREs actually impact T cell proliferation remains unknown. To systematically link variant CREs with T cell activation and proliferation, we used bulk CRISPRi screens in primary human T cells (Fig. 5a). As these data could be broadly useful for classifying variant function, we also assessed ~1,000 additional autoimmune variant CREs (Extended Data Fig. 9a,b). We created a gRNA library targeting each variant CRE, along with positive controls known to affect T cell proliferation and non-targeting gRNAs (Fig. 5a and Supplementary Table 24), and performed a screen to assess how dCas9–ZIM3-mediated silencing of variant CREs affected T cell proliferation (Supplementary Note and Methods).

a, Proliferation screen experimental workflow. b, Volcano plot of significant positive control genes and variant CREs (blue and red) and non-significant targets (gray), with the log2(fold change) on the x axis and the −log10(FDR) on the y axis. c, STRING network based on 13 emVar CREs that are CRISPRi proliferation hits. The lymphocyte activation and mRNA processing clusters and the PPP5C gene are highlighted in color. d, Locus plot depicting the PPP5C locus. Disease-associated variants (dots) are depicted according to MPRA allelic skew: rs4802307 (red), an emVar in a T cell DHS site, rs62136101(blue), a probable emVar in a T cell DHS site and non-emVars (gray). Accessible chromatin data from T cells are depicted as read pileups (peaks) on the locus track from various T cell types. The pink lines represent the location of emVars in DHS sites. e, Heatmap of differentially expressed genes when targeting CRISPRi to the PPP5C TSS or to the rs62136101 CRE using gRNAs compared to cells containing a non-target gRNA. f, Scatterplot depicting the correlation between differentially expressed genes when targeting rs62136101 versus NT (y axis) and the PPP5C TSS versus NT (x axis). P value in c is determined through a two-sided hypergeometric test, and those in f are determined using a Wald test with DESeq2. Error bars in f represent the standard error 95% confidence interval.
Through analyzing the effect of targeting all ~1,000 autoimmune GWAS variants in T cell DHS sites, we identified known positive controls, including VAV1 and IL-2RB as positive regulators of T cell proliferation, and CBLB, a known negative regulator (Extended Data Fig. 9c and Supplementary Table 25)48,49. We identified 21 additional variant CREs that were significantly associated with T cell proliferation (Padj < 0.1; Extended Data Fig. 9c and Supplementary Table 25). Among the hits with the strongest effect were variant CREs that contact the MYC promoter according to promoter capture HiC in T cells, such as rs10098999 (rheumatoid arthritis PICS = 0.0014) and rs10113762 (rheumatoid arthritis PICS = 0.0145), both of which are 800 kb downstream of MYC (Extended Data Fig. 9d)50. However, we found that variants that were hits within this screen were not enriched for statistically fine-mapped variants even at moderate posterior probabilities (Supplementary Note).
Focusing our analysis on the 56 emVar CREs that we analyzed in our scCRISPRi screens, we analyzed gRNAs that were enriched or depleted at day 21 compared to day 2 (Fig. 5b and Supplementary Table 26), identifying 12 emVar CREs that reduce proliferation when targeted with CRISPRi and one emVar CRE that promotes proliferation. Interestingly, the most significant emVar CRE hit that reduced T cell proliferation when targeted, rs10932019 (rheumatoid arthritis PICS 0.007), is 50 kb downstream from CD28, required for T cell activation. Other emVar CREs of note that reduced T cell proliferation when targeted were rs307370 (IBD PICS = 0.018) in the TNFRSF4 (encoding OX40) locus, rs61839660 (type 1 diabetes PICS = 0.98) in the IL2RA locus and rs9610375 (multiple sclerosis PICS = 0.0028) in the MAPK1 locus; each of the proteins encoded by these genes has previously been shown to be a positive regulator of T cell proliferation51,52,53. Conversely, the emVar CRE that increased T cell proliferation when targeted, rs11757155 (IBD PICS = 0.017), is in an intron of BACH2, encoding a TF involved in suppressing T cell activation and effector T cell differentiation54. Surprisingly, we found a correspondence between emVar CRE effects on proliferation and the effects of targeting their putative target genes within a genome-wide Cas9 proliferation screen in primary T cells. (Extended Data Fig. 9e,f), suggesting that many of the emVars could be in enhancers for these genes.
Given that many of our emVar CRE hits appeared in the T cell signaling and proliferation networks highlighted in the MPRA STRING network (Fig. 3), we next sought to determine whether the V2G genes of emVar CRE proliferation hits were enriched for these network clusters compared to a background of V2G genes linked to all 56 emVar CREs tested in the proliferation screen (Supplementary Table 27). We again found the top represented clusters were those pertaining to lymphocyte activation and signaling along with mRNA processing (Fig. 5c, Extended Data Fig. 9g,h and Supplementary Table 28). Our scCRISPRi screens found that five of the 13 proliferation hits also had a significant effect on the expression of local genes, including rs61839660 with IL2RA, IL15RA and RBM17, rs7823393 with GRINA, rs56095240 with SESN3 and both rs4802307 and rs62136101 with PPP5C (Fig. 4b). Therefore, we found that 13 of the 56 tested emVar CREs (23%) had a significant effect on T cell proliferation, linking these putatively causal variants to T cell function and highlighting putative target genes that enrich for T cell activation pathways.
PPP5C is a regulator of T cell proliferation
Two emVar CREs that reduced T cell proliferation when targeted, rs4802307 (emVar) and rs62136101 (an emVar with allelic skew in six out of seven donors; Extended Data Fig. 9i), are in the PPP5C locus on the same haplotype. The rs4802307-A (IBD lead variant) and rs62136101-T (R2 = 0.96 to rs4802307 in Europeans) alleles are associated with reduced PPP5C expression in T cell expression quantitative trait locus data and are protective from IBD (IBD PICS = 0.1 and 0.06, respectively) (Fig. 5d)28. From our single-cell screens, we found that PPP5C is the only significantly differentially expressed gene locally, suggesting that this is the target gene. PPP5C promotes RAF–MEK–ERK signaling and cancer cell proliferation47,55, but its function in T cells has not been assessed. Given that we find targeting both rs4802307 and rs62136101 to affect T cell proliferation but that PPP5C is not directly within the main T cell activation network, we reasoned that PPP5C could be a distal regulatory node to the T cell activation cluster. To assess whether silencing of PPP5C affects transcriptional programs in T cells, we targeted CRISPRi to both the rs62136101 CRE and the PPP5C TSS in T cells and compared the effects on global transcription to cells containing a non-targeting gRNA using RNA-seq. We found an upregulation of genes associated with T cell metabolism and function (Fig. 5e, Supplementary Tables 29 and 30 and Supplementary Note)56,57,58,59,60,61. Although targeting CRISPRi to rs62136101 had a more subtle effect than the TSS, both led to consistent differentially expressed genes involved in T cell biology (Fig. 5f). As PPP5C has been shown to act on MAPK signaling in cancer cell lines47,55,62, we sought to define how PPP5C shapes MAPK signaling in CD4 T cells. We performed protein blotting of phosphoproteins in the ERK–MAPK pathway in unstimulated and stimulated CD4 T cells with Cas9-mediated PPP5C ablation versus those containing non-target control sgRNAs, as well as on CD4 T cells transfected with a PPP5C-overexpression vector versus vector-only transfected CD4 T cells from two human donors (Extended Data Fig. 10a–c). We found that PPP5C knockout increased the phosphorylation of multiple components of the MAPK signaling pathway, including AKT, CREB, ERK1/2, GSKA3 and JNK, particularly in the unstimulated condition, while PPP5C overexpression reduced phosphorylation of these same signaling components in unstimulated conditions (Extended Data Fig. 10b–e). Therefore, PPP5C is a previously unappreciated component of T cell signaling that controls the baseline phosphorylation of several key members of the MAPK signaling pathway, and that IBD protective alleles downregulate, leading to tuning of T cell metabolic and effector programs.
Discussion
Identifying variants that underlie complex traits and defining their effects on disease-relevant cell types continues to be a longstanding challenge. Resources such as Open Targets Genetics and other online databases have been essential for providing observational and correlative data that aid in prioritizing variants as likely causal on a haplotype, defining the tissue in which the variant may promote the effect and the target gene. However, although these databases further refine variants that are likely causal, there often remain many potentially causal variants per haplotype with many putative target genes. To better understand variants that act within disease-relevant conditions, it would be ideal for online databases to include high-throughput perturbations of variants in many cell types, which could help identify variants that have effects on cis-regulatory regions and connect cis-regulatory regions to target genes across both disease-relevant and irrelevant contexts to better contextualize variant and haplotype activity. MPRA has proven to be a powerful approach that can enrich for causal variants and define the cell type and state in which the variant functions, suggesting its utility to define contexts for variant effects and to map likely causal variants genome-wide.
Identification of causal variants requires testing their effects in relevant cell types and assessing the broad biological functions of variants. T cells underlie the pathogenesis of many autoimmune diseases63,64,65. However, primary T cells have been notoriously difficult to genetically engineer and perturb until recent advances49,66. The Jurkat cell line has served as a tractable model for primary T cells for decades, although this cell line contains thousands of mutations, including within key tumor suppressors such as P53, the PI3K pathway and large structural variants23,67. Through testing the same MPRA library in both primary T cells and Jurkat cells, we find that both conditions identify likely causal variants, but the identified emVars largely differ, probably because of differences in TF usage within each setting. Given these data, we believe more variants that affect CRE activity would be discovered by assaying the same MPRA library in other primary T cell populations and states and other disease-relevant cell types, as well as assaying other potential functions of variants. MPRAs have been conducted in the context of other primary cells such as neuronal progenitors68, differentiated glutamatergic neurons69, brain organoids and human cortical tissue70, but only small MPRA libraries have been implemented in primary immune cells, such as one within primary human monocytes strongly implicating ETS2 regulation in monocytes in IBD pathogenesis71. Further technological development is required to implement MPRAs across other relevant cell types. Additionally, causal variants could function through alternate splicing or modulating transcript stability. MPRAs have been developed to test these functions72,73, and in future studies should be implemented within disease-relevant primary human cells.
Once putatively causal variants are identified, there remains a key challenge in defining their target genes and pathways. To link variants to genes, scCRISPRi screens have been performed, particularly within the K562 cell line20,22. Our scCRISPRi screens in primary T cells identify emVar target genes, many of which are relevant to T lymphocyte activation. However, we were surprised to find that our CRISPRi screens often did not agree with the Open Targets Genetics data with regard to the most likely target gene or the number of genes affected by a given variant. For example, SESN3 is not predicted to be a top target for rs56095240, but we found it to be the only differentially expressed gene in the 1 Mb region in our scCRISPRi screen when targeting this variant CRE. This brings into question whether variants or their haplotypes can have pleiotropic effects depending on the cell type and state in which it is tested, an idea supported by our MPRA in two cellular settings. However, additional experiments, including genomic editing of these loci in each setting, will be needed to determine whether this is indeed the case. Single-cell screens still lack sensitivity for smaller effects on expression; therefore, we probably missed disease-relevant genes. Additional target genes might be identified through increasing the number and efficacy of gRNAs and increasing the number of cells assayed. However, the effect size of a given enhancer on a gene and the specific context in which it functions will also affect whether a target gene is identified in these screens74. For some loci, we do find emVars CREs that regulate more than one gene. Thus, disentangling causal variant effects for these loci will be more complex than focusing on singular target genes within each locus, which has been the focus of genetic knockout studies for many years.
Although we expected causal variants to have larger effects on T cell function, the hits from our genome-wide CRISPRi screen targeting ~1,000 variant CREs to determine their effects on T cell proliferation did not enrich for causal variants. We suspect that variants that impose large effects on regulatory regions important for directly impacting disease processes are more likely to be rarer or eliminated in populations through purifying selection. Inversely, common disease variants may be more likely to reside in low-impact enhancers of key disease genes or higher-impact enhancers of non-critical genes with modest effects on cellular function, although more evidence will be needed to support this theory. Our data are in line with the recent observations showing systematic differences between expression quantitative trait loci and GWAS loci, whereby disease variants that have a high impact on a trait tend to have lower effects on local gene expression75. Through integrating our MPRA, scCRISPRi and proliferation-based CRISPRi screens, we identified a number of known and previously unappreciated target genes that control T cell activation, including PPP5C, a protein phosphatase that regulates ERK signaling in cancer cells47 but was previously unknown to affect T cells. Through testing variant CRE effects across other cellular functions, we can begin to better understand how common variants across cellular networks affect gene expression and function, and how variants may work together to lead to disease. Base editing, a method that can more precisely assess variant effects on biological functions, will still be required before definitively concluding variant mechanisms.
Our genomic screens in primary human T cells connect likely causal variants to their putative effects on T cell expression networks and function. These data can be used to propose mechanisms of risk and protection from autoimmune diseases mediated by primary T cells and begin to determine convergent properties of variants, which could be useful for stratifying polygenic risk scores and modifying treatments for individuals to target specific pathways.
Methods
Ethical regulations
This research complies with study protocols approved by the Benaroya Research Institute Institutional Review Board under protocol number IRB07109-633. The protocol was conducted according to the principles expressed in the Declaration of Helsinki. All cohorts provided informed, written consent.
Human subjects
For MPRA and bulk CRISPRi experiments, 11 fully deidentified donor peripheral blood mononuclear cells (seven for MPRA experiments, four for bulk CRISPRi experiments) were isolated from fresh apheresis leukoreduction packs (BloodWorks) using Ficoll-Paque plus (GE, 17-1440-03) (no sequencing of identifiable information), and for scCRISPRi screens and bulk RNA-seq, we used frozen peripheral blood mononuclear cells from five healthy donors within the BRI Biorepository: three for scCRISPRi screens (a 57-year-old Asian male, a 30-year-old Caucasian female and a 35-year-old Caucasian female) and two for bulk RNA-seq (a 56- year-old Asian male and a 32-year-old Caucasian male).
Cell culture
CD4 T cells were magnetically isolated (BioLegend, 480130) and activated in T cell media (TCM), constituting either X-VIVO 15 (Lonza, 04-418Q) supplemented with 25 mM HEPES, 1 mM sodium pyruvate, 0.5% non-essential amino acids, 1% penicillin–streptomycin, 0.5% l-glutamine, 5% FBS and 55 mM 2-mercaptoethanol; or CTS OpTmizer T cell Expansion Medium (ThermoFisher, A1048501), supplemented with 5% FBS, 1% glutamine, 100 U ml−1 penicillin–streptomycin and 55 mM 2-mercaptoethanol. Cells were activated with human T cell activation beads (Miltenyi, 130-091-441) and recombinant human IL-2 (final concentration, 100 U ml−1; NCI Biological Resources Branch). Lenti-X 293T cells were maintained in DMEM supplemented with 10% FBS, 1% glutamine, 100 U ml−1 penicillin–streptomycin, 1 mM sodium pyruvate, 1× MEM non-essential amino acids and 10 mM HEPES. Cells were passaged every 2–3 days using trypsin-EDTA for dissociation and kept at a confluency of less than 60%.
MPRA library transfection and sequencing
Please see the Supplementary Note.
Lentivirus production
The lentiviral production protocol was modified from a previous publication49. For making lentivirus, 293T cells (ATCC CRL-3216) were seeded in Opti-MEM I Reduced Serum Medium, GlutaMAX Supplement (ThermoFisher, 51985034) supplemented with 5% FBS, 1 mM sodium pyruvate and 1× MEM non-essential amino acids (as cOPTI-MEM) at 4 × 106 cells per 10 cm petri dish 1 day before the transfection. Cells were transfected at 80% confluency using 41.4 μl of Lipofectamine 3000 transfection reagent (ThermoFisher, L3000015) in 1,250 μl of plain OPTI-MEM (ThermoFisher, 31985070) at 21–25 °C. Next, 11 μg of transfer plasmid (dCas9-ZIM3-mCherry, Addgene, 154473; sgRNA libraries cloned into CROP-seq-opti, Addgene, 106280), 7.5 μg of psPAX2 (Addgene, 12260), 3.3 μg of pCMV-VSVG (Addgene, 8454) and 36.5 μl of p3000 reagent were added to 1,250 μl of room temperature plain OPTI-MEM in a separate tube and mixed by gentle pipetting. The plasmid and Lipofectamine 3,000 mixes were combined, mixed by gentle pipetting to a 2.5 ml volume of transfection mixture and incubated for 15 min at room temperature. Following incubation, 5 ml of medium was removed from the 10 cm dish and 2.5 ml of the transfection mixture was added. After 6 h, the transfection medium was replaced with 15 ml of cOPTI-MEM containing 1× ViralBoost (Alstem Bio, VB100). Lentivirus supernatant was collected and kept at 4 °C for 24 h after transfection (first collection) and replaced with 15 ml fresh cOPTI-MEM. The second collection was done 48 h after transfection. The two collections were pooled and spun down at 500g for 5 min at 4 °C to clear cell debris. Lenti-X concentrator (Takara Bio, 631232) was used to concentrate the virus, following the manufacturer’s instructions, and resuspended in plain OPTI-MEM at 100-fold less than the original volume. Concentrated virus was subsequently aliquoted and frozen at −80 °C.
Generation of CRISPRi libraries
Please see the Supplementary Note.
scCRISPRi screens
Five million CD4+ T cells were thawed and cultured in 5 ml of TCM supplemented with human T cell activation beads for 48 h. T cells were then infected with a 2–4% (v/v) solution of 100× concentrated dCas9-ZIM3-mCherry lentivirus to introduce the CRISPRi machinery. Then, 24 h later, the cells were washed twice with PBS and subsequently infected with an 8% (v/v) solution of 100× concentrated CRISPR-QTL virus, which targets specific variants of interest. A subset (5%) of gRNAs contained within the library targeted CD45 as a positive control to evaluate gene suppression efficacy. By day 2 post-library infection, puromycin was added to the culture at a final concentration of 2.0 µg ml−1 in fresh TCM to select for library transduced cells, and the cell density was adjusted to 0.5 million cells per ml. At 4 days post infection (4 dpi), the cells were resuspended in 20 ml of TCM containing 1.0 µg ml−1 of puromycin. At 10–12 dpi, the cells were collected, divided into six groups and stained with anti-CD3, anti-CD4 and anti-CD45 antibodies, as well as a live/dead dye (see Supplementary Table 31 for antibody dilutions). Six hashtag antibodies were also used to separate donors and to enable superloading of the 10× controller (Supplementary Table 31). Before sorting, six groups of cells were pooled and then sorted. A total of 150,000 (v1, one donor) and 330,000 (v2, two donors) mCherryhi/GFPhi cells (CD3+/CD4+/live/CD45+ pre-gated) were sorted using a FACS cell sorter (Supplementary Fig. 1). The sorted cells were promptly loaded onto two (v1) or six (v2) channels on the 10× Chromium X controller (10× Genomics) according to the manufacturer’s protocol, with a target capture of 20,000 (library 1) and 12,500 (library 2) cells per channel. Sequencing libraries were generated using the Chromium Next GEM Single Cell 5′ Kit v2 (10× Genomics, 1000265). Gene expression, CRISPR and feature barcoding libraries were pooled at a 4:1:1 ratio and treated with Illumina Free Adapter Blocking Reagent (Illumina, 20024144). Sequencing of pooled libraries was carried out on a NextSeq 2000 sequencer (Illumina), using a NextSeq P3 flowcell (Illumina) for v1 or sequenced on a Nova-seq X Plus (Illumina) 25B flow cell. Basecalls were processed to FASTQs on BaseSpace (Illumina).
Bulk CRISPRi screen for T cell proliferation
A total of 6 × 107 CD4+ T cells were activated in 30 ml of TCM for 24 h. T cells were then infected with 2% v/v 100× concentrated dCas9-ZIM3-mCherry lentivirus. Then, 24 h later, the cells were infected again with 0.25% v/v 100× concentrated 1,000 variant library lentivirus (multiplicity of infection, 0.5 ~ 1). At 1 dpi, cells were counted to ensure that there were at least 30 × 106 live cells. A total of 15 × 106 cells were collected as the time zero control (day 2) of the proliferation screens. For the remaining cells, fresh Th0 media (TCM with recombinant human IL-2 (final concentration, 500 U ml−1)) and puromycin (final concentration, 2.5 μg ml−1) were added to bring cells to 1 × 106 cells per ml. At 2 dpi, cells were collected, spun down and resuspended at 0.5 × 106 cells per ml in Th0 media. Cells were maintained between 0.5 and 1 × 106 cells per ml until 10 dpi, when cells were collected and live ZIM3–mCherry+ cells were FACS-sorted (typically ~10–30% of total cells; Supplementary Fig. 1 and see Supplementary Table 31 for antibody dilutions). Sorted ZIM3–mCherry+ cells were maintained at 0.5 × 106–1 × 106 cells per ml in Th0 media until day 21. At least 15 million cells were then collected per donor and stored at −80 °C until genomic DNA (gDNA) extraction.
Bulk CRISPRi screen sequencing library preparation
gDNA from cells (in ten million cell increments) was resuspended in 50 μl ChIP lysis buffer (1% SDS, 10 mM EDTA, in 50 mM Tris-HCl pH 8.1) and pipetted up and down. Lysed cells were transferred to a 96-well plate or eight-well strip, then incubated at 65 °C for 10 min. The sample was cooled to 37 °C, and 1 μl RNase cocktail (Ambion, AM2286) was added, mixed by pipetting and spun down, followed by incubation at 37 °C for 30 min. Next, 5 μl proteinase K (NEB, P8107) was added, and the sample was mixed by pipetting. The sample was then incubated at 37 °C for 2 h, then at 95 °C for 20 min to denature the proteinase K. To isolate gDNA, we added 36.4 μl Ampure XP to the sample, mixed thoroughly by pipetting, incubated for 5 min, and used a magnet to isolate magnetic beads and gDNA from the lysed sample. We pipetted off the supernatant and washed the sample three times with 80% ethanol while on the magnet. After drying the pellet for 5 min, gDNA was then eluted in 45 μl double-distilled H2O, yielding on average 3–6 pg per cell.
We then used 0.6 μg of gDNA from each sample for qPCR to determine the optimal PCR cycle number for library preparation. Each 10 μl qPCR reaction contained 5 μl of NEBNext Q5 Hotstart HiFi PCR master mix (NEB, M0543L), FwdInnerSeq and RevInnerSeq at 500 nM each (final concentration; Supplementary Table 31), 0.6 μg of gDNA, 1.7 μl of SYBR (diluted 1:10,000) and water to a final volume of 10 μl. Once the optimal PCR cycle was determined, 50 μl PCR reactions were used to amplify the amplicon, and the number of reactions was scaled to the total available gDNA. The PCR reactions from each sample were pooled, and 50 μl was taken for amplicon purification. Amplicons were purified using a two-step Ampure XP method. A 0.65× volume of Ampure XP was added to the sample, followed by mixing via pipette and incubating for 5 min. The sample was applied to the magnet, and the supernatant was isolated for further purification. To the supernatant, an additional 1.0× Ampure XP was added to the sample to capture the PCR amplicon. The sample was incubated for 5 min, and then the magnetic beads were captured by a magnet. The supernatant was removed and discarded, and the captured beads were washed three times with 80% ethanol. The sample was dried for 5 min and eluted with 25 μl of H2O. Samples were analyzed on a TapeStation to assess amplicon purity and size estimation, and the concentration of amplicon DNA was measured using Qubit. Samples were pooled based on the concentration of the specific target amplicon percentage and sequenced on a NextSeq 2000 with custom read1 primer hU6_R1 and custom index primer sgPuro_I (Supplementary Table 31).
Bulk RNA-seq
A total of 1 × 107 frozen CD4+ T cells were thawed and activated using human T cell activation beads in 10 ml of TCM. Then, 24 h later, the T cells were infected with a 10–15% v/v solution of 100× concentrated dCas9-ZIM3-mCherry lentivirus, facilitating the introduction of the CRISPRi machinery. At 24 h after CRISPRi infection, the cells were further infected with a 10–15% v/v solution of 100× concentrated gRNA virus designed to target specific variants of interest (see Supplementary Table 31 for gRNA sequences). As a positive control, we used a CD45 sgRNA to assess the efficacy of gene suppression. On day 2 post guide infection, puromycin was added into the culture at a final concentration of 2.0 µg ml−1 in fresh TCM to select for transduced cells and adjust the cell density to 0.5 million cells per ml. On day 4, the puromycin concentration was reduced to 1.0 µg ml−1 to maintain selection. Cells were expanded for an additional 3–4 days, and 1 day before cell sorting, flow cytometry analysis was performed to confirm the efficiency of CD45 knockdown (>85%) in mCherryhi/GFPhi cell populations (Supplementary Fig. 1 and see Supplementary Table 31 for antibody dilutions). On day 7 or 8, FACS was used to isolate at least 300,000 cells exhibiting high expression levels of mCherryhi/GFPhi. After sorting, cells were centrifuged to remove supernatant and immediately lysed using Trizol reagent. Cell lysates were subsequently frozen at −80 °C before RNA extraction. RNA extraction was carried out using Direct-zol RNA Microprep (ZYMO, R2063), following the manufacturer’s instructions, to obtain RNA for bulk RNA-seq. Total RNA was added to the reaction buffer from the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Takara, 634891), and reverse transcription was performed, followed by PCR amplification to generate full-length amplified cDNA. Sequencing libraries were constructed using the NexteraXT DNA library preparation kit with unique dual indexes (Illumina, FC-131-1096) to generate Illumina-compatible barcoded libraries. Libraries were pooled and quantified using a Qubit. Sequencing of pooled libraries was carried out on a NextSeq 2000 sequencer (Illumina) with paired-end 59-base reads, using a NextSeq P2 sequencing kit with a target depth of five million reads per sample.
Phospho-antibody arrays
A total of 10 × 106 activated CD4 T cells from two human donors were split and transduced with lentivirus containing PPP5C-targeting guides or non-targeting guides and Cas9. Then, 48 h post-transduction, cells were selected with puromycin (2 µg ml−1) and hygromycin (200 µg ml−1). After 14 days, cells were either left unstimulated or stimulated for 30 min with PMA (50 ng ml−1) and ionomycin (1 µg ml−1), and cell lysates were collected from 10 × 106 cells per condition. Separately, 100 million activated CD4 T cells from two human donors were split and nucleofected using a Neon Transfection System (1,600 V, 10 ms, three pulses) with a PPP5C-overexpression vector (VectorBuilder, VB900177-7632ksm) or a control vector (VectorBuilder, VB010000-9857ehb), rested for 48 h in TCM without IL-2, sorted based on BFP+ indicating successful transfection of the vector (Supplementary Fig. 2) and left unstimulated or stimulated for 30 min with PMA and ionomycin. The cell lysates were then collected. Lysates were mixed with protease and phosphatase inhibitors and frozen at −80 °C. Then, 20 µg of lysate was incubated on pre-blocked MAPK phospho arrays (RayBiotech, AAH-MAPK-1) overnight at 4 °C. Samples were washed with diluted wash buffer I and II for a total of five washes. Next, 1 ml of prepared detection antibody was added to each blot for 1.5–2 h at room temperature. Membranes were washed again as above with wash buffers I and II. Then, 2 ml of 1× horseradish peroxidase-anti-rabbit IgG was added to each blot and incubated for 2 h at room temperature. Samples were washed for a third time. Blots were exposed to detection buffer C + D and immediately imaged on a chemiluminescence system. We used ImageJ to obtain the density of each dot on the array and normalized each dot to the positive controls on the blot. We then compared the density of each dot from the PPP5C knockout and overexpression blots for each donor and stimulation condition by dividing the density value of the knockout or overexpression condition by that of the non-target or vector-only controls.
Analysis methods
Please see the Supplementary Note.
Statistics and reproducibility
We chose to perform our primary T cell MPRAs in seven human donors because we have found that this number is sufficient for identifying 10% effect sizes with 90% power assuming an activity standard deviation of 1.1, a Bonferroni-corrected alpha of 0.05 and 1,000 barcodes per SNP (https://andrewghazi.shinyapps.io/designmpra). We chose to perform our bulk CRISPRi screens with four donors because our simulation analyses found that we could successfully identify 10% effects with 80% power (https://zenodo.org/records/14847208). We chose to perform scCRISPRi screens with 150 cells per gRNA, as this was previously found to be reasonable power to detect differences in gene expression in cis (within 1 Mb)76. Two replicate samples were used for single gRNA CRISPRi RNA-seq experiments, which allowed us to detect 20% differences with 80% power assuming an alpha of 0.05. No data were excluded from the analyses. Given the unbiased nature of the experiments, we did not require the samples within experiments to be randomized or the investigators to be blinded to sample allocation during experiments and outcome assessment. All data met the assumptions of the statistical tests used.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data generated in this study from all figures are available in the Gene Expression Omnibus (GSE297472) and Database of Genotypes and Phenotypes (phs004072.v1.p1). Controlled access is required owing to personal sensitive information. Information on how to request access, as well as submission of sample requests, can be found at the link. The average time to decision once an application is submitted is 2 weeks. Jurkat MPRA data were obtained from GSE197538. The EMBL GWAS catalog (https://www.ebi.ac.uk/gwas/) was accessed on 10 August 2020. GWAS catalog data used in this study included type 1 diabetes (GCST005536), IBD (GCST003045), rheumatoid arthritis (GCST002318), psoriasis (GCST005527) and multiple sclerosis (GCST009597). The 1000 Genomes Phase 3 reference panel was obtained from ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502. DHS data across 733 samples were obtained from https://zenodo.org/record/3838751#.X_IA7-lKg6U. Histone ChIP-seq data were downloaded from ENCODE (encodeproject.org, https://hgdownload.cse.ucsc.edu/goldenpath/hg19/encodeDCC/wgEncodeRegTfbsClustered/wgEncodeRegTfbsClusteredV3.bed.gz). CAGE-based enhancer annotations were downloaded from https://fantom.gsc.riken.jp/5/datafiles/latest/extra/Enhancers. chromHMM were obtained from https://egg2.wustl.edu/roadmap/data/byFileType/chromhmmSegmentations/ChmmModels/core_K27ac/jointModel/final/. HOCOMOCO transcription factor position-weighted matrices were obtained from https://hocomoco11.autosome.org/downloads_v10 and https://hocomoco11.autosome.org/downloads_v11. ATAC-seq allelic skew data were obtained from a previous publication13 (https://www.nature.com/articles/s41588-019-0505-9). Chromatin accessibility quantitative trait loci were downloaded from a previous publication77 (https://www.nature.com/articles/s41588-018-0156-2). DeltaSVM precomputed weights for naive CD4+ T cells and Jurkat cells were obtained from http://www.beerlab.org/deltasvm_models/downloads/deltasvm_models_e2e.tar.gz. UK Biobank fine-mapping data were obtained from https://www.finucanelab.org/data. Jurkat single-cell data were obtained from https://www.10xgenomics.com/datasets/jurkat-cells-1-standard-1-1-0. DICE data were obtained from https://dice-database.org/downloads. Bulk CRISPR screen simulations are available at https://zenodo.org/records/14847208. Original blot MAPK images in Extended Data Fig. 10 are available in the source data. Source data are provided with this paper.
Code availability
Code supporting this manuscript is available at https://zenodo.org/records/15831587 (ref. 78).
References
Zhu, J., Yamane, H. & Paul, W. E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 28, 445–489 (2010).
Dominguez-Villar, M. & Hafler, D. A. Regulatory T cells in autoimmune disease. Nat. Immunol. 19, 665–673 (2018).
Sakaguchi, S. et al. Regulatory T cells and human disease. Annu. Rev. Immunol. 38, 541–566 (2020).
Pisetsky, D. S. Pathogenesis of autoimmune disease. Nat. Rev. Nephrol. 19, 509–524 (2023).
Farh, K. K. et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2015).
Ye, C. J. et al. Intersection of population variation and autoimmunity genetics in human T cell activation. Science 345, 1254665 (2014).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Schaid, D. J., Chen, W. & Larson, N. B. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 19, 491–504 (2018).
Alsheikh, A. J. et al. The landscape of GWAS validation; systematic review identifying 309 validated non-coding variants across 130 human diseases. BMC Med. Genomics 15, 74 (2022).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019).
Finucane, H. K. et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015).
Finucane, H. K. et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–629 (2018).
Calderon, D. et al. Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat. Genet. 51, 1494–1505 (2019).
Ray, J. P. et al. Prioritizing disease and trait causal variants at the TNFAIP3 locus using functional and genomic features. Nat. Commun. 11, 1237 (2020).
Ulirsch, J. C. et al. Systematic functional dissection of common genetic variation affecting red blood cell traits. Cell 165, 1530–1545 (2016).
Tewhey, R. et al. Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell 165, 1519–1529 (2016).
Siraj, L., et al. Functional dissection of complex and molecular trait variants at single nucleotide resolution. Preprint at bioRxiv https://doi.org/10.1101/2024.05.05.592437 (2024).
Mouri, K. et al. Prioritization of autoimmune disease-associated genetic variants that perturb regulatory element activity in T cells. Nat. Genet. 54, 603–612 (2022).
Ghoussaini, M. et al. Open targets genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res. 49, D1311–D1320 (2021).
Morris, J. A. et al. Discovery of target genes and pathways at GWAS loci by pooled single-cell CRISPR screens. Science 380, eadh7699 (2023).
Gasperini, M. et al. A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell 176, 1516 (2019).
Fulco, C. P. et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669 (2019).
Gioia, L., Siddique, A., Head, S. R., Salomon, D. R. & Su, A. I. A genome-wide survey of mutations in the Jurkat cell line. BMC Genomics 19, 334 (2018).
Onengut-Gumuscu, S. et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet. 47, 381–386 (2015).
International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365, eaav7188 (2019).
Okada, Y. et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381 (2014).
Tsoi, L. C. et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 44, 1341–1348 (2012).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015).
The ENCODE Project Consortium, et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020).
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Smith-Garvin, J. E., Koretzky, G. A. & Jordan, M. S. T cell activation. Annu. Rev. Immunol. 27, 591–619 (2009).
Doan, L. L. et al. Growth factor independence-1B expression leads to defects in T cell activation, IL-7 receptor alpha expression, and T cell lineage commitment. J. Immunol. 170, 2356–2366 (2003).
Van de Sande, B. et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat. Protoc. 15, 2247–2276 (2020).
Szklarczyk, D. et al. The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646 (2023).
Chen, E. Y. et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013).
Alerasool, N., Segal, D., Lee, H. & Taipale, M. An efficient KRAB domain for CRISPRi applications in human cells. Nat. Methods 17, 1093–1096 (2020).
Barry, T., Wang, X., Morris, J. A., Roeder, K. & Katsevich, E. SCEPTRE improves calibration and sensitivity in single-cell CRISPR screen analysis. Genome Biol. 22, 344 (2021).
Simeonov, D. R. et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 549, 111–115 (2017).
Liu, L. et al. The splicing factor RBM17 drives leukemic stem cell maintenance by evading nonsense-mediated decay of pro-leukemic factors. Nat. Commun. 13, 3833 (2022).
Mok, C. L. et al. Bad can act as a key regulator of T cell apoptosis and T cell development. J. Exp. Med. 189, 575–586 (1999).
Chen, Y. et al. The functions and roles of sestrins in regulating human diseases. Cell. Mol. Biol. Lett. 27, 2 (2022).
Lanna, A. et al. A sestrin-dependent Erk–Jnk–p38 MAPK activation complex inhibits immunity during aging. Nat. Immunol. 18, 354–363 (2017).
Stevenson, C. et al. Essential role of Elmo1 in Dock2-dependent lymphocyte migration. J. Immunol. 192, 6062–6070 (2014).
Chen, E. L. Y., Thompson, P. K. & Zuniga-Pflucker, J. C. RBPJ-dependent Notch signaling initiates the T cell program in a subset of thymus-seeding progenitors. Nat. Immunol. 20, 1456–1468 (2019).
Wang, Q. et al. Stage-specific roles for Zmiz1 in Notch-dependent steps of early T-cell development. Blood 132, 1279–1292 (2018).
Muthusamy, N., Barton, K. & Leiden, J. M. Defective activation and survival of T cells lacking the Ets-1 transcription factor. Nature 377, 639–642 (1995).
Lv, J. M. et al. PPP5C promotes cell proliferation and survival in human prostate cancer by regulating of the JNK and ERK1/2 phosphorylation. Onco Targets Ther. 11, 5797–5809 (2018).
Naramura, M. et al. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat. Immunol. 3, 1192–1199 (2002).
Schmidt, R. et al. CRISPR activation and interference screens decode stimulation responses in primary human T cells. Science 375, eabj4008 (2022).
Javierre, B. M. et al. Lineage-specific genome architecture links enhancers and non-coding disease variants to target gene promoters. Cell 167, 1369–1384.e19 (2016).
Schreiber, T. H., Wolf, D., Bodero, M., Gonzalez, L. & Podack, E. R. T cell costimulation by TNFR superfamily (TNFRSF)4 and TNFRSF25 in the context of vaccination. J. Immunol. 189, 3311–3318 (2012).
D’Souza, W. N., Chang, C. F., Fischer, A. M., Li, M. & Hedrick, S. M. The Erk2 MAPK regulates CD8 T cell proliferation and survival. J. Immunol. 181, 7617–7629 (2008).
Yang, S. Y., Denning, S. M., Mizuno, S., Dupont, B. & Haynes, B. F. A novel activation pathway for mature thymocytes. Costimulation of CD2 (T,p50) and CD28 (T,p44) induces autocrine interleukin 2/interleukin 2 receptor-mediated cell proliferation. J. Exp. Med. 168, 1457–1468 (1988).
Roychoudhuri, R. et al. BACH2 represses effector programs to stabilize Treg-mediated immune homeostasis. Nature 498, 506–510 (2013).
von Kriegsheim, A., Pitt, A., Grindlay, G. J., Kolch, W. & Dhillon, A. S. Regulation of the Raf–MEK–ERK pathway by protein phosphatase 5. Nat. Cell Biol. 8, 1011–1016 (2006).
Cook, M. E. et al. The ZFP36 family of RNA binding proteins regulates homeostatic and autoreactive T cell responses. Sci. Immunol. 7, eabo0981 (2022).
Zandhuis, N. D. et al. Regulation of IFN-γ production by ZFP36L2 in T cells is time-dependent. Eur. J. Immunol. 54, e2451018 (2024).
Brugarolas, J. et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 18, 2893–2904 (2004).
Maruhashi, T. et al. LAG-3 inhibits the activation of CD4+ T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat. Immunol. 19, 1415–1426 (2018).
Voskoboinik, I., Whisstock, J. C. & Trapani, J. A. Perforin and granzymes: function, dysfunction and human pathology. Nat. Rev. Immunol. 15, 388–400 (2015).
Cao, X. et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 27, 635–646 (2007).
Hsieh, F. S. et al. Inhibition of protein phosphatase 5 suppresses non-small cell lung cancer through AMP-activated kinase activation. Lung Cancer 112, 81–89 (2017).
Herold, K. C. et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N. Engl. J. Med. 346, 1692–1698 (2002).
Verstockt, B. et al. IL-12 and IL-23 pathway inhibition in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 20, 433–446 (2023).
Ghoreschi, K., Balato, A., Enerback, C. & Sabat, R. Therapeutics targeting the IL-23 and IL-17 pathway in psoriasis. Lancet 397, 754–766 (2021).
Shifrut, E. et al. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell 175, 1958–1971.e15 (2018).
Li, X. et al. The c-Rel–c-Myc axis controls metabolism and proliferation of human T leukemia cells. Mol. Immunol. 125, 115–122 (2020).
Lee, S. et al. Massively parallel reporter assay investigates shared genetic variants of eight psychiatric disorders. Cell 188, 1409–1424.e21 (2025).
Retallick-Townsley, K. G. et al. Dynamic stress- and inflammatory-based regulation of psychiatric risk loci in human neurons. Preprint at bioRxiv https://doi.org/10.1101/2024.07.09.602755 (2024).
Deng, C. et al. Massively parallel characterization of regulatory elements in the developing human cortex. Science 384, eadh0559 (2024).
Stankey, C. T. et al. A disease-associated gene desert directs macrophage inflammation through ETS2. Nature 630, 447–456 (2024).
Soemedi, R. et al. Pathogenic variants that alter protein code often disrupt splicing. Nat. Genet. 49, 848–855 (2017).
Griesemer, D. et al. Genome-wide functional screen of 3′UTR variants uncovers causal variants for human disease and evolution. Cell 184, 5247–5260.e19 (2021).
Bock, C. et al. High-content CRISPR screening. Nat. Rev. Methods Prim. 2, 9 (2022).
Mostafavi, H., Spence, J. P., Naqvi, S. & Pritchard, J. K. Systematic differences in discovery of genetic effects on gene expression and complex traits. Nat. Genet. 55, 1866–1875 (2023).
Chardon, F. M. et al. Multiplex, single-cell CRISPRa screening for cell type specific regulatory elements. Nat. Commun. 15, 8209 (2024).
Gate, R. E. et al. Genetic determinants of co-accessible chromatin regions in activated T cells across humans. Nat. Genet. 50, 1140–1150 (2018).
Dippel, M. A. Primary T cell MPRA Ho et al. analysis code. Zenodo https://doi.org/10.5281/zenodo.15831587 (2025).
Acknowledgements
We thank H. Finucane and J. Ulirsch for UK Biobank fine-mapping data, and S. Kales for plasmid preparation for MPRAs. We thank V. Green, A. Lacy-Hulbert, J. Hamerman, K. Cerosaletti, M. Dufort and M. Lawrence for critical review of the paper. Thanks to N. Doni Jayavelu and M. Altman for advice on network approaches. We acknowledge the Benaroya Research Institute’s Innovation fund, the BRI Clinical Core, the BRI Genomics Core and the BRI CATA Core, which enabled completion of this work. We thank the M.J. Murdock Charitable Trust for the purchase of scientific instrumentation, which enabled completion of the experiments. This study was funded by the National Institutes of Health grant DP2AI183504 (J.P.R.); National Institutes of Health grant U01AI176320 (J.H.B., J.P.R.); National Institutes of Health grant K22AI153648 (J.P.R.); National Institutes of Health grant R01DK140972 (J.P.R); Crohn’s & Colitis Foundation grant 1158945 (J.P.R.); National Institutes of Health grant R01AI151051 (R.T.); National Institutes of Health grant R35HG011329 (R.T.); National Institutes of Health grant K08AG086591 (M.H.G.); Michael Smith Health Research British Columbia Scholar (C.G.D.); Natural Sciences and Engineering Research Council of Canada, Banting Postdoctoral Fellowship (T.A.M.).
Author information
Authors and Affiliations
Contributions
C.H.H. and J.P.R. conceived the study. C.H.H., M.A.D., M.S.M., A.M., H.C., F.M.C., T.A.M., J.S., R.T. and J.P.R. developed the methodology. C.H.H., M.A.D., M.S.M., A.M., L.P.N. and S.H. performed the investigation. M.A.D., S.P., H.C., A.M. and J.P.R. visualized the project. R.T. and J.P.R. acquired funding. C.H.H. and J.P.R. were responsible for project administration. H.A.D., J.H.B., J.S., C.G.B., M.H.G., R.T. and J.P.R. supervised the research. J.P.R. wrote the original draft of the paper.
Corresponding author
Ethics declarations
Competing interests
R.T. holds patents related to the application of MPRA. J.S. is a scientific advisory board member, consultant and/or co-founder of Cajal Neuroscience, Guardant Health, Maze Therapeutics, Camp4 Therapeutics, Phase Genomics, Adaptive Biotechnologies, Scale Biosciences, Sixth Street Capital, Pacific Biosciences, Somite Theraputics and Prime Medicine. J.H.B. is a Scientific Co-Founder and Scientific Advisory Board member of GentiBio, a consultant for Bristol Myers Squibb and Moderna and has past and current research projects sponsored by Amgen, Bristol Myers Squibb, Janssen, Novo Nordisk and Pfizer. J.H.B. also has a patent for tenascin-C autoantigenic epitopes in rheumatoid arthritis. The other authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks Leah Kottyan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Primary T cell MPRAs have robust replication.
a, Scatterplot showing correlation between two replicates of plasmid barcode prevalence according to normalized read counts. b, Scatterplot showing correlation between two replicates of primary T cell barcode prevalence according to normalized read counts. c, Pie charts indicate pairwise Pearson correlation between plasmid and primary T cell replicates. d, Scatterplot showing correlation between primary T cell (y-axis) versus plasmid (x-axis) barcode prevalence according to normalized read counts.
Extended Data Fig. 2 Identification of active putative cis-regulatory regions with primary T cell MPRAs.
a, Scatterplot showing normalized tag count (x-axis) by expression fold change of barcode counts in RNA versus plasmid libraries. b, Same scatterplot as (a) but indicating spiked in positive (red) and negative (blue) controls. Variant library is indicated in green. c and d, Grid search assessing enrichment of expressed elements for primary T cell DHS sites at the given thresholds of expression fold-change (log2 mRNA/plasmid; y-axis) and expression significance (log10 adjusted P-value for element expression over baseline; x-axis). In red are the chosen cutoffs for calling putative CREs for primary T cell (c) and Jurkat cell (d) MPRAs. P-values for (c) and (d) are calculated using a two-sided Fisher’s exact test.
Extended Data Fig. 3 Primary T cell MPRA prioritizes variants in hundreds of loci.
a, Total number of GWAS loci tested (green) and number of loci with at least one emVar identified (orange) for each disease GWAS. b, Histogram of the number of emVars within each GWAS locus.
Extended Data Fig. 4 Variant locations relative to cis-regulatory features.
a, Location relative to TSSs of all MPRA tested variants, active elements (pCRE), and emVars. b, Enrichment of variants within pCREs (light blue) and emVars (dark blue) within chromHMM-defined genomic regions in human T cells. (P-value from two-sided Fisher’s exact test Bonferroni-corrected for 36 independent tests). c, Functional enrichment of variants within pCREs and emVars (nominal P-value threshold of 0.05 from two-sided Fisher’s exact test Bonferroni-corrected for 8 independent tests). d, Proportion of inactive element, pCRE variants, and emVars that have allelic bias in ATAC-seq. e, Scatter plot comparing MPRA log2 allelic bias (y-axis) with allelic bias in ATAC-seq from hematopoietic cells (x-axis)13. Red dots are emVars (n = 10) and gray dots are pCRE variants (n = 15). f, Proportion of MPRA inactive and pCRE variants, and emVars that are chromatin accessibility QTLs (caQTLs) from T cells77. g, Scatterplot comparing caQTL effect size (beta; x-axis) and MPRA log2 allelic bias (y-axis). Red dots are emVars (n = 4) and gray dots are pCREs (n = 10). h, Scatterplot comparing deltaSVM score (x-axis) with MPRA log2 allelic bias (y-axis). i, Proportion of MPRA inactive and pCRE variants and emVars that overlap TF motifs. j, Scatterplot comparing allele-specific TF binding scores (y-axis) and MPRA allelic bias (x-axis) for emVars predicted to perturb TF binding (n = 9). Calculations for (b and c) are risk ratios (see Supplementary Methods) with Fisher’s exact test P-values and Bonferroni correction (see Supplementary Tables 2 and 3 for exact P-values). (d, f, and i) P-values calculated using two-sided two proportions z test with no multiple comparisons adjustment. (e, g, h and j) R-squared and P-values are from linear regression F statistic and error bars represent standard error 95% confidence interval.
Extended Data Fig. 5 Primary T cell emVars enrich for causal variants.
a-c, Bar plot showing emVar enrichment for high posterior inclusion probability variants. Graphs in (a) and (b) consider all loci tested for PICS (a) and UKBB (b) fine mapping. The graph in (c) considers only loci with at least one emVar for UKBB finemapping. Each set of bar graphs is broken into three, with enrichment of DHS sites alone for fine-mapped variants (left), enrichment of primary T cell emVars for fine-mapped variants (middle), and enrichment of emVars in T cell DHS sites for fine-mapped variants (right), with the minimum posterior inclusion probability threshold indicated on the x-axis and fold enrichment shown on the y-axis. Details of PICS and UKBB enrichment results are shown in Supplementary Tables 7 and 8. Numbers below each bar show the number of emVars that are statistically fine-mapped at a given posterior probability threshold. Shade of each bar is the -log10 of the enrichment P-value. Enrichment in (a-c) was calculated as a risk ratio (see Methods), and P-values were determined through a two-sided Fisher’s exact test.
Extended Data Fig. 6 Primary T cell and Jurkat MPRAs identify different emVars modulated by distinct transcription factors.
a and b, Transcription factors whose motifs are predicted to be disrupted and the effect on allele-specific expression in (a) primary T cell MPRAs and (b) Jurkat MPRAs. Cohen’s d on the x-axis shows the collective effect size of variant alleles that disrupt a given TF motif and -log10 P-value on the y-axis. c, TF motif disruption of variants by disease, with disease on the y-axis, TF motif on the x-axis, dot size the -log10 P-values, and effect size, Cohen’s d, is the fill color. The motifs whose disruption caused the most significant upregulation and downregulation of expression for each disease and were hierarchically clustered according to TF and disease. For (a) and (b), effect size is calculated using Cohen’s d for variant alleles predicted to disrupt a given TF motif and P-values are calculated using a two-sided t test comparing effect on expression of variants that disrupt a given motif versus all other variants. The -log10 P-value in (a-c) is calculated using a two-sided t test comparing the effect on expression of variant alleles that disrupt a given motif compared to all other alleles tested.
Extended Data Fig. 7 Network analysis of predicted target genes of Jurkat emVars.
a, STRING network showing V2G genes linked to 31 emVars in T cell DHS sites (nodes) and edges representing the strength of gene-gene interactions. Network subclusters are represented by color. b, The subclusters with the most genes from the larger network in (a) with labeled gene nodes. c, Connectivity Map perturbagen class enrichment based on genes from STRING clusters identified from both primary T cell and Jurkat emVars in T cell DHS sites. -log10(FDR) is indicated by shade. d, Top 5 Jurkat network clusters with each putative emVar on the x-axis and target gene on the y-axis. e-g, The antigen processing (e), mRNA processing (f), and mRNA splicing (g) primary T cell network clusters from Fig. 3 with each putative emVar on the x-axis and target gene on the y-axis. Fill color indicates that the gene is a V2G gene of the indicated emVar. P-value in (a) is calculated using a hypergeometric test and those in (c) are calculated using a permutation test with FDR correction computed as the fraction of the ‘null signatures’ where the absolute normalized connectivity score exceeds reference signature.
Extended Data Fig. 8 Single cell CRISPRi screens identify emVar target genes.
a and b, QQ plots showing the expected (x-axis) versus observed (y-axis) Benjamini-Hochberg corrected two-sided SCEPTRE P-value, with dotted line indicating the significance cutoff for v1 (a) and v2 (b) libraries. Error bars represent standard error 95% confidence interval. c and d, Locus plots of the BAD (c) and ELMO1 (d) loci. pcHiC loops from primary human T cells are depicted below genes in the locus plot. Disease-associated variants (dots) are red if they are emVars in DHS, blue if they are emVars not within DHS, and gray if they are non-emVars. Accessible chromatin data from T cells are depicted as read pileups (peaks) on the locus track from various T cell types. The pink lines represent the location of emVars in DHS. Violin plots depicting the normalized expression of differentially expressed genes (y-axis) and the respective gRNA targets (x-axis). Benjamini-Hochberg corrected two-sided SCEPTRE P-values are provided for each tested gene in all panels. NT = non-target.
Extended Data Fig. 9 Genome-wide CRISPRi screen targeting variants in accessible chromatin.
a, Library makeup of genome-wide screen. b, Screen workflow. c, Rank order plot depicting targets of the CRISPRi screen, with positive control genes VAV1, IL2RB, TBX21, and CBLB, other tested genes, and variants indicated by rsid. Targets with MAGeCK permutation test P-values that are FDR corrected using the Benjamini-Hochberg with a value of less than 0.1 are indicated in blue. d, Locus plot of the MYC locus showing two variants that are proliferation hits in the screen (rank of hit in blue) in a distal enhancer that contacts the MYC promoter as determined by pcHiC in T cells. e and f, comparison of proliferation effects of targeting CRISPRi to emVar-CREs and effects of ablating local V2G-associated genes in a genome-wide CRISPR screen66 (e) with (f) showing only the most significant V2G gene. g and h, The lymphocyte activation (g) and mRNA processing (h) clusters made up from the hits from the 56 emVar proliferation screen with each putative emVar target gene represented with color. i, Normalized counts of rs62136101 variant alleles tested in primary T cell MPRAs across 7 donors. Each line indicates one donor.
Extended Data Fig. 10 PPP5C ablation and overexpression modulate ERK/MAPK phosphorylation.
a, MAPK phosphorylation array layout. b, MAPK array blots of CD4 T cell lysates from two donors in which PPP5C was Cas9 ablated (knock out or KO) or intact (non-target control or NTC) in unstimulated (0 min) or stimulated (30 min) conditions. c, MAPK array blots of CD4 T cell lysates from two donors in which PPP5C is overexpressed (OE) or intact (vector only) in unstimulated (0 min) or stimulated (30 min) conditions. d, Normalized blot density of PPP5C-ablated setting divided by that of the same probe in the NTC setting. Dark orange dots indicate unstimulated and green dots indicate the stimulated condition. e, Normalized blot density of PPP5C-overexpression setting divided by that of the same probe in the NTC setting. Light orange dots indicate unstimulated and grey dots indicate the stimulated condition. (d and e), n = 2 donors, with each dot indicating one donor.
Supplementary information
Supplementary Information
Supplementary Note, Supplementary Methods, Supplementary Figs. 1 and 2, Supplementary Table Legends 1–35.
Supplementary Tables 1–35
Supplementary Tables 1–35
Source data
Source Data Extended Data Fig./Table 10
Unprocessed dot blots for Extended Data Fig. 10.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ho, CH., Dippel, M.A., McQuade, M.S. et al. Genetic and epigenetic screens in primary human T cells link candidate causal autoimmune variants to T cell networks. Nat Genet 57, 2536–2545 (2025). https://doi.org/10.1038/s41588-025-02301-3
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41588-025-02301-3
