
Abstract
KRAS is one of the most frequently mutated oncogenes in cancer. Targeting mutant KRAS directly has been challenging because of minor structural changes caused by mutations. Despite recent success in targeting KRAS-G12C, targeted therapy for another hotspot mutant, KRAS-G12V, has not been described. We used CRISPR–Cas9 genome-wide knockout screens to identify genes that specifically modulate mutant KRAS harboring the G12V substitution. Our top hit, a fatty acid elongase (ELOVL6), showed remarkable selectivity in diminishing KRAS-G12V protein expression and aberrant oncogenic signaling associated with mutant KRAS. Our studies reveal that ELOVL6 can be targeted to control the production of phospholipids exploited by KRAS mutants for function-targeted and trigger-targeted degradation of the protein. Our results demonstrate the basis for a first-in-class small-molecule inhibitor to selectively clear KRAS-G12V from cancer cells.

This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

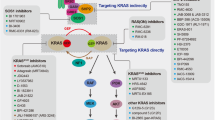

Data availability
The FASTQ files of sequencing reads were deposited to the National Center for Biotechnology Information Sequence Read Archive under BioProject PRJNA1275601. Source data are provided with this paper.
References
Moore, A. R., Rosenberg, S. C., McCormick, F. & Malek, S. RAS-targeted therapies: is the undruggable drugged? Nat. Rev. Drug. Discov. 19, 533–552 (2020).
Lu, S., Jang, H., Nussinov, R. & Zhang, J. The structural basis of oncogenic mutations G12, G13 and Q61 in small GTPase K-RAS4B. Sci. Rep. 6, 21949 (2016).
Martinko, A. J. et al. Targeting RAS-driven human cancer cells with antibodies to upregulated and essential cell-surface proteins. eLife 7, e31098 (2018).
Tolani, B. et al. RAS-mutant cancers are sensitive to small molecule inhibition of V-type ATPases in mice. Nat. Biotechnol. 40, 1834–1844 (2022).
Kim, D. et al. Pan-KRAS inhibitor disables oncogenic signalling and tumour growth. Nature 619, 160–166 (2023).
Holderfield, M. et al. Concurrent inhibition of oncogenic and wild-type RAS–GTP for cancer therapy. Nature 629, 919–926 (2024).
Schulze, C. J. et al. Chemical remodeling of a cellular chaperone to target the active state of mutant KRAS. Science 381, 794–799 (2023).
Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A. & Shokat, K. M. K-RAS(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 (2013).
Ostrem, J. M. & Shokat, K. M. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat. Rev. Drug Discov. 15, 771–785 (2016).
Lito, P., Solomon, M., Li, L. S., Hansen, R. & Rosen, N. Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351, 604–608 (2016).
Xiong, Y. et al. Covalent guanosine mimetic inhibitors of G12C KRAS. ACS Med. Chem. Lett. 8, 61–66 (2017).
Canon, J. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019).
Hallin, J. et al. The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and Patients. Cancer Discov. 10, 54–71 (2020).
Hong, D. S. et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 383, 1207–1217 (2020).
Skoulidis, F. et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med. 384, 2371–2381 (2021).
Matsuzaka, T. & Shimano, H. Elovl6: a new player in fatty acid metabolism and insulin sensitivity. J. Mol. Med. (Berl.) 87, 379–384 (2009).
Hart, T. et al. Evaluation and design of genome-wide CRISPR/SpCas9 knockout screens. G3 (Bethesda) 7, 2719–2727 (2017).
Wang, Z. et al. Efficient recovery of potent tumour-infiltrating lymphocytes through quantitative immunomagnetic cell sorting. Nat. Biomed. Eng. 6, 108–117 (2022).
Mair, B. et al. High-throughput genome-wide phenotypic screening via immunomagnetic cell sorting. Nat. Biomed. Eng. 3, 796–805 (2019).
Li, W. et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014).
Matsuzaka, T. et al. Crucial role of a long-chain fatty acid elongase, ELOVL6, in obesity-induced insulin resistance. Nat. Med. 13, 1193–1202 (2007).
Nagase, T. et al. Synthesis and biological evaluation of a novel 3-sulfonyl-8-azabicyclo[3.2.1]octane class of long chain fatty acid elongase 6 (ELOVL6) inhibitors. J. Med. Chem. 52, 4111–4114 (2009).
Shimamura, K. et al. 5,5-Dimethyl-3-(5-methyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-1-phenyl-3-(trifluoromethyl)-3,5,6,7-tetrahydro-1H-indole-2,4-dione, a potent inhibitor for mammalian elongase of long-chain fatty acids family 6: examination of its potential utility as a pharmacological tool. J. Pharmacol. Exp. Ther. 330, 249–256 (2009).
Shimamura, K. et al. Discovery and characterization of a novel potent, selective and orally active inhibitor for mammalian ELOVL6. Eur. J. Pharmacol. 630, 34–41 (2010).
Moon, Y. A., Ochoa, C. R., Mitsche, M. A., Hammer, R. E. & Horton, J. D. Deletion of ELOVL6 blocks the synthesis of oleic acid but does not prevent the development of fatty liver or insulin resistance. J. Lipid Res. 55, 2597–2605 (2014).
Su, Y. C. et al. Elovl6 is a negative clinical predictor for liver cancer and knockdown of Elovl6 reduces murine liver cancer progression. Sci. Rep. 8, 6586 (2018).
Kyselova, L., Vitova, M. & Rezanka, T. Very long chain fatty acids. Prog. Lipid Res. 87, 101180 (2022).
Zhou, Y. et al. Lipid-sorting specificity encoded in K-RAS membrane anchor regulates signal output. Cell 168, 239–251 (2017).
Tracey, T. J., Steyn, F. J., Wolvetang, E. J. & Ngo, S. T. Neuronal lipid metabolism: multiple pathways driving functional outcomes in health and disease. Front. Mol. Neurosci. 11, 10 (2018).
Jacquemyn, J., Cascalho, A. & Goodchild, R. E. The ins and outs of endoplasmic reticulum-controlled lipid biosynthesis. EMBO Rep. 18, 1905–1921 (2017).
Zhou, Y. et al. Signal integration by lipid-mediated spatial cross talk between RAS nanoclusters. Mol. Cell. Biol. 34, 862–876 (2014).
Zhou, Y., Prakash, P. S., Liang, H., Gorfe, A. A. & Hancock, J. F. The KRAS and other prenylated polybasic domain membrane anchors recognize phosphatidylserine acyl chain structure. Proc. Natl Acad. Sci. USA 118, e2014605118 (2021).
Zhou, Y. et al. Membrane potential modulates plasma membrane phospholipid dynamics and K-RAS signaling. Science 349, 873–876 (2015).
Liu, J. et al. Glycolysis regulates KRAS plasma membrane localization and function through defined glycosphingolipids. Nat. Commun. 14, 465 (2023).
Arora, N., Mu, H., Liang, H., Zhao, W. & Zhou, Y. RAS G-domains allosterically contribute to the recognition of lipid headgroups and acyl chains. J. Cell Biol. 223, e202307121 (2024).
Yeung, T. et al. Membrane phosphatidylserine regulates surface charge and protein localization. Science 319, 210–213 (2008).
Lu, A. et al. A clathrin-dependent pathway leads to KRAS signaling on late endosomes en route to lysosomes. J. Cell Biol. 184, 863–879 (2009).
Zhukovsky, M. A., Filograna, A., Luini, A., Corda, D. & Valente, C. Phosphatidic acid in membrane rearrangements. FEBS Lett. 593, 2428–2451 (2019).
Shimano, H. Novel qualitative aspects of tissue fatty acids related to metabolic regulation: lessons from Elovl6 knockout. Prog. Lipid Res. 51, 267–271 (2012).
Zhao, H. et al. Elovl6 deficiency improves glycemic control in diabetic db/db mice by expanding β-cell mass and increasing insulin secretory capacity. Diabetes 66, 1833–1846 (2017).
Matsuzaka, T. et al. ELOVL6 promotes nonalcoholic steatohepatitis. Hepatology 56, 2199–2208 (2012).
Fuentes, N. R. et al. Long-chain n-3 fatty acids attenuate oncogenic KRAS-driven proliferation by altering plasma membrane nanoscale proteolipid composition. Cancer Res. 78, 3899–3912 (2018).
Shin, J. J. & Loewen, C. J. Putting the pH into phosphatidic acid signaling. BMC Biol. 9, 85 (2011).
Hoshino, F. & Sakane, F. Polyunsaturated fatty acid-containing phosphatidic acids selectively interact with l-lactate dehydrogenase A and induce its secondary structural change and inactivation. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 1865, 158768 (2020).
Ghosh, S., Strum, J. C., Sciorra, V. A., Daniel, L. & Bell, R. M. RAF-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of RAF-1 in 12-O-tetradecanoylphorbol-13-acetate-stimulated Madin–Darby canine kidney cells. J. Biol. Chem. 271, 8472–8480 (1996).
Zhou, Y. & Hancock, J. F. Lipid profiles of RAS nanoclusters regulate RAS function. Biomolecules 11, 1439 (2021).
Kattan, W. E. et al. Components of the phosphatidylserine endoplasmic reticulum to plasma membrane transport mechanism as targets for KRAS inhibition in pancreatic cancer. Proc. Natl Acad. Sci. USA 118, e2114126118 (2021).
Koundouros, N. & Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 122, 4–22 (2020).
Singh, A. et al. A gene expression signature associated with ‘K-RAS addiction’ reveals regulators of EMT and tumor cell survival. Cancer Cell 39, 441–442 (2021).
Sunami, Y., Rebelo, A. & Kleeff, J. Lipid metabolism and lipid droplets in pancreatic cancer and stellate cells. Cancers (Basel) 10, 3 (2017).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019).
Surma, M. A. et al. Mouse lipidomics reveals inherent flexibility of a mammalian lipidome. Sci. Rep. 11, 19364 (2021).
Herzog, R. et al. A novel informatics concept for high-throughput shotgun lipidomics based on the molecular fragmentation query language. Genome Biol. 12, R8 (2011).
Acknowledgements
We thank S. Angers and J. Moffat for helpful discussions in designing the genome-wide CRISPR screens, S. Lin at the University of Toronto for providing library materials for the CRISPR screens, T. Ketela at the PMGC for help with NGS, R. Bolin, M. Castillo and A. Yabes at the Center for Comparative Medicine, Northwestern University for oral gavage injections and mouse body weight measurements and H. Fan and B. Shmaltsuyeva at Pathology Core, Northwestern University for help with immunohistochemistry. This study was supported by the Chicago Biomedical Consortium (C201612025 to S.O.K) and startup funds from Northwestern University to S.O.K.
Author information
Authors and Affiliations
Contributions
S.O.K., R.S.A. and X.H. contributed to the conceptualization and design of this work. X.H. performed most of the experiments in the study. S.X., C.Z. and H.W. assisted with the western blot experiments. S.A., S.X., C.Z. and X.H. performed the in vivo experiments. Z.W. fabricated the microfluidic cell sorting devices. X.H., R.S.A. and S.O.K. were the main writers of the manuscript. All authors discussed the results, analyzed the data and reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors have filed intellectual property related to the results reported in this study. Two patents have been submitted, entitled “Fatty acid elongase attenuator compounds and their uses” and “Genetic modulators of KRAS protein expression and their uses”.
Peer review
Peer review information
Nature Chemical Biology thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Overview of magnetic ranking cytometry (MagRC)-based cell sorting and endogenous KRAS protein level-dependent gRNA enrichment analysis of pooled genome-wide CRISPR-edited cells.
a, Schematic design of the microfluidic chip utilized for high-throughput cell sorting of TKOv3 CRISPR KO gRNA library transduced cells. The fluidic channel incorporates X-shaped microfabricated structures of different heights forming capture pockets to trap the magnetic nanoparticle labeled cells. Cells carrying high levels of immunomagnetic nanoparticles acquire a stronger magnetic force that can overcome the fluidic drag forces. Cells captured in zone 1 have high KRAS protein expression, and those in zone 2 have medium KRAS expression. The outlet connects to a syringe for the collection of cells with low KRAS expression. b, Representative flow cytometry profile of KRAS protein expression after sorting and cell sorting profiles of the screened cells (200 million cells per replicate). c, d, Scatter plots of individual gene enrichment scores and fold change over the unsorted population for (c) WT KRAS-expressing HT29 and (d) homozygous G12V KRAS-expressing SW480 cells.
Extended Data Fig. 2 ELOVL6 inhibition lowers mCherry-G12V KRAS protein expression and phosphorylated ERK signaling.
a, Dose-dependent effects of ELOVL6i treatment on KRAS protein levels after 72 h in mCherry-G12V-expressing SW480 cells from three biological replicates (P = 0.0015; P = 0.00018). b, Assessment of the impact of CRISPR-edited polyclonal ELOVL6 KO cells in HT29 cells transduced with mCherry-G12V mutant KRAS protein. Low and high G12V KRAS-expressing cells have different levels of ELOVL6 KO determined by mRNA expression from three biological replicates (P = 0.0074; P = 6.43e-6). c, Analysis of phosphorylated ERK expression in CRISPR-edited polyclonal ELOVL6 KO HT29 cells expressing mCherry-G12V KRAS from three biological replicates (P = 0.0014). Statistical significance was determined by a two-tailed unpaired T-test. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, data are represented as mean ± s.d.
Extended Data Fig. 3 ELOVL6i treatment alters the cellular lipid composition.
a, b, c, Untargeted lipidomic analysis of lipid species in G12V KRAS-expressing NCI-H727 control and ELOVL6i-treated cells after 1-week of treatment from three biological replicates. Data are shown as molar fractions of (a) glycerolipids, (b) sphingolipids, and (c) glycerophospholipids in total lipids detected. Statistical significance was determined by a two-tailed unpaired T-test. *P < 0.05, **P < 0.01, ***P < 0.001, data are represented as mean ± s.d. DAG: Diacylglycerol (P = 0.0036); TAG: Triacylglycerol (P = 0.0086); SM: Sphingomyelin (P = 0.0058); HexCer: Hexosylceramide (P = 0.0073); Cer: Ceramide (P = 0.37); CL: Cardiolipin (P = 0.026); PA: Phosphatidic acid (P = 0.024); PC: Phosphatidylcholine (P = 0.96); PE: Phosphatidylethanolamine (P = 0.022); PG: Phosphatidylglycerol (P = 0.0004); PI: Phosphatidylinositol (P = 0.003); PS: Phosphatidylserine (P = 0.0011).
Extended Data Fig. 4 Lipidomic profiles following 24-hour ELOVL6i treatment.
a, Untargeted lipidomic analysis of PS levels in NCI-H727 and HT29 cell lines after 24 h of treatment with 40 µM ELOVL6i from three biological replicates. Data are shown as molar fractions. b, KEGG pathway enrichment analysis of enriched lipids in ELOVL6i-treated NCI-H727 cells calculated using two-sided Fisher’s exact test. c, Molar fractions of asymmetrical PS species in NCI-H727 control and ELOVL6i treated cells (P = 0.044). d, Molar fractions of asymmetrical PS species in HT29 control and ELOVL6i treated cells. Statistical significance was determined by a two-tailed unpaired T-test. *P < 0.05, data are represented as mean ± s.d. FA: fatty acid.
Extended Data Fig. 5 Mixed-chain PS supplementation rescues KRAS membrane localization and protein levels following ELOVL6 inhibition.
a, Confocal imaging of SW480 cells expressing mCherry-G12V KRAS and Lact-C2-GFP or HEK293 cells expressing mCherry-WT KRAS and Lact-C2-GFP. Cells were serum-deprived and treated with 40 µM ELOVL6i for 24 h. PS 16:0/18:1(10 µM) was added back for 1-hour. b, Quantification of colocalization between G12V KRAS and Lact-C2 in SW480 cells with PS add-back (PS 18:0, PS 18:1, or PS 16:0/18:1). c, Quantification of colocalization between G12V KRAS and Lact-C2 in ELOVL6i-treated SW480 cells with PS add-back (P = 2.73e-8). d, Quantification of colocalization between WT KRAS and Lact-C2 in HEK293 cells with PS add-back. e, Quantification of colocalization between WT KRAS and Lact-C2 in ELOVL6i treated HEK293 cells with PS add-back. Colocalization measurements were collected from five fields of view in one independent experiment. Statistical significance was calculated by a two-tailed unpaired T-test. f, Immunoblot of HT29 cells expressing mCherry-G12V with different PS species add-back. g. Immunoblot of HT29 ELOVL6 KO cells expressing mCherry-G12V with different PS species add-back. The experiments f-g were repeated two times with similar results and representative results are shown. Data are shown as percent of normalized protein expression relative to control band density. ****P < 0.0001, data are represented as mean ± s.d.
Extended Data Fig. 6 ELOVL6i treatment induces attenuation of oncogenic signaling and KRAS-driven tumor growth in a heterozygous G12V KRAS mutant SW403 colon cancer model.
a, Effect of ELOVL6i treatment (300 mg/kg) on tumor volume and body weight in SW403(KRASG12V/WT) xenografts (Vehicle: n = 8; ELOVL6i: n = 7). Two-way ANOVA with Tukey’s multiple comparisons test was used to determine statistical significance (P = 0.001). ***P < 0.001, data are represented as mean ± s.e.m. b, Western blotting analysis of the impact of ELOVL6i treatment on KRAS effector signaling pathways in SW403 tumors collected from mice (n = 3) after 3 weeks of treatment. c, Flow cytometry analysis of cell populations with high pERK expression in tumors collected after 3 days, 1 week, and 3 weeks post-ELOVL6i treatment. d, MFI of pERK expression level relative to vehicle treated samples at each timepoint from three biological replicates (P = 0.007; P = 0.019). Two-tailed unpaired T-test was used to determine statistical significance. *P < 0.05, **P < 0.01, data are represented as mean ± s.d.
Extended Data Fig. 7 ELOVL6i suppresses tumor growth and prolongs survival in G12V KRAS mutant xenograft models.
a, Representative images of NCI-H441 tumor-bearing mice treated with vehicle or ELOVL6i (100 mg/kg) on day 10 and day 20. b, Tumor growth curves (P = 0.0002) and body weight for mice treated with vehicle (n = 8) or ELOVL6i (n = 8). c, Survival rate (P = 0.0154) in the vehicle (n = 8) and ELOVL6i treated groups (n = 8). d, Representative images of CFPAC-1 tumor-bearing mice treated with vehicle or ELOVL6i (100 mg/kg) on day 14 and day 21. e, Tumor growth curves (P = 3e-17) and body weight for mice treated with vehicle (n = 9) or ELOVL6i (n = 7). f, Survival rate (P = 0.0011) in the vehicle (n = 9) and ELOVL6i treated groups (n = 7). Two-way ANOVA with Bonferroni’s multiple comparisons test was used to determine statistical significance. Log-rank test was used to determine the p-value for survival curve. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, data are represented as mean ± s.e.m.
Extended Data Fig. 8 Cell viability assessment of ELOVL6i treated cells across human cancer cell lines expressing WT or mutant KRAS variants.
Cell viability was measured after a 7-day treatment with ELOVL6i at the indicated concentrations from three biological replicates. Data are represented as mean ± s.d.
Supplementary information
Supplementary Information
Supplementary Figs. 1–16 and Tables 1–4.
Supplementary Video 1
Supplementary Video 1a. Live-cell fluorescence imaging of SW480 cells expressing mCherry–KRAS-G12V upon treatment with ELOVL6i.
Supplementary Video 2
Supplementary Video 1b. Live-cell fluorescence imaging of SW480 cells expressing mCherry–KRAS-G12V labeled with cell membrane dye upon treatment with ELOVL6i.
Supplementary Video 3
Supplementary Video 2a. Live-cell fluorescence imaging of HT29 cells expressing mCherry–WT KRAS upon treatment with ELOVL6i.
Supplementary Video 4
Supplementary Video 2b. Live-cell fluorescence imaging of HT29 cells expressing mCherry–WT KRAS labeled with cell membrane dye upon treatment with ELOVL6i.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 2
Unprocessed western blots.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 3
Unprocessed western blots.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 4
Unprocessed western blots.
Source Data Fig. 5
Statistical source data.
Source Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 5
Unprocessed western blots.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 6
Unprocessed western blots.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hu, X., Atwal, R.S., Xiao, S. et al. ELOVL6 activity attenuation induces mutant KRAS degradation. Nat Chem Biol (2025). https://doi.org/10.1038/s41589-025-01998-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41589-025-01998-x

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}