
Abstract
HLA-bound peptides encoded by recurrent driver mutations are candidate targets for T cell-directed immunotherapy. Here we identify two neopeptides encoded by the CTNNB1S37F mutation presented on the frequent HLA-A*02:01 and HLA-A*24:02 molecules in cell lines naturally expressing the mutation and HLA alleles. This mutation leads to a gain of function in β-catenin and is estimated to occur in >7,000 new cancer cases annually in the United States. T cell receptors (TCRs) that specifically recognize the mutant peptides were isolated from naive healthy donor T cells. T cells redirected with CTNNB1-S37F TCRs efficiently killed CTNNB1S37F+ cell lines and patient-derived organoids in vitro and eradicated established tumors in a melanoma cell line mouse model and a patient-derived xenograft model of endometrial adenocarcinoma naturally expressing the mutation and the restricting HLA. We propose that TCR-T cells targeting CTNNB1-S37F can serve as a basis for solid cancer immunotherapy.
Similar content being viewed by others

Main
Somatic mutations can encode mutant peptides that are recognized by T cells in complex with HLA, called neoepitopes1,2,3. Independent studies have shown that tumor mutational burden is associated with clinical response to immune checkpoint blockade (ICB), indirectly pointing to a role for neoepitopes as rejection antigens3,4,5. However, only 1–3% of mutation-encoded candidate neoantigens in individuals with solid cancer are spontaneously recognized by the endogenous T cell pool following immunotherapy, such as treatment with tumor-infiltrating lymphocytes and ICB6,7. To enhance T cell antitumor reactivity, T cells can be genetically engineered in vitro to express T cell receptors (TCRs) that specifically target neoantigens. Because 99% of neoantigens are private7, originating from mutations specific to the individual tumor, major efforts have been dedicated to the identification of cognate TCRs from individuals with cancer. Such TCR-engineered T cells have been used in two recent clinical trials for solid cancers. In the first trial, no clinical responses were observed in 16 individuals8. In the second trial, partial objective clinical responses were seen in three of seven individuals with metastatic colorectal cancer (CRC) refractory to conventional therapies9. Targeting of private neoantigens, however, requires highly personalized approaches that are time and resource demanding. Public neoantigens are, on the other hand, shared among multiple individuals and are generated from recurrent mutations occurring in specific genomic hot spots. When presented on frequent HLA molecules, such mutations offer great potential for the development of tumor-specific therapies10. Driver mutations are particularly attractive targets. Due to their biological relevance in the initiation and progression of cancer, the mutations are more likely to be expressed in a substantial proportion of tumor cells, including metastases11. Clinical responses have been observed after TCR targeting of HLA-bound peptides encoded by recurrent driver mutations in KRAS and TP53 (refs. 12,13). However, these represent case reports, awaiting follow-up trials targeting these or other shared mutations.
We recently identified a shared driver mutation in FLT3 as an efficient rejection antigen14. This mutation leads to constitutive activation of FLT3, enhancing proliferation and survival of leukemic cells. Mass spectrometry (MS) analysis showed that the FLT3-D835Y peptide was presented on HLA-A*02:01 on primary leukemia cells. T cells genetically engineered to express a T cell receptor (TCR-T cells) specifically targeting the HLA-A*02:01-bound peptide efficiently eliminated myeloid leukemia cells in patient-derived xenograft (PDX) models with high disease burden, in a model of minimal residual disease and in a model investigating the cytotoxic effects on leukemia-propagating cells14. These results encouraged us to search for HLA-bound peptides encoded by recurrent gain-of-function mutations in solid cancer where there is a great unmet medical need.
The CTNNB1 gene, which encodes β-catenin, is mutated in 3.2% of all cancers (GENIE Cohort v. 15.1-public)15. Mutations are almost exclusively observed in the N-terminal exon 3 region of the gene and affect phosphorylation sites responsible for regulation of β-catenin degradation (Fig. 1a). These gain-of-function mutations lead to constitutive oncogenic signaling and elevated levels of β-catenin by preventing phosphorylation-dependent protein degradation (Fig. 1b)16,17. Due to the importance of β-catenin in cancer initiation and progression, the Wnt signaling pathway has been investigated as a potential target of treatment. However, attempts have resulted in poor therapeutic benefits due to insufficient efficacy, specificity and safety18,19.

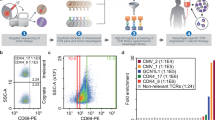
a, Illustration of the ꞵ-catenin protein domains. The amino acid sequence encoded by the CTNNB1S37F mutation and its immediate flanking sequence is shown. b, Schematic illustration of the Wnt–β-catenin signaling pathway with or without exon 3-mutated β-catenin. LRP, lipoprotein receptor-related protein. Images in a and b created with BioRender.com. c, Peptides encoded by mutant CTNNB1S37F predicted to bind to HLA-A*02:01 and HLA-A*24:02, corresponding WT peptides and their predicted binding affinity or predicted MS eluted ligand (EL) rank (%; strong binder: <0.500, weak binder: <2.000, nonbinder: >2.000) according to NetMHCpan-4.1 (ref. 21). d, Flow plots showing natural or transduced expression of HLA-A*02:01 and HLA-A*24:02 in CTNNB1S37F-mutated cell lines. e, Quantified surface expression per cell of CTNNB1-WT or CTNNB1-S37F peptides eluted from HLA-A*02:01 or HLA-A*24:02 of Mel888A24 + A2 and Hutu80A2 + A24 cells using targeted MS and isotope-labeled peptides. Numbers represent the mean of n = 3 independent experiments, and each data point represents one experiment with one technical replicate; ND, not detected. f, Sequence logos displaying preferred amino acids in positions 1–10 for HLA-A*02:01 and 1–9 for HLA-A*24:02 generated using Seq2Logo v.2.0 (ref. 51).
According to searches in the American Association for Cancer Research (AACR) Project GENIE data (GENIE Cohort v. 15.1-public)15 and the COSMIC database (https://cancer.sanger.ac.uk/cosmic)20, the three most frequent mutations in CTNNB1 are CTNNB1S37F, CTNNB1S45F and CTNNB1T41A. CTNNB1S37F mutation-encoded peptides are predicted to bind strongly to the frequent HLA alleles HLA-A*02:01 and HLA-A*24:02 compared to peptides encoded by the other two mutations, predicted to be weakly binding or nonbinding by NetMHCpan v.4.1 (ref. 21). A study published in 1996 showed that tumor-infiltrating lymphocytes from an individual with melanoma could recognize the CTNNB1-S37F peptide SYLDSGIHF when bound to HLA-A*24:02, and a cell line (Mel888) expressing this mutation was generated from the individual22. However, it was suggested that the level of expression of this epitope may be insufficient to allow efficient recognition by T cells.
Here, we revisited the possibility that CTNNB1-S37F may represent a therapeutic target. By use of MS, we demonstrated presentation of the HLA-A*24:02–SYLDSGIHF peptide and identified HLA-A*02:01–YLDSGIHFGA as a shared neopeptide. CTNNB1-S37F-reactive TCRs were identified from healthy donors. CTNNB1-S37F-engineered TCR-T cells eradicated tumors in an in vivo melanoma model and in an in vivo PDX model of endometrial adenocarcinoma, both models naturally expressing the mutation and restricting HLA. The lack of effective therapeutic modalities currently targeting the β-catenin pathway, and a clear unmet medical need in CTNNB1-mutated cancers, present an opportunity for TCR-T cell therapy.
Results
Prevalence and incidence of CTNNB1 S37F-mutated cancers
We investigated the prevalence of the CTNNB1S37F mutation by analyzing next-generation sequenced primary and metastatic cancers from the Memorial Sloan Kettering Metastatic Events and Tropisms (MSK-MET) study23. Although endometrial, cervical, small bowel and non-small cell lung cancers had the highest CTNNB1S37F mutation frequencies (3.35%, 2.91%, 1.06% and 0.76%, respectively), the analysis supported that the cancer types with the highest number of mutated samples were endometrial, non-small cell lung and prostate cancer (Extended Data Fig. 1a,b and Supplementary Data 1a). Among the cancer types that are reported most frequently in the MSK-MET study, each year, 7,155 new individuals in the United States would be estimated to get diagnosed with a cancer expressing the mutation (Supplementary Data 1a).
CTNNB1 S37F-encoded peptides are presented by HLA-A*02:01 and HLA-A*24:02
To identify neopeptides presented on frequent HLA class I molecules, we generated a minigene containing the CTNNB1S37F hot spot mutation flanked by the wild-type (WT) sequence in the genome. The minigene was transduced into Epstein–Barr virus (EBV)-transformed B-LCL 721.221 cell lines (B721.221) with stable HLA class I monoallelic expression. Next, we used immunopeptidomics to identify the naturally processed and presented peptides. Two peptides covering the CTNNB1S37F mutation were identified by MS, a 10-mer presented on HLA-A*02:01 (YLDSGIHFGA) and a 9-mer presented on HLA-A*24:02 (SYLDSGIHF; Extended Data Fig. 2a), consistent with strong predicted binding in NetMHCpan v4.1 (ref. 21; Fig. 1c).
We next investigated endogenous processing and presentation in two cell lines that naturally express the mutation: Mel888 and Hutu80. The Mel888 melanoma cell line naturally expresses HLA-A*24:02 (hereafter Mel888A24). It was transduced to also express HLA-A*02:01 (Mel888A24 + A2). The Hutu80 small intestine adenocarcinoma cell line naturally expresses HLA-A*02:01 (Hutu80A2), whereas HLA-A*24:02 was genetically introduced (Hutu80A2 + A24; Fig. 1d and Extended Data Fig. 2b). Endogenous processing and presentation of the two CTNNB1-S37F neopeptides was confirmed, and the amount of peptides eluted from Mel888A24 + A2 and Hutu80A2 + A24 was quantified using targeted MS and isotope-labeled peptides (Extended Data Fig. 2c–e and Supplementary Table 1). On a peptides per cell basis, the CTNNB1-S37F neopeptide presented on HLA-A*02:01 was quantified to 101 on Mel888 and 16 on Hutu80, whereas the CTNNB1-S37F neopeptide presented on HLA-A*24:02 was quantified to 17 on Mel888 and 20 on Hutu80 (Fig. 1e). These results corresponded well with the HLA expression levels for the two cell types shown in Fig. 1d. Although Mel888 and Hutu80 cells were transduced to express HLA-A*02:01 and HLA-A*24:02, respectively, expression levels for these introduced proteins were comparable to those found on naturally expressing cell lines (Extended Data Fig. 2f). The WT CTNNB1–HLA-A*02:01 peptide was not identified on either Mel888 or Hutu80 cells, whereas the WT CTNNB1–HLA-A*24:02 peptide was identified at low levels (Fig. 1e and Extended Data Fig. 2c–e). This was contrary to the stronger predicted HLA binding for the HLA-A*02:01 WT peptide, retaining the favored leucine at the P2 anchoring position of HLA-A*02:01, whereas the HLA-A*24:02 WT peptide was predicted as a nonbinder, suggesting that the HLA-A*02:01 WT peptide is not efficiently processed and presented (Fig. 1c,f).
We previously demonstrated that the stability of the peptide–MHC complex is a strong predictor of neoantigen immunogenicity14,24 and therefore experimentally measured half-lives of mutant and WT CTNNB1 peptides. For HLA-A*02:01, the neopeptide displayed a half-life almost twice that of the WT peptide (7.3 h versus 4.1 h, respectively; Extended Data Fig. 2g). For HLA-A*24:02, only the CTNNB1-S37F neopeptide was predicted to bind strongly, consistent with the preferred P9 phenylalanine anchor of HLA-A*24:02 (Fig. 1c,f). Experimental half-life measurements confirmed increased neopeptide–MHC stability. The difference in observed half-life was, however, likely underestimated by our assay due to the higher stability of the HLA-A*24:02 UV peptide than the HLA-A*02:01 UV peptide (HLA-A*02:01 UV peptide mean: 3.6 h versus HLA-A*24:02 UV peptide mean: 6.8 h; Extended Data Fig. 2g). Overall, in silico peptide binding predictions and experimental HLA half-life results were in good agreement with the targeted MS analysis.
Identification of HLA-A*02:01-restricted and HLA-A*24:02-restricted CTNNB1-S37F TCRs
To identify TCRs reactive to the two CTNNB1-S37F neopeptides, naive CD8+ T cells from HLA-A*02:01+ or HLA-A*24:02+ healthy donors were primed for 10 days as previously described14,25 (see also Methods). Dual-colored CTNNB1-S37F HLA-multimer+ T cells were single-cell sorted for TCR sequencing (Extended Data Fig. 3a). Three T cell populations from two HLA-A*02:01+ donors yielded three unique TCR sequences: TCRA2-1, TCRA2-2 and TCRA2-3 (Fig. 2a). One CD8+ T cell population from an HLA-A*24:02+ donor yielded one TCR sequence, TCRA24 (Fig. 2a). All four TCRs were efficiently expressed in third-party peripheral blood (PB) CD8+ T cells (TCR-T cells), as demonstrated by staining with anti-mouse TCRβ antibody reactive to the mouse constant region introduced into the TCRs (Fig. 2b and Extended Data Fig. 3b). The TCR-T cells stained positively with the relevant CTNNB1-S37F HLA multimers, but not with the corresponding CTNNB1-WT HLA multimers (Fig. 2c).

a, Naive CD8+ T cells were primed in cocultures for 10 days with CTNNB1S37F-encoded peptides, and wells that stained positively for relevant CTNNB1-S37F peptide–HLA-A*02:01 or peptide–HLA-A*24:02 multimers at the end of culture are shown. b,c, CD8+ T cells transduced with the CTNNB1-S37F TCRs stained with anti-mouse TCRβ chain antibody (b) or the relevant CTNNB1-S37F or CTNNB1-WT pMHC multimers (c; gating strategy in Extended Data Fig. 3a,b). d, CTNNB1-S37F TCR-T cells were co-incubated with HLA class I monoallelic B721.221 cell lines pulsed with the indicated concentrations of relevant peptides. Activation was measured as the percentage of CD137+ cells, and the mean EC50 values of the indicated CTNNB1-S37F TCRs were calculated. Data are pooled from n = 3 independent experiments, and each data point represents mean ± s.e.m. of n = 3 different PBMC donors with n = 2 technical replicates. e, Activation of CD8+ CTNNB1-S37F TCR-T cells measured by upregulation of CD137 after co-incubation with Mel888A24, Hutu80A2, Mel888A24 + A2 or Hutu80A2 + A24 cells with or without loading of the cognate CTNNB1-S37F peptide (100 nM). f, Evaluation of the ability of TCRA2-1, TCRA2-2 and TCRA2-3 T cells to persistently kill the Hutu80A2 cell line after three repeated challenges with tumor cells using IncuCyte live-cell imaging. In e and f, data from one experiment using n = 2 different T cell donors are shown; bars show the mean, and dots represent the average of n = 3 technical replicates.
The CTNNB1-S37F TCR-T cells recognized their cognate neoepitopes loaded onto monoallelic B721.221 cells with high to very high sensitivity (TCRA2-3 half-maximal effective concentration (EC50) = 11.8 nM, TCRA2-1 EC50 = 2.84 nM, TCRA2-2 EC50 = 499 pM and TCRA24 EC50 = 227 pM), whereas the corresponding WT peptide was not recognized by any of the TCRs (Fig. 2d and Extended Data Fig. 3c). CTNNB1-S37F TCR EC50 values were compared to two previously identified TCRs: the clinically applied and effective HLA-A*02:01-restricted NY-ESO-1 TCR 1G4 (TCR1G4)26 and a TCR targeting the highly immunogenic Nef134-10 peptide in individuals with HLA-A*24:02+ HIV (TCRHIV (ref. 27); Extended Data Fig. 3d). The three HLA-A*02:01-restricted CTNNB1-S37F TCRs showed 1.6- to 38-fold higher sensitivity than TCR1G4, with an EC50 of 24.6 nM. This was in the same range as previously reported EC50 values for TCR1G4 in similar assays but with different target cells (5.5 nM (ref. 14) and 7.7 nM (ref. 28)). The HLA-A*24:02-restricted CTNNB1-S37F TCR showed somewhat lower sensitivity than TCRHIV T cells (EC50 = 363 pM versus EC50 = 95.8 pM, respectively). TCRHIV exhibited EC50 values of approximately 3 nM (ref. 27) when previously tested in a different lysis-based assay, highlighting the variability between assays measuring functional avidity. The TCR-T cells also recognized endogenously processed and presented peptide in an HLA-restricted manner on the Mel888 and Hutu80 cell lines, naturally expressing the mutation and HLA-A*24:02 (Mel888A24) or HLA-A*02:01 (Hutu80A2; Fig. 2e and Extended Data Fig. 3e). Mel888A24 and Hutu80A2 + A24 cells were recognized by TCRA24, whereas Hutu80A2 and Mel888A24 + A2 cells were recognized by all HLA-A*02:01-restricted TCRs (TCRA2-1, TCRA2-2 and TCRA2-3). All identified CTNNB1-S37F TCRs demonstrated CD8 dependency, although low-level CD4+ T cell reactivity to peptide-loaded targets was observed (Extended Data Fig. 4a).
To evaluate killing efficacy of the HLA-A*02:01-restricted TCRs, coculture experiments were conducted with Hutu80A2 cells for 48 h. Cytotoxicity was evaluated using real-time live-cell imaging (Incucyte, Essen Biosciences) and analyzing changes in green fluorescent protein+ (GFP+) area. TCRA2-2 T cells demonstrated potent killing at an effector:target (E:T) ratio of 4:1 and consistently outperformed the other HLA-A*02:01-restricted TCRs in repeated killing assays (Fig. 2f and Extended Data Fig. 4b). Persistent in vitro killing was recently demonstrated to be the best predictor of TCR efficacy in vivo29.
Given its picomolar sensitivity, stronger recognition of Mel888A24 + A2 and Hutu80A2 + A24 and efficient, persistent killing of Hutu80A2 cells (Fig. 2d–f), TCRA2-2 was chosen among the HLA-A*02:01-restricted CTNNB1-S37F TCRs, together with HLA-A*24:02-restricted TCRA24, for further in vitro and in vivo characterization.
Activation of TCRA2-2 and TCRA24 T cells is peptide and HLA restricted
The killing capacity of TCRA2-2 and TCRA24 T cells was further evaluated through coculture experiments with the Mel888A24 + A2 and Hutu80A2 + A24 cell lines. Cytotoxicity was compared to that seen with mock- and TCR1G4-transduced T cells. Both TCRs showed potent killing at E:T ratios as low as 2:1 for Mel888A24 + A2 and 4:1 for Hutu80A2 + A24 (Fig. 3a). HLA-restricted killing by TCRA2-2 and TCRA24 T cells was confirmed by the absence of killing after coculture with untransduced Mel888A24 and Hutu80A2 cells, respectively (Extended Data Fig. 4c).

a, Evaluation of TCRA2-2 and TCRA24 T cell killing of the Mel888A24 + A2 or Hutu80A2 + A24 cell lines using IncuCyte live-cell imaging. Data were pooled from n = 3 independent experiments with different T cell donors and n = 3 technical replicates in each experiment. Individual data points represent the average of all experiments, and error bars represent s.e.m. b, Activation of TCRA2-2 and TCRA24 T cells following coculture with HLA-A*02:01 (top) or HLA-A*24:02 cell lines of different tissue origins in the absence or presence of the cognate CTNNB1-S37F peptide (100 nM). The suffix +A2 or +A24 indicates cell lines transduced with HLA-A*02:01 or HLA-A*24:02, respectively; n = 1–2 independent experiments using n = 3 different T cell donors; bars show the means, and each dot represents the average of n = 3 technical replicates. S/F, CTNNB1S37F-mutated cell lines. c, Heat maps indicating activation as measured by CD137 staining of TCRA2-2 and TCRA24 T cells after co-incubation with B721.221 cells expressing HLA-A*02:01 or HLA-A*24:02, loaded separately with each substituted peptide (10 nM) from the mimotope library (gating strategy in Extended Data Fig. 5a). White circles indicate reactivity to the cognate peptide sequences. The threshold for positive reaction (CD137) was set to 5% of the mean reactivity seen toward the cognate peptide. d, Percentage of CD137+ TCRA2-2 (left) or TCRA24 (right) T cells following co-incubation with B721.221 cells pulsed with peptides (10 nM) predicted by ScanProsite as potentially cross-reactive. For TCRA2-2, reactivity for peptide 4 was tested against mRNA encoding a 30-mer sequence of the peptide-encoding gene (AT8B3) containing the potential cross-reactive peptide (middle). B721.221 cells were electroporated with mock or AT8B3 mRNA and co-incubated with TCRA2-2 T cells. Dashed, horizontal lines represent the cutoff threshold (CD137 > 5% of mean reactivity to the cognate peptide) to determine hits. List of peptides are available in Supplementary Table 3a,b. Data in c and d are from one of two independent experiments with different T cell donors, and individual data points represent the mean of one (c) or two (d) technical replicates.
To assess HLA and peptide specificity, TCRA2-2 or TCRA24 T cells were cocultured with a diverse panel of 27–29 human CTNNB1WT cell lines from various tissues (Fig. 3b and Extended Data Fig. 5a) that express a variety of HLA alleles in addition to HLA-A*02:01 or HLA-A*24:02 (Supplementary Table 2). Neither TCR showed reactivity, unless the CTNNB1WT cells were preloaded with the corresponding CTNNB1-S37F peptide, suggesting high peptide and HLA specificity.
To further uncover potential cross-reactivity, peptide mimotope libraries were synthesized by systematically substituting each amino acid in the neoepitopes with all possible natural amino acids, resulting in 190 and 171 peptides for the HLA-A*02:01 and HLA-A*24:02 CTNNB1-S37F peptides, respectively. Each peptide in the relevant mimotope library was preloaded onto HLA-A*02:01+ or HLA-A*24:02+ B721.221 cells before incubation with TCRA2-2 or TCRA24 T cells. T cell activation was assessed by CD137 expression (Fig. 3c and Extended Data Fig. 5a–c). All combinations of amino acid substitutions that triggered ≥5% activation of the TCRs were queried in the human proteome databases UniProtKB/Swiss-Prot using the ScanProsite tool (https://prosite.expasy.org/scanprosite/; Extended Data Fig. 5d). This search identified 4 additional 10-mer peptides for TCRA2-2 and 47 9-mer peptides for TCRA24 in the human proteome that could potentially be recognized by the TCRs (Supplementary Table 3a,b). However, none of these candidate off-target peptides activated the TCRs (Fig. 3d). For TCRA2-2, predicted peptide number 4 could not be synthesized. Reactivity was therefore tested against mRNA encoding a 30-mer sequence of the peptide-encoding gene (AT8B3) containing the potential cross-reactive peptide in the middle and linked to a GFP tag to control for expression. B721.221 cells electroporated with AT8B3 mRNA did not activate TCRA2-2 T cells (Fig. 3d).
We next demonstrated that PB mononuclear cell (PBMC), B cell and T cell viability from three HLA-A*02:01+ and HLA-A*24:02+ donors remained unaffected after 48 h of coculture with autologous mock- or CTNNB1-S37F TCR-T cells unless loaded with the cognate peptide (Extended Data Fig. 6a,b). CTNNB1-S37F TCR-T cell expansion also matched that of mock T cells (Extended Data Fig. 6c). These results support the expected absence of fratricide, as healthy PBMCs express CTNNB1WT.
CTNNB1-S37F TCR-T cells eradicate melanoma and prevent relapse in vivo
To investigate the killing efficacy of TCRA2-2 and TCRA24 T cells in vivo, NOD-Prkdcscid-Il2rgTm1/Rj (NXG) mice were engrafted with Mel888A24 + A2 melanoma cells transduced with a firefly luciferase–enhanced GFP (ffluc–eGFP) fusion gene (Mel888A24 + A2 + ffluc–eGFP). Tumor engraftment was monitored by bioluminescence imaging (BLI; Spectrum In Vivo Imaging System) to determine the T cell injection time point and treatment grouping (Supplementary Table 4). Mice were either left untreated (n = 6) or were treated with mock-transduced (n = 7), control TCR1G4 (n = 7), TCRA24 (n = 7) or TCRA2-2 T cells (n = 7) 8 days after tumor injection (day 0), following tumor establishment confirmed by BLI in all mice (Fig. 4a,b).

a, Schematic overview of the Mel888A24 + A2 + ffluc–eGFP CTNNB1S37F in vivo melanoma tumor model; n = 6–7 mice per group. Figure created with BioRender.com; 2 M, 2 million. b, BLI of Mel888A24 + A2 + ffluc–eGFP melanoma-engrafted mice on days –1, 1, 8, 27 and 101 after T cell injection. c,d, Survival analysis for each treatment group (c) and tumor size (d) measured by calipers in individual mice at the indicated time points after T cell injection. e, Percentage of hCD45+ T cells in mouse PB at the indicated time points (gating strategy in Extended Data Fig. 8a). Survival analysis in c was performed by log-rank (Mantel–Cox) test. Data in d and e are from one experiment, and each data point represents an individual mouse.
All mice treated with TCRA2-2 T cells were tumor free by day 8 after T cell injection (Fig. 4b). TCRA24 T cells eliminated melanoma in three of seven mice by day 27, with one additional mouse achieving a complete response by day 45. During the 115-day observation period, only one CTNNB1-S37F TCR-T cell-treated mouse had to be killed due to tumor burden (TCRA24 day 43). Four CTNNB1-S37F TCR-T cell-treated mice were killed for nontumor reasons (graft-versus host disease (GvHD) = 2, weight loss = 1, eye injury = 1; Supplementary Table 5). Thus, among a total of 14 mice receiving the therapeutic TCRs (n = 7 in each group), 9 survived for the whole duration of the experiment (TCRA24 = 3; TCRA2-2 = 6; Extended Data Fig. 7a), demonstrating efficient in vivo elimination of CTNNB1S37F-mutated melanoma.
Among relapsing CTNNB1-S37F TCR-T cell-treated mice surviving to the experimental endpoint, all relapses occurred after day 90 when T cell numbers had dropped drastically due to ceased interleukin-2 (IL-2) injections (TCRA24 = 2, TCRA2-2 = 1; Fig. 4d,e and Supplementary Table 5). Tumor analysis showed complete HLA-A*02:01 loss in the relapsed TCRA2-2 T cell-treated mouse, but no HLA-A*24:02 loss in TCRA24 T cell-treated relapsing tumors (Extended Data Fig. 7b,c). Mock and TCR1G4 T cell-treated tumors contained high human CD3+ and CD8+ T cell frequencies, whereas relapsed TCRA24 and TCRA2-2 T cell-treated tumors showed very low T cell frequencies (Extended Data Fig. 7d,e). This difference might be explained by the different sampling times, as most control mice were killed before or right after IL-2 cessation. No substantial differences in PD-1, TIGIT and TIM-3 expression were observed on tumor-infiltrating T cells between control and TCRA24 T cell-treated mice (Extended Data Fig. 7f–h). Although CTLA-4 expression was significantly higher on TCRA24 T cells than in control cells (Extended Data Fig. 7i), the overall percentages of CTLA-4+ cells were low compared to the other exhaustion markers.
All control mice (untreated, mock and TCR1G4) had tumors at the time of euthanasia (days 28–50 after T cell injection). For most (15/20), the reason for euthanasia was a tumor size of >300 mm3, whereas the remaining 5 were euthanized due to weight loss (4) or GVHD (1; Fig. 4b–d and Supplementary Table 5). Repeated PB analysis confirmed sustained CTNNB1-S37F TCR-T cell presence (Fig. 4e and Extended Data Fig. 8a,b), with similar exhaustion marker levels (PD-1, TIGIT, TIM-3 and CTLA-4) across groups (Extended Data Fig. 8c–f). Due to risk of xenoreactivity from endogenous TCR repertoires, IL-2 frequency was reduced on day 24 and stopped on day 34 to limit T cell expansion (Fig. 4e). In total, three mice were killed for GvHD (mock = 1, TCRA24 = 1 and TCRA2-2 = 1). Importantly, the TCRA24 and TCRA2-2 T cell-treated mice that were killed due to GvHD were tumor free at the time of euthanasia. The three mice with GvHD symptoms had the highest blood human CD45+ (hCD45+) cell levels (hCD45+ cells >20%) in their groups (Extended Data Fig. 8g–j and Supplementary Table 5).
CTNNB1-S37F TCR-T cells eradicate organoids and PDX tumors in vivo
To evaluate T cell-mediated killing of primary tumor material, TCRA2-2 and TCRA24 T cells were cocultured with two patient-derived tumor organoids from large intestine adenocarcinoma (CRC) and endometrial endometrioid adenocarcinoma (EAC; Supplementary Table 2). Both organoids naturally express the CTNNB1S37F mutation, and the EAC organoid naturally expresses HLA-A*02:01, whereas the CRC organoid was transduced to express HLA-A*02:01 and HLA-A*24:02 (CRC+A2+A24). Both TCRA2-2 or TCRA24 T cells eradicated the CRC+A2+A24 organoids, whereas TCRA2-2 T cells efficiently eliminated the EAC organoids (Fig. 5a and Extended Data Fig. 9a,b).

a, Killing of CRC+A2+A24 and EAC organoids by TCRA2-2- or TCRA24-transduced T cells relative to mock-transduced or TCR1G4-transduced T cells (control T cells). Bars show mean ± s.e.m. of one or two independent experiments with n = 3 different PBMC donors, and each dot represents the average of one donor with n = 3 technical replicates. b, Schematic overview of the EAC PDX in vivo endometrial adenocarcinoma model; n = 7 mice per group. Figure created with BioRender.com; 15 M, 15 million. c, Tumor volume measured by calipers at the indicated time points after implantation of PDX fragments. Data are presented as mean ± s.e.m. Endpoint tumor volumes were compared among the tumor-only- and TCR1G4- and TCRA2-2-T cell-treated groups using one-way analysis of variance (ANOVA). If significant, pairwise differences were assessed by Tukey’s post hoc test; *P < 0.05; tumor only versus TCRA2-2 P = 0.0183; TCR1G4 versus TCRA2-2 P = 0.0352. d, Percentage of hCD45+ cells among all CD45+ cells (both mouse CD45+ and hCD45+) in mouse PB at the indicated time points (gating strategy in Extended Data Fig. 8a). Each data point represents an individual mouse. The data in c and d are from one experiment.
To assess the in vivo killing capacity of the CTNNB1-S37F TCR-T cells against primary tumor material, NXG mice were subcutaneously implanted with EAC PDX fragments naturally expressing CTNNB1S37F and HLA-A*02:01. Tumor engraftment was monitored by calipers, and mice were left untreated (n = 7) or were treated with control TCR1G4 (n = 7) or TCRA2-2 T cells (n = 7) on day 9 after implantation, once palpable tumors had developed in all mice (Fig. 5b and Supplementary Table 6).
Tumors in TCRA2-2 T cell-treated mice did not grow after treatment, except for slow expansion observed in one mouse, in contrast to tumors in the control groups (tumor only versus TCRA2-2 P = 0.0183; TCR1G4 versus TCRA2-2 P = 0.0352; Fig. 5c and Extended Data Fig. 9c). Longitudinal PB analysis demonstrated a marked increase in hCD45+ cells on day 46 and a drop in hCD8+ or mouse TCRꞵ+ (mTCRꞵ+) cells, indicating the onset of xenoreactivity in several mice across both the TCRA2-2- and TCR1G4-treated groups (Fig. 5d and Extended Figs. 8a and 9d–f). Consequently, the experiment was terminated on day 56 (day 47 following T cell injection).
At the experimental endpoint, tumors were completely eliminated in six of seven mice receiving TCRA2-2 T cells, whereas all control mice had tumors (Fig. 5c and Supplementary Table 6). These results demonstrate that TCR-T cells targeting a single recurrent mutation can eradicate in vivo primary patient-derived tumors naturally expressing both the target mutation and the restricting HLA allele.
Considerations for clinical application of CTNNB1-S37F TCRs
Considering phenotypic frequencies for HLA-A*02:01 and HLA-A*24:02 in the US population (35.48% and 18.82%, respectively), 3,885 new individuals in the United States would have cancers expressing the CTNNB1S37F mutation annually and either of the two relevant HLA alleles restricting the identified CTNNB1-S37F-targeting TCRs (Supplementary Data 1a). The corresponding numbers for the previously identified TCRs targeting the recurrent mutations TP53R175H, presented by HLA-A*02:01 (ref. 13), and KRASG12V, presented by HLA-A*11:01 or HLA-A*03:01 (ref. 30), would be 11,638 and 16,593 individuals, respectively (Supplementary Data 1b,c). Screening for targetable mutations is increasingly made possible by different precision medicine initiatives, such as DRUP-like trials31,32, and identification of HLA type is possible from sequencing data33.
However, the eligibility for a phase 1 trial with a novel cellular therapy depends on several additional factors, like performance status, comorbidities and organ function. To provide preliminary estimates of the potential phase 1 populations in endometrial, non-small cell lung and prostate adenocarcinoma, representing the groups with the highest total number of CTNNB1S37F-mutated tumors, we consulted with experts in gynecological, thoracic and urological oncology. First, the proportion of individuals who develop refractory or metastatic disease was estimated based on 2024 cancer incidence and mortality data reported by the Cancer Registry of Norway34. Based on these data, it can be estimated that 15% of individuals with endometrial adenocarcinoma will develop refractory or metastatic disease. Among these, another 50% of individuals are estimated to be alive, in good performance status (ECOG 0-1) and otherwise suitable for a phase 1 trial after exhausting standard-of-care systemic therapy. The estimated phase 1 population for endometrial adenocarcinoma would thus be 7.5% of the annual incidence. For non-small cell lung cancer, the corresponding numbers were estimated to be 65%, 30% and 19.5%, respectively, and for prostate adenocarcinoma 15%, 75% and 11.25%. Considering the frequency of the HLA-A*02:01 and HLA-A*24:02 alleles restricting the CTNNB1-S37F TCRs, the estimated annual numbers of individuals with CTNNB1S37F-mutated endometrial, non-small cell lung and prostate adenocarcinoma in the United States eligible for a phase 1 trial would be 92, 161 and 84, respectively (Supplementary Data 1a). By contrast, the estimated numbers of individuals eligible for treatment with a TCR recognizing the shared TP53R175H mutation presented on HLA-A*02:01 (ref. 13) in these same cancer types are 47, 120 and 154, respectively (Supplementary Data 1b). For the KRASG12V mutation, presented on HLA-A*11:01 or HLA-A*03:01, the corresponding estimates are 81, 662 and 5, respectively (Supplementary Data 1c).
Discussion
Shared neoantigens are attractive as targets for T cell-directed cancer immunotherapy. However, to be clinically useful, the peptides must be presented for recognition by T cells. Here, we show that two peptides encoded by a shared driver mutation in CTNNB1 (S37F) are presented in the context of HLA-A*02:01 and HLA-A*24:02 on tumor cells, and we isolate four reactive TCRs from naive healthy donor T cells. T cells engineered to express two of these TCRs efficiently killed patient-derived organoids of colorectal and endometrial origin and elicited a complete response in 11 of 14 mice with established melanoma. At termination of the experiment on day 115, only 1 of 14 mice treated with cognate TCRs was killed due to high tumor burden. By contrast, all control mice had to be killed between days 28 and 50, mostly due to tumor size. TCRA2-2 T cells also achieved complete tumor elimination in six of seven mice in a PDX model of endometrial adenocarcinoma, whereas all control mice showed sustained tumor progression at termination of the experiment on day 56. These results demonstrate that T cells engineered with neoantigen-specific TCRs can eliminate established solid tumors in vivo, including patient-derived tumors with natural expression of the cognate peptide–MHC (pMHC) complex, and prevent relapse during long-term follow-up.
The CTNNB1S37F mutation was selected because it is prevalent and known to promote oncogenesis and cancer progression. To identify HLA-bound peptides, we took a targeted approach35. In the first step, MS was used to detect HLA-bound peptides presented on monoallelic EBV-transformed B cells transduced with a minigene encoding a 30-mer peptide. The S37F mutation was positioned in the center flanked by 14–15 amino acids corresponding to the CTNNB1WT sequence to allow for natural processing. Results from step 1 were next used to design a targeted MS assay for tumor cells with endogenous expression of candidate pMHC complexes. The results showed that the tumor cells presented, on average, 16–101 S37F pMHC complexes on the cell surface. A similar approach was recently used to characterize T cell epitopes for mutant KRAS and TP53 (refs. 30,36,37).
The TCRs showed exquisite specificity for the CTNNB1S37F mutation. The corresponding WT sequences were not recognized even when target cells were loaded with nonphysiologically high peptide concentrations. The TCRs also passed stringent tests for off-target reactivity recently developed in our laboratory38. To this end, TCR specificity was mapped against target cells loaded with libraries of peptides wherein each amino acid in the cognate epitope was replaced with all other amino acids. The results did not reveal cross-reactive peptides in the human proteome. To test for potential recognition of peptides unrelated to the neoantigen, we cocultured TCR-engineered T cells with a large panel of cell lines of various tissue origins. These cell lines, coexpressing CTNNB1WT and HLA-A*02:01 or HLA-A*24:02 in addition to a multitude of other HLA alleles, did not activate TCR-engineered T cells. Collectively, the results from in vitro experiments supported that the TCRs are highly peptide and HLA specific.
To assess the prevalence of the CTNNB1S37F mutation in human cancer, we accessed data from the MSK-MET study. This dataset contains gene sequence information from 25,775 samples of primary and metastatic cancers23. The highest number of cases was found in endometrial cancer, non-small cell lung cancer and prostate cancer. Considering frequencies of HLA-A*02:01 and HLA-A*24:02 in the US population (35.48% and 18.82%, respectively), 3,885 individuals in the United States are annually estimated to express pMHC that could be targeted by CTNNB1S37F-reactive TCRs (Supplementary Data 1a). These alleles are expressed at even higher frequencies in individuals of European ancestry (HLA-A*02:01: 47.8%) and in several Asian populations (HLA-A*24:02: Japan = 59.6%, South Korea = 39.2%, China = 28.2% and India = 24.6%). In a phase 1 trial, however, only individuals with metastatic, refractory cancer in good performance status after exhausting standard therapies would be eligible for inclusion, considerably reducing participant numbers. Recruiting individuals for clinical trials with TCR-T cell therapies targeting shared mutations, like CTNNB1S37F, TP53R175H or KRASG12V, is therefore challenging, as the frequencies of these mutations in combination with the HLA alleles that the corresponding mutated peptides are presented on are low in the overall population. However, recent trends and initiatives in precision medicine, such as DRUP-like trials31,32, could facilitate participant inclusion. Such trials aim to evaluate the toxicity and efficacy of commercially targeted anticancer drugs repurposed in individuals with advanced cancer where a potentially actionable mutation is detected by next-generation sequencing, which also facilitates extraction of HLA type33. Within such infrastructures, individuals expressing mutation/HLA combinations for which TCRs have been identified could also be assigned to TCR-T cell trials for treatment with one or more TCRs.
Shared driver mutations typically occur at an early stage in tumorigenesis and are subject to positive selection during disease progression39,40. If expressed as mutated peptides presented on HLA molecules, they represent attractive targets for off-the-shelf immunotherapy because the proteins are ubiquitously expressed in the tumor11. Although a recent clinical trial using personalized cancer vaccines targeting both private and recurrent driver mutations in renal cell carcinoma demonstrated no recurrence at a median follow-up of 40.2 months (ref. 41), another study in individuals with advanced/metastatic solid tumors showed limited clinical benefit from the combination of a vaccine encoding 20 shared driver mutations and ICB42. By comparison, vaccines encoding participant-specific neoantigens have shown promise in reducing metastatic events in melanoma43,44,45 and delaying relapses in pancreatic cancer46. The occurrence of negative selection and neoantigen depletion in untreated cancers was recently investigated with little evidence pointing at negative selection of either driver or passenger mutations, suggesting that either few predicted neoantigens are presented and truly immunogenic or that efficient immune evasion mechanisms enable tumor development47,48. It seems possible that shared driver mutations are more prone to induce immune tolerance or exhaustion because they occur at a stage when the tumor induces little or no inflammation and have the possibility to interact with the immune system the longest. Poor immunogenicity or tolerance may explain why the mutations are shared in the first place and why they are not subject to immune editing. Indeed, although evidence is challenging to obtain in humans, mouse studies have provided direct evidence for cancer-induced tolerance of tumor-specific T cells49,50. It is worth noting that we had to screen 16 healthy donors (6 HLA-A*24:02+ and 10 HLA-A*02:01+ donors) to identify TCRs reactive to the CTNNB1-S37F mutation. A high number of donors was needed to also identify TCRs targeting the FLT3-D835Y mutation in acute myeloid leukemia14. Collectively, these results show that tolerance can be overcome by ‘outsourcing’ the immune response to healthy donors who have not been tolerized to neoantigens24. However, screening methods with higher throughput are needed.
Therapeutic targeting of β-catenin has proven challenging. Small-molecule inhibitors have been identified, but none of these have progressed beyond the preclinical stage18,19. Results from this study show that TCR-engineered T cells may be an attractive alternative. Together, TCR-T cells targeting the shared CTNNB1-S37F mutation can efficiently recognize and eradicate mutant cancer cells, targeting a pathway that is important for initiation and propagation of a multitude of solid cancers. Because no mutation is likely to be present in all cancer cells, such TCR-T cells in combination with other mutation-specific TCR-T cells or other therapeutic modalities could provide a promising immunotherapeutic strategy.
Methods
Healthy blood donor cells and cell lines
HLA-typed PBMCs from healthy donor buffy coats were obtained from the Blood Bank, Oslo University Hospital. Mononuclear cells were isolated by density-gradient centrifugation (Axis-Shield). The study was approved by the Regional Committee for Medical and Health Research Ethics (REK) South-East Norway, the Institutional Review Board and the Data Protection Officer, Oslo University Hospital. Informed consent was obtained per the Declaration of Helsinki and institutional guidelines (REK 2018/2006 and 2018/879). Cell lines were from ATCC (CFPAC-1, CHP-212, KLE, K562, HEK293, HEL92.1.7, HCT116, Hutu80, HepG2, RD, PANC-1, EA.hy926, Daoy, T2 and Phoenix AMPHO), German Collection of Microorganisms and Cell Cultures (Jurkat, MOLM-13, MOLT-4, EOL-1, MV4-11, Reh, RS4;11, THP-1, SET2, OCI-M2 and UT-7) or Korean Cell Line Bank (SNU-638) or were kindly gifted (BV-173, J. Myklebust; HaCat, F. Jansen; Colo668, F. Lund-Johansen; Caco2, R. A. Lothe (all Oslo University Hospital); Mel888, D. Peeper, (Netherlands Cancer Institute)). CTNNB1WT or CTNNB1S37F status was obtained from Cellosaurus (RRID: SCR_013869) and Depmap (https://depmap.org/portal/). HLA-typed lymphoblastoid and EBV-transformed B-LCL 721.221 (B721.221) cells were from Fred Hutchinson Cancer Center Research Cell Bank. Cell lines were cryopreserved, with only vials from generation 1–4 used. Cell lines that were not first generation were short tandem repeat profiled (Labcorp DNA Identification Lab, https://celllineauthentication.labcorp.com/). Cultures were maintained as per the supplier’s instructions at 37 °C with 5% CO2 and regularly tested for mycoplasma.
Organoids
The HCM-SANG-0270-C20 organoid, generated by the Human Cancer Models Initiative and obtained from ATCC (PDM-47), was cultured in Cell Basement Membrane (ATCC) with ATCC Organoid Media Formulation 4. The medium was supplemented with 10 µM ROCK Inhibitor Y-27632 (ATCC) for 2–3 days after subculture. The 922882-316-R organoid was generated in-house from the 922882-316-R PDX, obtained from the National Cancer Institute Patient-Derived Models Repository, in vivo. Tumor tissue was dissociated into single cells using DMEM with 1 mg ml−1 hyaluronidase (Sigma, H3506), 1 mg ml−1 collagenase type IV (Sigma, C5138-1G), 10% fetal bovine serum (FBS) and 50 µg ml−1 DNase I (Sigma, DN25-100MG), with mechanical disruption. Tumor samples were incubated at 37 °C and 200 rpm for 30 min with pipetting every 10 min. Digestion was stopped with PBS supplemented with 2% FBS, followed by centrifugation and incubation in 2 mg ml−1 Dispase II Buffer (Sigma, D4693-1G) for 10 min at 37 °C. After a final wash with PBS/2% FBS and centrifugation, cells were filtered through a 40-µm strainer, counted and plated in Cultrex Reduced Growth Factor Basement Membrane Extract, Type 2 (R&D Systems) with NCI complete medium type 6E. Organoids were maintained at 37 °C and 5% CO2 and were regularly tested for mycoplasma.
Minigene design
The minigene was constructed to identify naturally presented neopeptides containing CTNNB1-S37F encoded by the amino acid sequence SHWQQQSYLDSGIHFGATTTAPSLSGKGNP, validated by protein alignment using the NCBI web-based tool. A full-length FASTA file for the WT protein sequence was retrieved from UniProt, and the mutation sequence was obtained from the COSMIC database20. The minigene was codon optimized, synthesized and subcloned into pMSCV-IRES-EGFP vector via EcoRI and BamHI sites by GenScript.
MS analysis of peptides presented on HLA
B721.221 cells with stable monoallelic expression of HLA-A*24:02 or HLA-A*02:01 were transduced to express the CTNNB1S37F minigene. Sorting by FACS ensured high HLA and minigene expression before expansion to 100 × 106 cells for immunopeptidomics analysis. HLA-bound peptides were purified as previously described14. Briefly, Strep-Tactin magnetic XT beads (200 µl; IBA Lifesciences) were washed with 500 µl each of 0.1 M Tris-HCl (pH 8) and PBS (pH 7.4), followed by a 1-h incubation with 1 ml of cell lysate at 4 °C. Beads were washed with 500 µl of PBS (2×) and 0.1 M Tris-HCl (1×), and HLA-bound peptides were eluted with 1% trifluoroacetic acid and desalted14.
Peptide solutions (5 µl) were analyzed on an Ultimate 3000 nano-UHPLC system (Dionex) connected to a Q Exactive mass spectrometer (ThermoElectron) with a nanoelectrospray source. Separation used an Acclaim PepMap 100 C18 column (3-µm beads, 100 Å, 75-μm inner diameter, 50-cm length) at 300 nl min−1. The gradient was 10–35% B for 95 min, 50% B for 18 min and 80% B for 5 min (A: 0.1% formic acid; B: 0.1% formic acid/90% acetonitrile). The mass spectrometer operated in data-dependent mode, alternating MS and MS/MS acquisition. Full MS spectra (m/z of 350–1,650) were acquired at a resolution of 70,000 (m/z of 200), with a 3 × 106 ion accumulation target and 20-ms max accumulation time. The top 12 ions (>1 × 105) were isolated (m/z = 2 without offset) and fragmented using higher-energy collision dissociation at a resolution of 17,500 and normalized collision energy of 27, with 5 s of dynamic exclusion and 120 ms of maximum MS/MS ion accumulation. Data were analyzed using PEAKS (Bioinformatics Solutions).
Quantification of CTNNB1 epitopes by parallel reaction monitoring
A total of 2.7 × 108 Mel888 and 4 × 108 Hutu80 cells were processed as previously described14. Peptides that were pulled down were purified and dissolved in 25 μl of 3% acetonitrile/0.1% trifluoroacetic acid. Each sample was spiked with 20 ng of heavy isotope-labeled peptides (SYLDSGI[13C6,15N]HF, SYLDSGI[13C6,15N]HS, YLDSGI[13C6,15N]HFGA and YLDSGI[13C6,15N]HSGA). Samples were analyzed using a Q Exactive mass spectrometer (ThermoElectron) and a parallel reaction monitoring method14. For determining the limit of detection/quantification, a serial dilution of heavy isotope-labeled peptides (10 fg to 10 ng) targeted the [M + 2H]2+ ions of the target peptides (Supplementary Table 1). The following parallel reaction monitoring settings were used: resolution of 30,000, target accumulation of 2 × 105, normalized collision energy of 27, isolation window of 2.0 m/z and 120-ms max ion accumulation for MS/MS. Raw data were analyzed using Skyline (MacCoss Lab Software), and peptide quantification was performed using different sets of MS2 transitions per peptide (Supplementary Table 1). Molecules per cell were calculated using Avogadro’s number30.
Induction of antigen-specific T cells
Monocyte-derived dendritic cell priming and isolation of HLA-A*24:02–CTNNB1-S37F-reactive T cells were performed as previously described14,52,53. Naive CD8+ T cells were primed using artificial antigen-presenting cells (APCs) coated with anti-CD28 (clone CD28.2, BioLegend) and in-house biotinylated HLA-A*02:01 pMHC molecules on DynaBeads M-450 streptavidin (Thermo Fisher), as previously described24,54. Coculture was performed in TexMACS medium with 1% penicillin–streptomycin and IL-21 (30 ng ml−1; PeproTech). After 10–12 days, cultures were screened for pMHC multimer+ cells. pMHC multimers, labeled with dual streptavidin fluorochromes (PE, PE-Cy7, CF594, BV605, BV421 or APC-R700), were prepared in-house54,55. Neoepitope-reactive CD8+ T cells were identified as live, CD8+ T cells double-positive for two distinct pMHC multimers and negative for all others.
Sorting pMHC multimer+CD8+ T cells
Wells with pMHC multimer+ cells were collected and stained with LiveDead Fixable Near-IR, anti-human CD8, PE- and APC-conjugated neoantigen pMHC multimers and BV421- and BV605-conjugated UV pMHC multimers. Live CD8+ T cells, double positive for neoantigen and negative for UV pMHC multimers, were sorted by FACS into single-cell preparations in 96-well PCR plates containing lysis buffer for TCR sequencing.
TCR sequencing and cloning
Paired TCRα and TCRβ sequences from single cells were obtained by three nested PCRs using multiplexed primers covering all TCR V genes, following Han et al.56 with minor modifications57. Briefly, cDNA synthesis and the first PCR reaction were performed in two separate steps. Primer sequences and cycling conditions are provided in the original protocol56. The single-cell TCR library was sequenced using paired-end 300-bp Illumina MiSeq sequencing. MiXCR was used for data analysis, and ImmuneScape VDJassembler was used for full-length TCR reconstruction. Each TCR was manually verified in the IMGT database. Variable TCRα and TCRβ fragments were codon optimized, synthesized and cloned into pMP71 retroviral vector (GenScript).
Gene transfer to human PBMCs, cell lines and organoids
TCRs specific for the CTNNB1S37F mutation were transduced into healthy donor-derived PBMCs for both in vitro and in vivo experiments as previously described58. T cells from the same donor were transduced with the NY-ESO-1-specific TCR (TCR1G4) as an irrelevant TCR control for in vivo experiments. Briefly, PBMCs were resuspended at 2 × 106 cells per ml in TexMACS with 1% penicillin–streptomycin, IL-7 and IL-15 (5 ng ml−1 each; PeproTech), added to tissue-culture-treated plates coated with anti-CD3 (OKT3, eBioscience) and anti-CD28 (28.6, eBioscience) and incubated at 37 °C and 5% CO2. Retroviral supernatants were generated as previously described58. After 72 h, PBMCs were collected, resuspended in fresh supplemented TexMACS (1% penicillin–streptomycin, IL-7 and IL-15 (5 ng ml−1 each; PeproTech)) and transduced by mixing 1:1 with retroviral supernatant on RetroNectin-coated (20 µg ml−1; Takara) nontreated six-well plates, followed by spinoculation at 3,000 rpm for 60 min, repeated the next day. TCR-transduced PBMCs were used fresh or frozen for later use. Before experiments, cells were cultured for 48–72 h after feeding, and a fraction was stained with anti-mTCRβ and pMHC multimer to assess transduction efficiency by flow cytometry.
HLA monoallelic B721.221 cell lines were generated by retroviral transduction of HLA class I-null cells to stably express single HLA class I alleles, as previously described59. Cell lines BV-173, HaCat, UT-7, OCI-M2, K562, HEL 92.1.7, COLO668, T2, HCT116, RD, Daoy, HEK293, Caco2, MV4-11, MOLM-13, SET2, Jurkat, MOLT-4, Reh, THP-1 and Hutu80 were transduced with HLA-A*24:02 tagged with eGFP. Cell lines EA.hy926, HaCat, K562, HEL 92.1.7, COLO668, RD, RS4;11, MV4-11, EOL-1, MOLM-13, Jurkat, MOLT-4, Reh, SNU-638 and Mel888 were transduced with the HLA-A*02:01 allele, with or without eGFP tagging. Transductions used retroviral supernatants encoding full-length HLA alleles. For in vivo experiments, Mel888 cells were stably transduced to express HLA-A*02:01, ffluc and eGFP, designated Mel888A24 + A2 + ffluc–eGFP. B721.221 monoallelic cell lines were also transduced with an eGFP-tagged minigene. All transduced cell lines were sorted by FACS based on GFP expression or with anti-HLA-A/HLA-B/HLA-C (W6/32) or anti-HLA-A2, expanded and cryopreserved.
The HCM-SANG-0270-C20 organoid was transduced to express both HLA-A*02:01 and HLA-A*24:02 (tagged with eGFP). Organoids were isolated from Matrigel and digested with TrypLE into small clusters of 5–10 cells, washed and resuspended in complete organoid medium with 10 µM ROCK inhibitor Y-27632 to prevent detachment-induced cell death. Transductions followed the same procedure as described above, with spinoculation at 600g for 1 h. After 24–48 h, the supernatant was removed, and organoids were collected, washed, resuspended in Matrigel and plated as domes in six-well plates. Fresh medium with 10 µM ROCK inhibitor was added after solidification.
pMHC stability assay
The pMHC stability assay was performed as previously described14,24. Briefly, UV-mediated peptide exchange was performed for 1 h, and products were incubated at 4 °C overnight. DynaBeads M-450 streptavidin (Thermo Fisher) were washed twice in PBS supplemented with 1% Tween and coated with pMHC monomers at room temperature with shaking at 1,850 rpm (Mixmate). After coupling, beads were washed twice, resuspended in 200 µl of TexMACS and incubated at 37 °C. At 0, 3, 6 and 12 h, 20-µl aliquots were stained with anti-β2-microglobulin (B2M-01, MediQip/Exbio) for 10 min at room temperature, shaken at 1,850 rpm (Mixmate), washed and immediately analyzed by flow cytometry.
Antibodies, dyes and flow cytometry
Flow cytometry was performed on a FACSymphony A5, LSR II or LSR Fortessa (all BD Biosciences), bulk cell sorting was performed on a SONY SH800 (SONY Biotechnology), and single-cell sorting was performed on a FACSAria II cell sorter (BD Biosciences) for single-cell sorting. Data analysis was performed with FlowJo v10.6.2 (TreeStar). For surface staining, cells were incubated with antibodies for 15–20 min at 4 °C and washed. For mouse blood, red blood cells were lysed using ammonium–chloride–potassium buffer (Life Technologies) before staining. Mouse tumors were dissociated into single-cell suspensions by enzymatic digestion with 1 mg ml−1 hyaluronidase (Sigma, H3506), 1 mg ml−1 collagenase IV (Sigma, C5138-1G), 50 µg ml−1 DNase I (Sigma, DN25-100MG) and 10% FBS in DMEM/PBS, followed by mechanical disruption. Tumors were incubated at 37 °C for a maximum of 1 h, filtered, washed and Fc blocked (Nordic Biosite) for 15 min at room temperature before antibody staining. Fluorescent antibodies (BD Biosciences or BioLegend, unless otherwise specified) targeted human CD45 (HI30), CD3 (UCHT1), CD4 (SK3), CD8 (RPA-T8), CD279 (PD-1; J105, eBioScience), CD366 (TIM-3; 7D3), TIGIT (741182), CD112 (TX31), CD155 (SKII.4), CD274 (PD-L1; MIH1), HLA-A*02 (BB7.2), CD326 (EpCAM; MPC-11), CD24 (ML5), β2-microglobulin (B2M-01, MediQip AB/Exbio), HLA-A/HLA-B/HLA-C (W6/32), CD137 (4B4-1, eBioscience) and HLA-A*24 (17A10, MBL/Nordic Biosite) and mouse CD45 (30-F11) and TCRβ chain (H57-597). Live/Dead Fixable Near-IR (Life Technologies) excluded dead cells. Carboxyfluorescein succinimidyl ester (CFSE) or CellTrace Violet (CTV; Life Technologies) labeled target cells for T cell activation assays.
T cell activation assays
TCR-transduced T cell reactivity was evaluated by the upregulation of CD137 expression via flow cytometry. Transduced T cells were cocultured with target cell lines or autologous PBMCs in U-bottom 96-well plates at an E:T ratio of 1:2 (50,000:100,000 cells per well). For peptide titration assays, B721.221 cells expressing HLA-A*02:01 or HLA-A*24:02 were peptide loaded. For comparison with TCR1G4 and TCRHIV, BV-173 cells were used, as TCR1G4 showed background reactivity to B721.221 cells. GFP− target cells were labeled with 0.75 µM CFSE or CTV to separate them from effectors. Where indicated, target cells were electroporated with mRNA encoding a GFP-tagged 30-mer containing the relevant peptide or were peptide loaded for 2 h, followed by two washes before coculture. After 16–18 h of co-incubation, cells were washed twice and stained to measure CD137 upregulation on live CD3+GFP−CFSE−CTV−CD8+mTCRβ+ cells. Results are presented as the percentage of CD137+ CD8+ mock-transduced or mTCRβ+CD8+ T cells.
Incucyte-based cytotoxicity assay using cell lines as targets
For cytotoxicity assays, triplicate wells of GFP-transduced target cells (8,000 cells per well) were plated in 96-well flat-bottom plates with appropriate medium and incubated overnight. The following day, TCR-transduced T cells in TexMACS were added at E:T ratios from 8:1 to 1:1, assuming tumor cell doubling overnight. Plates were placed in an Incucyte (Essenbiosciences) for real-time imaging over 48 h, with GFP and phase read every 2 h. Cytotoxicity was measured as the reduction in GFP+ area over time. For repetitive killing assays, cocultures were repeated every 42 h. After each cycle, T cells were collected by gentle pipetting (ensuring that adherent cancer cells did not detach), washed with PBS and added to fresh targets at the same E:T ratio.
Flow cytometry-based cytotoxicity assay using organoids as targets
Tumor organoid–T cell cocultures followed a modified Cattaneo et al.60 protocol. Organoids were isolated from Matrigel, and a fraction was dissociated for cell counting. Organoids (30,000–100,000 single-cell equivalents per well) were plated in complete organoid medium with 10 µM ROCK inhibitor (Y-27632, ATCC) in 96-well flat-bottom plates. TCR-transduced T cells in TexMACS were added at a 2:1 E:T ratio, with a 1:1 mix of organoid and TexMACS medium. After 48 h, cells were washed twice with PBS, and organoids were dissociated in TrypLE for 5–15 min, with mixing every 5 min. Dissociated cells were washed, stained to distinguish organoids from T cells and resuspended in FACS buffer containing CountBright beads (Thermo Fisher), with 3,500 bead events recorded per well. HCM-SANG-0270-C20 organoids were gated as live CD3−GFP+ cells, and 922882-316-R organoids were gated as live CD3−EpCAM+CD24+ cells. Viable tumor cell counts were normalized to mock- or TCR1G4-transduced controls and are reported as percent viability.
In vivo activity of TCRA2-2 and TCRA24 T cells in a melanoma xenograft model
Animal work was approved by the Norwegian Food Safety Authority (application ID: 29569) and conducted following institutional guidelines and the 2010/63/EU directive on animal welfare. Mice (six per cage) were housed in Eurostandard type III macrolone cages with a 7 a.m. to 7 p.m light cycle at 22 ± 1 °C with 62 ± 5% humidity. In-house-bred 8- to 10-week-old female NXG mice were subcutaneously injected with 2 × 106 Mel888A24 + A2 + ffluc–eGFP cells. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications14,30,58. Tumor engraftment was followed by BLI (Spectrum In Vivo Imaging System) to allocate the mice to control or treatment groups (tumor only or T cells transduced with mock, TCR1G4, TCRA2-2 or TCRA24), ensuring similar mean engraftment before T cell infusion. Freshly TCR-transduced T cells were fed every 2–3 days with fresh TexMACS medium plus 1% penicillin–streptavidin and IL-7/IL-15 (5 ng ml−1) for 10 days before treatment. A total of 10 × 106 T cells were injected per mouse via tail vein. All mice received daily intraperitoneal injections of 2,500 IU human IL-2 (R&D Systems) until day 24 after T cell injection; injections were then reduced to three times per week from day 24 and were stopped on day 34. TCR-transduced T cell numbers in blood were tracked by weekly flow cytometry. Tumor size and T cell infiltration were assessed at the time of euthanasia. Tumors were sampled at euthanasia or experimental termination (day 115) for flow cytometry. Data collection and analysis were not performed blind due to the conditions of the experiments. Mice were monitored for pain, with humane endpoints defined as >20% weight loss or a tumor size of >400 mm3. All mice were humanely killed.
In vivo activity of TCRA2-2 T cells in an endometrial adenocarcinoma PDX model
Animal work was approved by the Norwegian Food Safety Authority (application ID: 31025) and was conducted under institutional and 2010/63/EU animal welfare guidelines. Mice (six to eight per cage) were housed in Eurostandard type III macrolone cages under the same light, temperature and humidity conditions described above. The PDX 922882-316-R was obtained from the Patient-Derived Models Repository. In-house-bred 8- to 10-week-old female NXG mice were subcutaneously implanted with 17ꞵ-estradiol pellets (Innovative Research of America, NE-121) and 2- to 3-mm tumor fragments using aseptic surgery. Sample sizes were determined as detailed above. Tumor engraftment was followed by palpation and caliper measurement. On day –1, tumor volume was measured, and nine mice were evenly allocated into control or treatment groups (tumor only or T cells transduced with TCR1G4 or TCRA2-2). Freshly TCR-transduced T cells were prepared as described above. Each mouse received 15 × 106 T cells via tail vein injection. To reduce bias, injections were performed by a blinded operator. All mice received intraperitoneal injections of 2,500 IU human IL-2 (R&D Systems) three times a week for 2 weeks and then twice weekly until day 33. Two mice in each group were excluded from analysis due to insufficient engraftment. TCR-transduced T cell percentages in blood were monitored weekly by flow cytometry. All mice were humanely killed at the experimental endpoint.
Estimation of mutation prevalence
CTNNB1S37F prevalence was estimated from 25,775 primary (n = 15,632) and metastatic (n = 10,143) human samples across 50 tumor types sequenced using the MSK-IMPACT panel (MSK-MET study23). Mutation prevalence by cancer type is displayed as percentage. Analysis was performed in R v4.3.1. The same approach was used for KRASG12V and TP53R175H. Annual US patient numbers by cancer type were estimated from 2024 American Cancer Society data61. HLA genotype frequencies were obtained from http://www.allelefrequencies.net, with phenotype frequencies calculated using the Hardy–Weinberg equation. Estimated phase 1 trial-eligible participant numbers were based on the 2024 Cancer in Norway report34 and discussions with urological, gynecological and thoracic oncology experts.
Software and statistical analysis
Statistical analyses were performed using GraphPad Prism v.9 (GraphPad Software). Survival was assessed by log-rank (Mantel–Cox) test. One-way ANOVA with a Tukey’s multiple comparisons test was used for comparing more than two groups. For the EAC PDX model, endpoint tumor volumes (day 56) were compared by one-way ANOVA, with a Tukey’s post hoc test for pairwise differences if significant. Statistical significance was set at P < 0.05. Binding affinity and MS eluted ligand rank were predicted using NetMHCpan v4.1, comparing eluted ligand ranks to a set of random natural peptides21. Sequence logos for HLA-A*02:01 and HLA-A*24:02 motifs were generated by exporting human 9- to 10-mer linear epitope datasets from the Immune Epitope Database (https://www.iedb.org/), filtered by HLA restriction. Logos were created with Seq2Logo v2.0, using the following color codes: polar (NQSGTY, green), acidic (DE, red), basic (RKH, blue) and hydrophobic (AVLIPWFM, black)51.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Datasets accessed for this study include UniProtKB/Swiss-Prot and the Protein Data Bank via the ScanProsite tool (https://prosite.expasy.org/scanprosite/), the COSMIC database (Tate et al.20; https://cancer.sanger.ac.uk/cosmic/login, CTNNB1), the pMHC class I binding prediction tool NetMHCpan v4.1 (ref. 21; https://services.healthtech.dtu.dk/services/NetMHCpan-4.1/), the AACR Project GENIE (GENIE v15.1-public15; https://www.cbioportal.org/), the Immune Epitope Database (10- and 9-mer epitopes for HLA-A*02:01 and HLA-A*24:02 from human hosts) accessed via https://www.iedb.org/result_v3.php, the Allele Frequency Net Database (http://www.allelefrequencies.net/hla6015a.asp?hla_locus=A&hla_country=United+States) and data from Nguyen et al.23 (https://www.cbioportal.org/study/summary?id=msk_met_2021), Siegel et al.61 and the Cancer Registry of Norway34. MS proteomics data were deposited in ProteomeXchange via PRIDE62 under accession code PXD054276. Source data are provided with this paper.
References
Matsushita, H. et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482, 400–404 (2012).
Castle, J. C. et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 72, 1081–1091 (2012).
Rizvi, N. A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).
Le, D. T. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017).
Snyder, A. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199 (2014).
Borch, A. et al. IMPROVE: a feature model to predict neoepitope immunogenicity through broad-scale validation of T-cell recognition. Front. Immunol. 15, 1360281 (2024).
Parkhurst, M. R. et al. Unique neoantigens arise from somatic mutations in patients with gastrointestinal cancers. Cancer Discov. 9, 1022–1035 (2019).
Foy, S. P. et al. Non-viral precision T cell receptor replacement for personalized cell therapy. Nature 615, 687–696 (2023).
Parkhurst, M. et al. Adoptive transfer of personalized neoantigen-reactive TCR-transduced T cells in metastatic colorectal cancer: phase 2 trial interim results. Nat. Med. 30, 2586–2595 (2024).
Pearlman, A. H. et al. Targeting public neoantigens for cancer immunotherapy. Nat. Cancer 2, 487–497 (2021).
Makohon-Moore, A. P. et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 49, 358–366 (2017).
Leidner, R. et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N. Engl. J. Med. 386, 2112–2119 (2022).
Kim, S. P. et al. Adoptive cellular therapy with autologous tumor-infiltrating lymphocytes and T-cell receptor-engineered T cells targeting common p53 neoantigens in human solid tumors. Cancer Immunol. Res. 10, 932–946 (2022).
Giannakopoulou, E. et al. A T cell receptor targeting a recurrent driver mutation in FLT3 mediates elimination of primary humanacute myeloid leukemia in vivo. Nat. Cancer 4, 1474–1490 (2023).
AACR Project GENIE Consortium. AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discov. 7, 818–831 (2017).
Rubinfeld, B. et al. Stabilization of β-catenin by genetic defects in melanoma cell lines. Science 275, 1790–1792 (1997).
Gao, C. et al. Exon 3 mutations of CTNNB1 drive tumorigenesis: a review. Oncotarget 9, 5492–5508 (2018).
Yu, F. et al. Wnt/β-catenin signaling in cancers and targeted therapies. Signal Transduct. Target. Ther. 6, 307 (2021).
Cui, C., Zhou, X., Zhang, W., Qu, Y. & Ke, X. Is β-catenin a druggable target for cancer therapy? Trends Biochem. Sci. 43, 623–634 (2018).
Tate, J. G. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019).
Reynisson, B., Alvarez, B., Paul, S., Peters, B. & Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 48, W449–W454 (2020).
Robbins, P. F. et al. A mutated β-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J. Exp. Med. 183, 1185–1192 (1996).
Nguyen, B. et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 185, 563–575 (2022).
Stronen, E. et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 352, 1337–1341 (2016).
Ali, M. et al. Induction of neoantigen-reactive T cells from healthy donors. Nat. Protoc. 14, 1926–1943 (2019).
Robbins, P. F. et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 29, 917–924 (2011).
Shimizu, A. et al. Structure of TCR and antigen complexes at an immunodominant CTL epitope in HIV-1 infection. Sci. Rep. 3, 3097 (2013).
Bethune, M. T. et al. Isolation and characterization of NY-ESO-1-specific T cell receptors restricted on various MHC molecules. Proc. Natl Acad. Sci. USA 115, E10702–E10711 (2018).
Rosenberger, L. et al. Selection of therapeutically effective T-cell receptors from the diverse tumor-bearing repertoire. J. Immunother. Cancer 13, e011351 (2025).
Bear, A. S. et al. Biochemical and functional characterization of mutant KRAS epitopes validates this oncoprotein for immunological targeting. Nat. Commun. 12, 4365 (2021).
van der Velden, D. L. et al. The Drug Rediscovery Protocol facilitates the expanded use of existing anticancer drugs. Nature 574, 127–131 (2019).
DRUP the Drug Rediscovery Protocol. https://drupstudy.nl/drup-international-public/ (2025).
Claeys, A., Merseburger, P., Staut, J., Marchal, K. & Van den Eynden, J. Benchmark of tools for in silico prediction of MHC class I and class II genotypes from NGS data. BMC Genomics 24, 247 (2023).
Cancer Registry of Norway. Cancer in Norway 2024—Cancer Incidence, Mortality, Survival and Prevalence in Norway (Norwegian Institute of Public Health, 2025).
Bollineni, R. C., Tran, T. T., Lund-Johansen, F. & Olweus, J. Chasing neoantigens; invite naive T cells to the party. Curr. Opin. Immunol. 75, 102172 (2022).
Hsiue, E. H. et al. Targeting a neoantigen derived from a common TP53 mutation. Science 371, eabc8697 (2021).
Wang, Q. et al. Direct detection and quantification of neoantigens. Cancer Immunol. Res 7, 1748–1754 (2019).
Foldvari, Z. et al. A systematic safety pipeline for selection of T-cell receptors to enter clinical use. npj Vaccines 8, 126 (2023).
Gerstung, M. et al. The evolutionary history of 2,658 cancers. Nature 578, 122–128 (2020).
Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
Braun, D. A. et al. A neoantigen vaccine generates antitumour immunity in renal cell carcinoma. Nature 639, 474–482 (2025).
Rappaport, A. R. et al. A shared neoantigen vaccine combined with immune checkpoint blockade for advanced metastatic solid tumors: phase 1 trial interim results. Nat. Med. 30, 1013–1022 (2024).
Sahin, U. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017).
Weber, J. S. et al. Individualised neoantigen therapy mRNA-4157 (V940) plus pembrolizumab versus pembrolizumab monotherapy in resected melanoma (KEYNOTE-942): a randomised, phase 2b study. Lancet 403, 632–644 (2024).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017).
Sethna, Z. et al. RNA neoantigen vaccines prime long-lived CD8+ T cells in pancreatic cancer. Nature 639, 1042–1051 (2025).
Van den Eynden, J., Jimenez-Sanchez, A., Miller, M. L. & Larsson, E. Lack of detectable neoantigen depletion signals in the untreated cancer genome. Nat. Genet. 51, 1741–1748 (2019).
Kherreh, N., Cleary, S. & Seoighe, C. No evidence that HLA genotype influences the driver mutations that occur in cancer patients. Cancer Immunol. Immunother. 71, 819–827 (2022).
Willimsky, G. & Blankenstein, T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature 437, 141–146 (2005).
Staveley-O’Carroll, K. et al. Induction of antigen-specific T cell anergy: an early event in the course of tumor progression. Proc. Natl Acad. Sci. USA 95, 1178–1183 (1998).
Thomsen, M. C. & Nielsen, M. Seq2Logo: a method for construction and visualization of amino acid binding motifs and sequence profiles including sequence weighting, pseudo counts and two-sided representation of amino acid enrichment and depletion. Nucleic Acids Res. 40, W281–W287 (2012).
Bartok, O. et al. Anti-tumour immunity induces aberrant peptide presentation in melanoma. Nature 590, 332–337 (2021).
Pataskar, A. et al. Tryptophan depletion results in tryptophan-to-phenylalanine substitutants. Nature 603, 721–727 (2022).
Toebes, M. et al. Design and use of conditional MHC class I ligands. Nat. Med. 12, 246–251 (2006).
Hadrup, S. R. et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat. Methods 6, 520–526 (2009).
Han, A., Glanville, J., Hansmann, L. & Davis, M. M. Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat. Biotechnol. 32, 684–692 (2014).
Risnes, L. F. et al. Disease-driving CD4+ T cell clonotypes persist for decades in celiac disease. J. Clin. Invest. 128, 2642–2650 (2018).
Ali, M. et al. T cells targeted to TdT kill leukemic lymphoblasts while sparing normal lymphocytes. Nat. Biotechnol. 40, 488–498 (2022).
Meyer, S. et al. Prevalent and immunodominant CD8 T cell epitopes are conserved in SARS-CoV-2 variants. Cell Rep. 42, 111995 (2023).
Cattaneo, C. M. et al. Tumor organoid–T-cell coculture systems. Nat. Protoc. 15, 15–39 (2020).
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 74, 12–49 (2024).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Acknowledgements
This work was supported by the Research Council of Norway through its Centers of Excellence scheme (332727; J.O. and F.L.-J.) and through grant 316060 (J.O.), the Norwegian Cancer Society grants 216135-2020 (J.O.) and 208247-2019 (F.L.-J.), South-Eastern Regional Health Authority Norway (2021074; J.O.), the European Research Council under the European Union’s Horizon 2020 Research and Innovation Program (grant agreement number 865805; J.O.), the University of Oslo and Oslo University Hospital and the Novo Nordisk Foundation. We are grateful to S.-W. Qiao at the University of Oslo for helpful advice in setting up the assay for single-cell TCR sequencing. We are also thankful for excellent technical assistance from the Oslo University Hospital Flow Cytometry Core Facility and to the Proteomics Core Facility, Department of Immunology, University of Oslo/Oslo University Hospital, supported by the Core Facilities program of the South-Eastern Norway Regional Health Authority, for MS-based proteomic analyses. The Proteomics Core facility is also a member of the National Network of Advanced Proteomics Infrastructure, which is funded by the Research Council of Norway INFRASTRUKTUR program (project number 295910). We thank Å. Helland (Oslo University Hospital and University of Oslo), who leads the ongoing national DRUP-like clinical trial in Norway, IMPRESS-Norway, O. T. Brustugun (Vestre Viken Hospital and the University of Oslo), K. Berner (Head of Section of Urological Oncology, Oslo University Hospital) and K. Lindemann (Oslo University Hospital and University of Oslo) for advice regarding estimates on the potential lung, prostate and endometrial adenocarcinoma populations eligible for a phase 1 trial.
Author information
Authors and Affiliations
Contributions
J.O., M.M.N., M.S.E. and F.L.-J. conceived and designed the study. M.S.E., H.P., N.C., M.M.N., M.L. and I.B. performed in vitro experiments. J.H., M.S.E. and Y.L. performed in vivo experiments. T.T.T. and R.C.B. performed MS experiments. W.Y. performed molecular biology experiments. E.H.R. and M.S.E. performed the analysis of human mutational data. M.D.-S. performed biobanking and administrative work. J.O., M.M.N. and F.L.-J. supervised the study. J.O., M.S.E., M.M.N. and F.L.-J. wrote the paper. All authors reviewed and revised the paper.
Corresponding authors
Ethics declarations
Competing interests
A patent application will be filed by the Oslo University Hospital institutional technology transfer office Inven2 protecting the TCR sequences (J.O., M.M.N. and M.S.E. are inventors). J.O. is on the scientific advisory board of Asgard Therapeutics, and J.O. and F.L.-J. are cofounders of T-Rx therapeutics. The other authors declare no competing interests.
Peer review
Peer review information
Nature Immunology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Nick Bernard, in collaboration with the Nature Immunology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Estimated prevalence of the CTNNB1S37F mutation.
a, Number of unique patient tumor samples in the different cancer types analyzed in the MSK-MET study23. b, Mutation prevalence by cancer type, estimated from cancer specimens reported in the MSK-MET study23. For each cancer type and group, the prevalence is displayed as percentage, with the number of CTNNB1S37F-mutated samples among the total the number of samples analyzed in brackets. In a and b, blue = metastatic cases, orange = primary cases.
Extended Data Fig. 2 Identification of HLA-A2- and HLA-A24-bound CTNNB1S37F-encoded peptides detected from both minigene-transduced and naturally mutated cell lines.
a, Mass to charge (m/z) ratio spectra of CTNNB1-S37F peptides eluted from HLA-A*02:01+ or HLA-A*24:02+ B721.221 cell lines transduced with a minigene containing the CTNNB1S37F mutation. b, Overview of natural and introduced expression of HLA-A*02:01 and HLA-A*24:02 in Mel888A24+A2 and Hutu80A2+A24 cell lines. c, d, Mirror image plots comparing the fragmentation pattern of endogenous CTNNB1-S37F and CTNNB1-WT peptides (blue) eluted from the two cell lines (c) Mel888A24+A2 and (d) Hutu80A2+A24, as compared to their isotopically labelled counterparts (pink). e, Representative retention time images showing targeted quantification analysis of CTNNB1-S37F and corresponding CTNNB1-WT peptides eluted from HLA-A*02:01 and HLA-A*24:02 of Mel888A24+A2 and Hutu80A2+A24 cell lines compared with their cognate spike-in standard peptides. Data shown from one experiment is representative of n = 3 individual experiments. Transition masses listed in Supplementary Table 1. f, Expression levels of HLA-A*02:01 (left) and HLA-A*24:02 (right) in Mel888A24+A2 and Hutu80A2+A24 transduced to express these alleles, as compared to a selection of naturally HLA-A*02:01- and HLA-A*24:02-expressing cell lines. The peak to the left represents background fluorescence whereas the peak to the right shows anti-HLA-staining for each cell line. g, Off-rates for CTNNB1-S37F and CTNNB1-WT peptides binding to HLA-A*02:01 or HLA-A*24:02 coupled to fluorescent particles, assessed by flow cytometry. Each datapoint represents mean fluorescence intensity (MFI) values at indicated timepoints (hours), reflecting the amount of intact pMHC complexes per particle. Vertical lines indicate calculated half-lives. Data shown for one experiment is representative of n = 3 independent experiments with one technical replicate per experiment, and each data point represents one technical replicate.
Extended Data Fig. 3 The CTNNB1-S37F-reactive HLA-A2- and HLA-A24- restricted TCRs recognize mutant but not the corresponding WT peptides.
a, Gating strategy used in Fig. 2a, to identify and sort CD8+ T cells staining double positive for dual-colored pMHC-multimers. b, Gating strategy used in Fig. 2b, c, to identify TCR-transduced cells. Pre-gating was performed as in Extended Data Fig. 3a (three left plots) and show viable CD8+ cells staining positively with anti-mouse TCRβ (middle plot), or with APC- and PE-labeled pMHC-multimers (right plot). c, Activation of CD8+ CTNNB1-S37F TCR-T cells following co-incubation with target cells expressing the relevant HLA in presence or absence of additional loading with the cognate CTNNB1-S37F or CTNNB1-WT peptide for the relevant TCR (1 µM). d, CTNNB1-S37F TCR and control TCR1G4 or TCRHIV T cells were co-incubated with target cells expressing the relevant HLA and pulsed with indicated concentrations of cognate peptide. Activation was measured as percentage of CD137+ cells and the mean EC50 values of the indicated TCRs were calculated. Data are pooled from n = 3 independent experiments and each datapoint represents mean ± s.e.m. of n = 3 different PBMC donors with n = 2 technical replicates. e, Related to Fig. 2e, flow plots showing activation of CD8+ CTNNB1-S37F TCR-T cells following co-incubation with Mel888A24, Hutu80A2, Mel888A24+A2 or Hutu80A2+A24 ± additional loading with the cognate CTNNB1-S37F peptide (100 nM). Data shown for one PBMC donor is representative of n = 2 donors in one experiment.
Extended Data Fig. 4 TCRA2-2 and TCRA24 T cells mediate HLA-restricted killing in vitro of cell lines naturally expressing the CTNNB1S37F mutation.
a, Activation of CTNNB1-S37F TCR-T cells staining negatively for CD8 (considered CD4 T cells) measured by upregulation of CD137 after co-incubation with Mel888A24 or Hutu80A2, or Mel888A24+A2 or Hutu80A2+A24 cells with or without pulsing with the cognate CTNNB1-S37F peptide (100 nM). Data are from the same experiments as shown for CD8+ T cells in Fig. 2e. One independent experiment using n = 2 different T cell donors; bars show mean, and dots represent the average of n = 3 technical replicates. b, Evaluation of the ability of TCRA2-1, TCRA2-2 and TCRA2-3 to persistently kill the Hutu80A2 cell line upon three repeated challenges with tumor cells using IncuCyte live cell imaging. T cells were rechallenged with fresh target cells each 42 h (final timepoint at 42×3 = 126 h). c, Evaluation of TCRA2-2, TCRA24 and TCR1G4 T cell killing of the Hutu80A2 and Mel888A24 cell lines using IncuCyte live cell imaging. Data in b and c represent mean ± s.e.m. of one experiment with n = 3 technical replicates and (b) n = 2 or (c) n = 1 PBMC donors.
Extended Data Fig. 5 Mapping of peptide reactivity does not reveal off-target peptide recognition by TCRA2-2 or TCRA24 in the human proteome.
a, Gating strategy to identify activated TCR-T cells as measured by positive staining with CD137. Target cell lines were either eGFP+ or labelled with CFSE, and CD137+ T cells were identified among hCD3+, hCD8+, eGFP−/CFSE− (mock-transduced T cells), and from hCD3+, hCD8+, mTCRβ+ (TCR-transduced T cells). b, c, Graphs show percentage of CD137+ (b) TCRA2-2 or (c) TCRA24 T cells in response to HLA-A*02:01+ or HLA-A*24:02+ B721.221 cells pre-loaded with individual peptides (10 nM) from the mimotope libraries consisting of 190 and 171 peptides for the two TCRs, respectively. Pink (TCRA2-2) and green (TCRA24) dots indicate response to the cognate CTNNB1-S37F peptide. The substituted amino acid in the original peptide is highlighted in the heading. The cut-off threshold to determine hits was set to 5% of the mean reactivity seen towards the target peptide (indicated by dashed, horizontal lines). Graphs show results presented as mean from n = 2 independent experiments with different T cell donors, and one technical replicate (dot) per peptide and experiment. d, Peptide reactivity patterns for the HLA-A*02:01 (pink) and HLA-A*24:02 (green) CTNNB1-S37F TCR-T cells. The recognized alternative amino acids are indicated in square brackets for each given position in the peptide motifs. The motifs were queried in the ScanProsite tool against the human proteome database.
Extended Data Fig. 6 TCRA2-2 and TCRA24 T cells mediate peptide-specific, HLA-restricted killing in vitro.
a, Gating strategy used in Extended Data Fig. 6b to identify viable PBMCs, B cells and T cells. Transduced T cells from the healthy HLA-A*02:01+ and HLA-A*24:02+ donors were labeled with CTV prior to coculture to distinguish them from the untransduced PBMCs, B cells and T cells from the same donor. CTV− events represent untransduced target cells, while CTV+ events denote effector cells transduced with CTNNB1-S37F TCR (mTCRꞵ+, CD8+) from healthy donor PBMCs. B cells were identified as CD20+, CD45+, and T cells as CD3+, CD45+. To normalize counts between wells, fluorescent beads (10,000) were added into each well and 3,000 beads were acquired for flow cytometry analysis (shown in FSC/SSC plot, upper left corner). b, Viable PBMCs, B cells and T cells after 48 h of coculture with autologous TCRA2-2, TCRA24 or mock-transduced T cells from three HLA-A*02:01+ and HLA-A*24:02+ healthy donors. PBMCs were loaded with peptides for 18 h before coculture with autologous T cells at an E:T ratio of 2:1. Flow cytometry was used to measure viability of target cells, normalized to the mean of mock treated samples. Data is presented as mean of n = 2 or 3 technical replicates of n = 1 experiment, and each dot represents one technical replicate. c, Expansion of healthy donor PBMCs at indicated days following transduction with mock, TCRA2-2 or TCRA24. Data are presented as mean ± s.e.m. of one independent experiment with n = 3 different PBMC donors, one technical replicate.
Extended Data Fig. 7 Frequencies and phenotypes of TCR-T cells in the tumors at point of euthanasia, and HLA expression on tumor cells.
a, Bioluminescence imaging of Mel888A24+A2+ffluc-eGFP melanoma-engrafted mice on day 115 after T cell injection (mock, TCR1G4, TCRA24 or TCRA2-2). Only mouse #2 and #6 (TCRA24), and mouse #7 (TCRA2-2) had tumors at experimental endpoint. GvHD = symptoms of GvHD; Other = eye splinter. b, Gating strategy to identify Mel888A24+A2+ffluc-GFP cells, T cells, and relevant markers in mouse tumor lysates. Mel888A24+A2+ffluc-GFP cells were identified as hCD45−, mDC45− (upper row), and eGFP+, CD155+ (second row). Mel888A24+A2+ffluc-GFP cells were analyzed for staining with anti-HLA-A2, -HLA-A24 and -HLA-A,B,C. T cells were identified as mCD45−, hCD45+, and hCD3+, hCD8+, mTCRβ+ (third row), and analyzed for PD-1, TIGIT, CTLA-4 and TIM-3 expression. Gates were set using FMO stained controls. c, Percentage of HLA-A*02:01+ (top), HLA-A*24:02+ or HLA-A,B,C+ (W6/32) Mel888A24+A2+ffluc-eGFP cells. All tumors were stained for HLA-A*02:01, while only relapsing tumors were stained for HLA-A*24:02 and HLA-A,B,C. d, Percentage of hCD3+hCD8+ T cells relative to Mel888A24+A2+ffluc-GFP cells in the tumor. d–i, Samples with ≥ 20 hCD3+hCD8+ T cells were included for further mTCRꞵ and T cell exhaustion marker analysis. The TCRA2-2- and two of the TCRA24 -treated tumors had too few T cells for analysis, while all control-treated mice were included. Therefore, data are shown for all controls and two TCRA24-treated tumors in e–i. Percentage of (e) mTCRβ+ among CD8+ T cells, and (f) PD-1+, (g) TIGIT+, (h) TIM-3+ or (i) CTLA-4+among mTCRβ+ cells in tumors. (c–i) Data is presented as mean and each dot represents one mouse. (f–i) Expression was compared between the different treatment groups using one-way ANOVA. If significant, pairwise differences were assessed using Tukey’s post hoc tests: P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001. TIGIT: Mock vs. TCR1G4 P = 0.0294. CTLA-4: Mock vs. TCRA24 P = 0.039; TCR1G4 vs. TCRA24 P = 0.0365.
Extended Data Fig. 8 TCRA2-2 and TCRA24 T cells persist in peripheral blood and upregulate exhaustion markers to similar levels as control TCR-T cells in vivo.
a, Gating strategy to identify percentage and numbers of hCD45+ cells, hCD8+ T cells, and exhaustion markers on mTCRβ+ T cells in mouse peripheral blood. The gates for mTCRβ+ cells were set based on mock transduced T cells and the gates for exhaustion markers were set based on fluorescent minus one (FMO) staining. b–f, Percentage of (b) mTCRβ+ cells among hCD8+ T cells, and percentage of (c) PD-1+, (d) TIGIT+, (e) TIM-3+ or (f) CTLA-4+ cells among mTCRβ+ cells in mouse PB at indicated days after T cell injection. (b–f) Each dot represents one individual mouse. A threshold of n = 30 hCD3+, hCD8+, mTCRβ+cells detected in the blood was set as a criterion for sample inclusion for analysis. Data shown are from one experiment. g–j, Plots showing percentages of hCD45+ among all cells in mouse PB at indicated days after T cell injection, in mice treated with (g) mock, (h) TCR1G4, (i) TCRA24, or (j) TCRA2-2 transduced T cells. Mice in g, i, j, with hCD45+ cell frequencies exceeding 20% of total blood cells (indicated by the dotted line) were euthanized due to symptoms of GvHD (Mock #5, TCRA24 #4, TCRA2-2 #6). IL-2 injections were reduced on day 24 and fully stopped on day 34. (g–j) Dots represent individual mice, and data shown are from n = 1 experiment.
Extended Data Fig. 9 TCRA2-2 T cells eliminate patient-derived endometrial adenocarcinoma xenografted tumors in a mouse model and persist in peripheral blood in vivo.
a, b, Gating strategy used in Fig. 5a to identify viable tumor cells: (a) CRC CD3−, eGFP+ cells and (b) EAC EpCAM+, CD24+ cells in single cell suspensions of organoids following coculture with TCR-T cells. Gates for beads used to determine absolute counts are shown in left plots (FSC versus SSC). c, Tumor volume (mm3) measured by caliper at indicated time points after implantation of EAC PDX fragments. Each line represents an individual mouse. d–f, Percentage of (d) hCD45+ among all CD45+ cells (both mCD45+ and hCD45+), (e) hCD8+ among hCD3+hCD45+, and (f) mTCRβ+ among hCD45+ hCD8+ in mouse PB at indicated days after T cell injection in mice treated with TCRA2-2 (top) or TCRA24 (bottom).
Supplementary information
Supplementary Information
Supplementary Tables 1–6.
Supplementary Data 1
a, Estimated annual number of individuals eligible for treatment with a TCR targeting the shared CTNNB1 S37F mutation presented by HLA-A*02:01 or HLA-A*24:02. b, Estimated annual number of individuals in the United States eligible for treatment with a TCR targeting the shared TP53R175H mutation presented by HLA-A*02:01. c, Estimated annual number of individuals eligible for treatment with a TCR targeting the shared KRASG12V mutation presented by HLA-A*03:01 or HLA-A*11:01.
Source data
Source Data Fig. 1
Statistical source data.
Source Data Fig. 2
Statistical source data.
Source Data Fig. 3
Statistical source data.
Source Data Fig. 4
Statistical source data.
Source Data Fig. 5
Statistical source data.
Source Data Extended Data Fig. 2
Statistical source data.
Source Data Extended Data Fig. 3
Statistical source data.
Source Data Extended Data Fig. 4
Statistical source data.
Source Data Extended Data Fig. 6
Statistical source data.
Source Data Extended Data Fig. 7
Statistical source data.
Source Data Extended Data Fig. 8
Statistical source data.
Source Data Extended Data Fig. 9
Statistical source data.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Eggebø, M.S., Heinzelbecker, J., Palashati, H. et al. TCR-engineered T cells targeting a shared β-catenin mutation eradicate solid tumors. Nat Immunol 26, 1726–1736 (2025). https://doi.org/10.1038/s41590-025-02252-1
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41590-025-02252-1
