
Abstract
Research on patients with advanced esophageal squamous cell carcinoma (ESCC) who have progressed on immunotherapy remains limited. BL-B01D1 is a first-in-class antibody–drug conjugate consisting of an EGFR–HER3 bispecific antibody bound to a topoisomerase I inhibitor (Ed-04) payload via a cleavable linker. Here, we present safety and efficacy data from a phase 1 study of BL-B01D1, in 82 patients previously treated for ESCC. The primary endpoint was the recommended phase 2 dose. Administered doses were 2.0 (n = 22) and 2.5 (n = 60) mg kg−1 D1D8 infusion every 3 weeks (Q3W). The confirmed objective response rate (cORR) was 29.3% (24 of 82) in all patients and 32.9% (24 of 73) among evaluable patients. For patients dosed at 2.5 mg kg−1, cORR was 39.6% (21 of 53) and disease control rate was 79.2% (42 of 53). In the 2.0 mg kg−1 group, cORR was 15.0% (3 of 20), and the disease control rate was 50.0% (10 of 20). The phase 2 dose was established at 2.5 mg kg−1 D1D8 Q3W. The incidence of G3 treatment-related adverse events at 2.5 mg kg−1 was 63.3%; most common adverse events were anemia (28.3%), leukopenia and thrombocytopenia (18.3%, each), and neutropenia (16.7%). Two cases of ≥G3 interstitial lung disease were observed. Overall, BL-B01D1 demonstrated promising efficacy and manageable safety in patients with metastatic ESCC. A further phase 3 clinical trial has already been initiated. ClinicalTrials.gov registration: NCT05262491.
Similar content being viewed by others

Main
Esophageal cancer is the eleventh most common malignancy and the seventh most common cause of cancer-related death1. Esophageal squamous cell carcinoma (ESCC) is the most common type of esophageal cancer worldwide2. Immune checkpoint inhibitors (ICIs), such as anti-programmed cell death protein 1 (PD-1), programmed cell death ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4), combined with platinum-based chemotherapy, have become the standard treatment for advanced ESCC and offer significant survival benefits3,4,5,6. However, only 10–20% of patients with ESCC experience long-term survival, as most develop resistance to these drugs7,8,9. Second-line treatment for advanced ESCC involves using irinotecan, which also shows limited efficacy. The objective response rate (ORR) with irinotecan does not exceed 10% (refs. 10,11). Therefore, there is an urgent need to develop new and effective treatment strategies to overcome the limitations of the current therapies and improve survival rates in patients with ESCC.
Epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase. It belongs to the v-erb-b2 erythroblastic leukemia viral oncogene (ErbB)/human epidermal receptor (HER) family, which includes four proteins: EGFR/HER1, ERBB2/HER2, ERBB3/HER3 and ERBB4/HER4 (ref. 12). EGFR overexpression is observed in 50–70% of patients with ESCC and is associated with tumor invasiveness and poor survival outcomes13,14,15,16. Despite the prevalence of EGFR overexpression in patients with ESCC, current anti-EGFR therapies, such as cetuximab, panitumumab and gefitinib, have shown limited benefits in improving overall survival (OS)17,18,19,20. Recent studies have indicated that EGFR and HER3 are frequently coexpressed in various gastrointestinal cancers and are associated with tumorigenesis and progression21,22. HER3 is a unique pseudokinase receptor of the ErbB family and known to mediate resistance to anti-EGFR therapies. Furthermore, preclinical study has found that simultaneous targeting of EGFR and HER3 enables the engagement of both EGFR–EGFR homodimers and EGFR–HER3 heterodimers, thereby blocking their downstream signaling pathways23,24. Thus, targeting both EGFR and HER3 should offer broad-spectrum and pan-tumor-killing therapies for patients with gastrointestinal cancer. Although several drugs targeting EGFR and/or HER3, including bispecific antibodies and antibody–drug conjugates (ADCs), have been reported and some have shown preliminary efficacy in phase 1 and phase 2 studies, none have yet achieved successful outcomes in the ESCC population25,26,27,28,29.
BL-B01D1 is a potential first-in-class ADC consisting of an EGFR×HER3 bispecific antibody bound to a topoisomerase I inhibitor (Ed-04) payload via a cleavable linker30. BL-B01D1 specifically binds to EGFR and/or HER3 on the surface of tumor cells, gains entry through endocytosis and releases Ed-04 into the lysosome after enzymatic cleavage. This, in turn, blocks DNA replication and RNA synthesis and damages the DNA structure, ultimately leading to tumor cell death. In addition, the Fc region of the antibody portion of BL-B01D1 can mediate antibody-dependent cell-mediated cytotoxicity to exert tumoricidal effects30. In cancer-patient-derived tumor xenograft models (derived from lung, colon, pancreas, and so on), the bispecific ADC drug BL-B01D1 showed stronger antitumor effects than any single ADC drug targeting EGFR or HER3 (ref. 30). Furthermore, preliminary data on its safety profile and antitumor activity have been observed in the phase 1 dose escalation and dose expansion study previously published by Ma et al.31. Also, a recommended phase 2 dose (RP2D) of 2.5 mg kg−1 on days 1 and 8 every 3 weeks was suggested for treating advanced solid tumors.
Due to its unique anatomical features and complications arising from surgery or the disease itself, ESCC often presents with a high incidence of malnutrition and poor tolerance to treatment32,33. Here, we selected two dose groups (2.0 mg kg−1 and 2.5 mg kg−1) and explored the suitability of RP2D for patients with ESCC. We present safety, tolerability and efficacy data of BL-B01D1 in previously treated metastatic ESCC from the dose-expansion phase of the BL-B01D1-103 study (ClinicalTrials.gov registration: NCT05262491). These data informed the determination of RP2D in patients with ESCC based on the results from the two dose cohorts.
Results
Participant and baseline characteristics
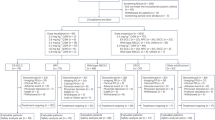
From 12 December 2022 to 4 December 2023, 82 patients with ESCC were enrolled in the study and treated with BL-B01D1. Among them, 22 patients received 2.0 mg kg−1 and 60 were treated with 2.5 mg kg−1 every 3 weeks on days 1 and 8 (Extended Data Fig. 1). The median follow-up time was 10.0 months. As of the data cutoff date on 30 September 2024, 11 patients continued to receive BL-B01D1 treatment. Of the 71 patients who terminated treatment, 40 experienced disease progression, eight experienced treatment-related adverse events (AEs) (TRAEs), one died, ten withdrew voluntarily and 12 had other reasons for discontinuation (Fig. 1). The median age was 62 years (range, 45–75 years) and 89% of the patients were male. Thirty-one (37.8%) patients experienced resistance to first-line treatment and 51 (62.2%) showed disease progression after two or more lines of systemic therapy. Additionally, 78 (95.1%) patients were resistant to combined treatment (anti-PD-1/L1 with chemotherapy) or administered sequentially. Demographic and baseline clinical characteristics of the patients are listed in Table 1.

Consort diagram showing patient enrollment into the study.
Safety
Safety analysis was performed for all 82 patients, with a summary of TRAEs provided in Table 2. Treatment-emergent adverse events (TEAEs) are summarized in Supplementary Table 1. All patients experienced one or more AEs, with common occurrences including anemia (70 (85.4%)), followed by leukopenia and thrombocytopenia (44 patients each (53.7%)), neutropenia and nausea (each with 35 patients (42.7%)) and asthenia (33 (40.2%)). Grade ≥3 TRAEs were noticed in 48 patients (58.5%), with the most frequent being anemia (23 (28.0%)), leukopenia (13 (15.9%)), and thrombocytopenia and neutropenia (12 patients each (14.6%)).
Among the 60 patients receiving the dosing regimen of 2.5 mg kg−1, the most common TRAEs included anemia (51 (85.0%)), followed by thrombocytopenia (35 (58.3%)), leukopenia (34 (56.7%)) and neutropenia and nausea (each with 28 (46.7%) patients). Grade ≥3 TRAEs occurred in 38 patients (63.3%), with the most frequent being anemia (17 (28.3%)), followed by leukopenia and thrombocytopenia (11 patients each (18.3%)) and neutropenia (10 (16.7%)). Due to TRAEs, 26 (43.3%) patients required a dose reduction.
Among the 22 patients receiving the dosing regimen of 2.0 mg kg−1, the most common TRAEs included anemia (19 (86.4%)), leukopenia and asthenia (ten patients each (45.5%)), thrombocytopenia (9 (40.9%)) and neutropenia and nausea (each with seven patients (31.8%)). Grade ≥3 TRAEs occurred in ten patients (45.5%), with the most frequent being anemia (6 (27.3%)), decreased lymphocyte count (3 (13.6%)) and leukopenia and neutropenia (each with two patients (9.1%)). Due to TRAEs, 4 (18.2%) patients required a dose reduction.
Interstitial lung disease (ILD) was observed in three (3.7%) patients. Two of these patients had a history of thoracic radiotherapy and were from the 2.5 mg kg−1 dose group, with one experiencing grade 2 and the other grade 3 ILD. Both of these cases resolved after oral or intravenous glucocorticoid treatment. The remaining one case of Grade 5 ILD occurred in a patient (from the 2.0 mg kg−1 dose cohort) without a history of thoracic radiotherapy. The patient declined further medical evaluations after developing fever and dyspnea. Later, the patient developed mycoplasma and viral infectious pneumonia, which ultimately resulted in severe respiratory failure and death.
Efficacy
Among 82 patients, the confirmed ORR (cORR) was 24 out of 82 (29.3%, 95% confidence interval (CI) 19.7–40.4). A total of 73 patients were evaluable for efficacy according to Response Evaluation Criteria in Solid Tumors (RECIST) v.1.1. The ORR was 35.6% (95% CI, 24.7–47.7) and the cORR was 32.9% (95% CI, 22.3–44.9). The disease control rate (DCR) was 71.2% (95% CI, 59.4–81.2). The median progression-free survival (PFS) (mPFS) was 4.2 months (95% CI, 3.6–5.6), the median overall survival (OS) (mOS) was 6.9 months (95% CI, 5.6–NR (not reached)) and the median duration of response (DOR) (mDOR) was 6.7 months (95% CI, 5.6–12.4; Table 3 and Extended Data Fig. 2). Of the 26 responders defined by the best overall response (BOR), 25 achieved a partial response (PR; 23 were confirmed to have PR) and one achieved a complete response (CR). Additionally, 26 patients had stable disease (SD) and 19 experienced disease progression (PD; Table 3).
A total of 53 out of 60 patients from the 2.5 mg kg−1 dose group were radiologically evaluable. In this group, the cORR was 39.6% (95% CI, 26.5–54.0) and the DCR was 79.2% (95% CI, 65.9–89.2). The mPFS was 5.4 months (95% CI, 4.0–6.9), the mOS was 11.5 months (95% CI, 6.1–NR) and the mDOR was 6.7 months (95% CI, 5.6–12.4) (Table 3). Among them, 23 achieved PR (21 confirmed PR), 19 had SD and ten experienced PD. The maximum change in the target lesion burden from baseline is shown in Fig. 2. The mPFS was 5.0 months (95% CI, 4.0–6.7) and the mOS was 8.3 months (95% CI, 5.6–NR) in all 60 patients of 2.5 mg kg−1 dose group (Fig. 3).

Waterfall plot showing best percentage changes in target lesions from baseline (n = 71), separated according to 2.0 mg kg−1 and 2.5 mg kg−1 dose groups. Dashed lines indicate a 20% increase in tumor size and a 30% decrease in tumor size.

a, Kaplan–Meier estimates of PFS for the 2.0 mg kg−1 (n = 22) and 2.5 mg kg−1 (n = 60) dose groups. b, Kaplan–Meier estimates of OS in the 2.0 mg kg−1 (n = 22) and 2.5 mg kg−1 (n = 60) dose groups.
In the group receiving a 2.0 mg kg−1 dose, 20 patients were evaluable. The cORR was 15.0% (95% CI, 3.2–37.9) and the DCR was 50.0% (95% CI, 27.2–72.8). The mPFS was 2.7 months (95% CI, 1.3–4.1), the mOS was 5.6 months (95% CI, 3.4–6.9) and the mDOR was 4.6 months (95% CI, 2.8–NR) (Table 3). Among these patients, one achieved CR, two achieved PR, seven achieved SD and nine experienced PD. The maximum change in the target lesion burden from baseline is shown in Fig. 2. The mPFS was 2.7 months (95% CI, 1.3–3.6) and the mOS was 5.5 months (95% CI, 3.3–6.6) in all 22 patients of 2.0 mg kg−1 dose group (Fig. 3). In Table 1, patients in the 2.0 mg kg−1 dose group (45.5% of patients had at least three previous lines of therapy) tended to receive treatment in later lines compared to those in the 2.5 mg kg−1 group (23.3% of patients had at least three previous lines of therapy). An additional analysis compared treatment efficacy across different previous lines of therapy between the two dosing groups (Supplementary Table 2).
The RP2D for BL-B01D1 in the ESCC population was selected based on an integrated clinical assessment of safety and efficacy. A comparison between the 2.5 mg kg−1 and 2.0 mg kg−1 groups showed that AEs were manageable in both groups. However, the 2.5 mg kg−1 group demonstrated a clinically meaningful improvement in cORR compared to the 2.0 mg kg−1 group (39.6% (95% CI, 26.5–54.0) versus 15.0% (95% CI, 3.2–37.9)). Therefore, RP2D was established at 2.5 mg kg−1, administered on day 1 and day 8 of a 3-week cycle.
Subgroup analyses were performed based on age, sex, Eastern Cooperative Oncology Group (ECOG) performance status, previous line of therapy, previous systematic treatment, metastatic organs and other variables. No differences in ORR and PFS were observed across the subgroups (Extended Data Fig. 3).
Pharmacokinetics
A total of 43 patients were included in the pharmacokinetics analysis. The immunogenicity data, including anti-drug antibodies, were immature for disclosure. The maximum concentration (Cmax) of total antibody (TAB), ADC and toxin (Ed04) increased with increasing doses (Extended Data Fig. 4). The time to maximum concentration (Tmax) of TAB, ADC and the toxin was relatively consistent between the two dose groups. The half-life (T1/2) of BL-B01D1 ranged from 19.9 h to 21.9 h across doses of 2.0 mg kg−1 to 2.5 mg kg−1 in a single dose. Other pharmacokinetic (PK) parameters included the area under the blood concentration-time curve from 0 to infinity (AUC0–∞) and to the last detectable concentration collection time t (AUC0–t), clearance, terminal elimination rate, lambda (λz) and volume of distribution (Vd) are summarized in the Supplementary Table 3.
Biomarker analysis
Exploratory analyses of EGFR and HER3 expression were included. Tumor tissue samples were collected when available and analyzed by immunohistochemistry (IHC). In the 2.5 mg kg−1 group, tumor specimens were available for 50 out of 60 patients, among which 44 cases were efficacy evaluable. For the remaining ten patients, samples were unable to provide a sufficient number of tumor tissue sections for IHC staining or inadequate tumor cellularity upon pathology review. Nearly all tumors expressed EGFR and HER3 among patients with available pretreatment specimens. No relationship was noticed between EGFR or HER3 expression and antitumor response (Extended Data Fig. 5). Details on sample availability, biomarker evaluation methods and comparative subgroup analyses are provided in Methods and Supplementary Tables 4 and 5.
Discussion
This phase 1 study demonstrated that BL-B01D1, the first-in-human EGFR–HER3 bispecific ADC, exhibited manageable safety and promising efficacy in patients with metastatic ESCC who had progressed after previous standard treatments, including ICIs and chemotherapy. RP2D was established at 2.5 mg kg−1, administered on day 1 and day 8 of a 3-week cycle (D1D8 infusion every 3 weeks (Q3W)).
Recent research on treatment options for patients with advanced ESCC who have become resistant to first-line immunotherapy has focused on exploring alternative combinations and therapies. Due to resistance to immunotherapy, the 5-year survival rate for patients with advanced ESCC receiving first-line ICIs is only 10–20% (refs. 7,8,9). Research on patients with advanced ESCC who have progressed on immunotherapy remains limited. The CAP 02 phase 2 study reported the efficacy and safety of camrelizumab plus apatinib with an ORR of 10.2% and a DCR of 69.4% in patients with advanced ESCC previously treated with first-line ICIs34. Another phase 2 study evaluated the efficacy of radiotherapy combined with camrelizumab and irinotecan in patients with oligometastatic ESCC after first-line immunotherapy plus chemotherapy failure, reporting an ORR of 40.8% and a DCR of 75.5% (ref. 35). In the current study, we present the clinical trial of BL-B01D1 in patients with metastatic ESCC that achieved promising results. At a dose of 2.0 mg kg−1, we observed an cORR of 15.0% and DCR of 50.0%. At the higher dose of 2.5 mg kg−1, the cORR and DCR increased to 39.6% and 79.2%, respectively. A clear correlation between efficacy and BL-B01D1 dosage was evident. Aside from the higher incidence (18.3%) of grade 3 or higher thrombocytopenia in the 2.5 mg kg−1 dose group compared to that in the 2.0 mg kg−1 dose group (4.5%), the remaining AEs were comparable between the two groups and were consistent with previously reported data31. Our findings establish 2.5 mg kg−1 as the RP2D for patients with ESCC. These results notably surpass the efficacy of traditional second-line therapies, such as irinotecan, which typically achieves an ORR of less than 10% (refs. 10,11). Therefore, BL-B01D1 offers an effective therapeutic option for patients who do not respond to, or develop resistance to, immunotherapy, thereby filling a crucial gap in the treatment of ESCC.
Regarding the safety profile, BL-B01D1 demonstrated manageable toxicity. Hematological toxicities such as anemia, leukopenia, thrombocytopenia and neutropenia were the most common AEs with incidence rates ranging from 40% to 85%, and grade 3 or higher AEs with incidence rates of 15% to 30% in all patients and those receiving the 2.5 mg kg−1 dose. These toxicities are similar to those observed for other monoclonal antibodies and ADCs26,28. In the early stages of treatment, myelosuppression was relatively common in patients with ESCC who were receiving BL-B01D1. Therefore, we implemented prophylactic treatment to elevate white blood cell counts during subsequent treatments, which alleviated the related AEs. Other commonly observed AEs include gastrointestinal symptoms (nausea, vomiting and diarrhea) and dermatomucosal toxicities (rash and oral mucositis). These findings are consistent with those reported in preclinical and clinical studies of already marketed small-molecule- and antibody-based EGFR inhibitors36,37. ILD, which was particularly concerning to clinicians, occurred in a total of three patients (3.7%). Two of these patients received the 2.5 mg kg−1 dose and recovered after standardized glucocorticoid therapy. Considering that both patients had a history of radiotherapy, we recommend intensive surveillance of patients with similar treatment history, as they may have increased the risk of complications. One patient who received the 2.0 mg kg−1 dose experienced an ILD-related death due to refusal of medical treatment, eventually succumbing to severe respiratory failure coinfected with mycoplasma and viral pathogens.
Although most patients were found positive for EGFR and HER3 expression, we found no correlation between the expression levels and treatment response to BL-B01D1. Notably, even in patients with EGFR 1+ or HER3 1+ status, the ORRs were 57.1% and 50.0%, respectively. This observation suggests that BL-B01D1 may serve as a potential treatment option for patients with advanced ESCC who do not respond to conventional EGFR monoclonal antibody or tyrosine kinase inhibitors (TKIs) such as cetuximab, panitumumab and gefitinib17,18,19,20. Future studies with larger sample size are needed to identify predictive biomarkers for the response to BL-B01D1 and to enhance the accuracy of patient population selection.
Despite the encouraging results, several limitations should be noted. First, this was an open-label, single-arm study without a control group and, as such, was exploratory by nature. This design limits the ability to directly compare the efficacy of BL-B01D1 with standard treatments. Second, the exploratory biomarker analyses were based on a limited number of evaluable tumor samples, which may restrict the generalizability of the findings. The ongoing phase 3 randomized controlled trial (NCT06304974) is expected to provide more definitive evidence regarding the efficacy of BL-B01D1 and its potential predictive biomarkers.
In summary, BL-B01D1 exhibited manageable safety and encouraging antitumor activity in patients with metastatic ESCC at an RP2D of 2.5 mg kg−1, especially in those who had failed immunotherapy. Future phase 3 clinical trials should further validate the effectiveness and safety of BL-B01D1. Careful attention must be paid to the management of AEs in subsequent investigations.
Methods
Trial design and participants
BL-B01D1-103 (NCT05262491) is an open-label, multicenter, dose escalation and dose-expansion phase 1a/1b study that evaluated BL-B01D1 monotherapy in patients with locally advanced or metastatic gastrointestinal tract tumors and other solid tumors. In the phase 1b (dose-expansion) stage, one or more safe and effective dose groups were selected for an expanded study based on the safety, pharmacokinetics and preliminary efficacy data obtained in phase 1a. The primary aim was to further evaluate the safety, preliminary efficacy and PK characteristics of BL-B01D1 across different tumor types, and ultimately to determine the RP2D.
For the ESCC population, the dose selection (2.0 mg kg−1 and 2.5 mg kg−1) during the dose-expansion phase was based on the following considerations: (1) existing evidence from the previous first-in-human phase 1 study31, in which the 2.5 mg kg−1 dose was proposed as the RP2D based on a comprehensive evaluation of safety, pharmacokinetics and preliminary efficacy across several solid tumor types; (2) clinical and biological factors specific to the ESCC population, including tolerability and disease characteristics (poor tolerance and malnutrition). Patients were sequentially enrolled into one of the two cohorts.
The study protocol and all amendments were approved by institutional review board or independent ethics committee at each center. This trial was performed in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines as defined by the International Conference on Harmonization. All the patients provided written informed consent before participating in the trial. Compensation for participants was provided in accordance with the principles prespecified in the informed consent. The full study protocol is provided in the supplementary file.
Patient eligibility
Each patient eligible for inclusion in this trial had to meet all the following criteria:
-
(1)
Signs the informed consent voluntarily and follows the program requirements.
-
(2)
Either sex.
-
(3)
Age ≥18 years and ≤75 years.
-
(4)
Has a life expectancy of ≥3 months.
-
(5)
Has a pathologically and/or cytologically documented locally advanced or metastatic solid tumor that cannot be cured or for which no standard treatment is available.
-
(6)
Agrees to provide archived or fresh tumor samples from primary or metastatic sites within 2 years. If the subject is unable to provide tumor samples, he/she may still be admitted after evaluation by the investigator, provided other inclusion criteria are met.
-
(7)
Stage 1a must have at least one evaluable lesion; Stage 1b must have at least one measurable lesion as defined by the RECIST v.1.1.
-
(8)
Has a performance status (ECOG PS) of 0–1.
-
(9)
Toxicity of previous antitumor therapy has returned to level ≤1 as defined by the National Cancer Institute (NCI)—Common Terminology Criteria for Adverse Events (CTCAE) v.5.0 (except for asymptomatic laboratory abnormalities such as elevated alkaline phosphatase hyperuricemia, elevated serum amylase/lipase and elevated blood glucose; except for toxicity that the investigator determined to have no safety risk, such as alopecia, grade 2 peripheral neurotoxicity, hypothyroidism stabilized by hormone replacement therapy, and so on).
-
(10)
Has no serious cardiac dysfunction, with a left ventricular ejection fraction ≥50%.
-
(11)
Has adequate organ function before registration, defined as:
-
(a)
Bone marrow function: absolute neutrophil count ≥1.5 × 109 −1, platelet count ≥100 × 109 l−1, hemoglobin ≥90 g l−1.
-
(b)
Hepatic function: total BIL ≤1.5 upper limit of normal (ULN), aspartate aminotransferase (AST) and alanine aminotransferase (ALT) without liver metastasis ≤2.5 ULN, AST and ALT with liver metastasis ≤5.0 ULN.
-
(c)
Renal function: creatinine ≤ 1.5 ULN, or creatinine clearance ≥50 ml min−1 (according to the Cockcroft and Gault).
-
(a)
-
(12)
Coagulation function: international normalized ratio ≤1.5 × ULN, and activated partial thromboplastin time (APTT) ≤1.5 ULN.
-
(13)
Urinary protein ≤2+ or ≤1,000 mg 24 h−1.
-
(14)
For premenopausal women of childbearing potential, a pregnancy test must be performed within 7 days before the start of treatment. Serum or urine pregnancy must be negative, and the patient must be nonlactating. Adequate barrier contraceptive measures should be taken during treatment and 6 months after the end of treatment for all participants (regardless of sex).
Patients screened for any of the following conditions were excluded from the study:
-
(1)
Chemotherapy, biological therapy, immunotherapy, radical radiotherapy, major surgery, targeted therapy (including small-molecule inhibitors of tyrosine kinase), and other antitumor therapies within 4 weeks or five half-lives (whichever is shorter) before the first administration; mitomycin and nitrosoureas treatment within 6 weeks before the first administration; and oral fluorouracil-like drugs such as S-1, capecitabine or palliative radiotherapy within 2 weeks before the first administration.
-
(2)
History of severe heart disease, such as New York Heart Association ≥grade 2 heart failure. History of transmural myocardial infarction, unstable angina pectoris and so on.
-
(3)
Prolonged QT interval (male QTc >450 ms or female QTc >470 ms), complete left bundle branch block and grade III atrioventricular block.
-
(4)
Active autoimmune and inflammatory diseases, such as systemic lupus erythematosus, psoriasis requiring systemic treatment, rheumatoid arthritis, inflammatory bowel disease and Hashimoto’s thyroiditis; except for type I diabetes, hypothyroidism that can be controlled only by alternative treatment and skin diseases that do not require systemic treatment (such as vitiligo and psoriasis).
-
(5)
Other malignancies diagnosed within 2 years before the first administration, with the following exceptions: basal cell carcinoma of the skin, squamous cell carcinoma of the skin and/or carcinoma in situ after radical resection.
-
(6)
Poorly controlled hypertension by two types of antihypertensive drug (systolic blood pressure >150 mmHg or diastolic blood pressure >100 mmHg).
-
(7)
A diabetic patient with poor glycemic control with the following conditions: (a) two fasting blood sugars >10 mmol l−1, (b) a glycosylated hemoglobin level of >8% or (c) concomitant diabetic gangrene.
-
(8)
History of ILD (including pulmonary fibrosis, and so on) or imaging during screening suggestive of such disease; current radiation pneumonitis grade ≥1 as defined by Radiation Therapy Oncology Group and the European Organization for Research and Treatment of Cancer .
-
(9)
Lung disease defined as grade ≥2 according to CTCAE v.5.0; concomitant lung disease resulting in clinically severe respiratory impairment, including, but not limited to, the following:
-
(a)
Any underlying lung disease (for example, pulmonary embolism within 3 months before randomization, severe asthma, severe chronic pulmonary obstructive disease)
-
(b)
Restrictive lung disease
-
(c)
Previous total lung resection
-
(a)
-
(10)
Patients with massive plasmapheresis, plasmapheresis with symptoms, or poorly controlled plasmapheresis (poorly controlled, defined as requiring two or more puncture drains in 1 month).
-
(11)
Except for imaging studies suggesting that the tumor had invaded or encircled large blood vessels in the chest, neck, or pharynx, which, in the opinion of the investigator, did not affect the patient’s enrollment in the medication.
-
(12)
Thrombotic events, such as unstable deep vein thrombosis, arterial thrombosis and pulmonary embolism, requiring therapeutic intervention within 6 months before screening, except for infusion-associated thrombosis.
-
(13)
Symptoms of active central nervous system metastasis. However, participants with stable brain metastasis were included. Stable is defined as:
-
(a)
With or without antiepileptic drugs, the seizure-free state lasted for >8 weeks.
-
(b)
There is no need to use glucocorticoids.
-
(c)
Continuous multiple magnetic resonance imaging scans (scanning interval of at least 8 weeks) showing a stable state on imaging.
-
(a)
-
(14)
A previous history of allergies to recombinant humanized antibodies or human–mouse chimeric antibodies or any of the components of BL-B01D1.
-
(15)
Previous history of autologous or allogeneic stem cell transplantation (Allo-HSCT).
-
(16)
In the adjuvant (or neoadjuvant) treatment of anthracyclines, the cumulative dose of anthracyclines exceeds 360 mg m−2.
-
(17)
Positive for human immunodeficiency virus antibody, active tuberculosis, active hepatitis B virus infection (hepatitis B virus DNA copy number >103 IU ml−1), or active hepatitis C virus infection (hepatitis C virus antibody positive and hepatitis C virus RNA > lower limit of detection).
-
(18)
Severe infections (CTCAE > grade 2) such as severe pneumonia, bacteremia, sepsis and tuberculosis within 4 weeks before the first use of the study drug; indication of active pulmonary infection within 2 weeks before the first use of the study drug.
-
(19)
Participated in another clinical trial within 4 weeks before participating in the study.
-
(20)
Other conditions that the investigator deems unsuitable for participation in this clinical trial.
-
(21)
Poorly controlled plasmapheresis (pleural effusion, abdominal effusion, pelvic effusion, pericardial effusion).
Procedures
The drug (BL-B01D1) was supplied by the sponsor (Sichuan Baili Pharmaceutical Chengdu). For the ESCC populations, dose expansion was performed in two dose groups: 2.0 mg kg−1 and 2.5 mg kg−1 D1D8 Q3W. The drug was administered intravenously. The first infusion was completed in 120 ± 10 min. If the infusion reaction was tolerable, the subsequent infusion was completed in 60–120 ± 10 min.
Outcomes
For the dose-expansion stage (1b), the primary endpoint was the RP2D of BL-B01D1. The secondary endpoints included TEAEs that occurred during BL-B01D1 treatment, ORR, DCR, DOR, PK parameters and immunogenicity. PFS, OS and biomarker analyses were exploratory endpoints.
Data on AEs were collected according to the NCI CTCAE (v.5.0). Dose-limiting toxicity (DLT) was observed during the first treatment cycle at each dose level in the dose-escalation phase. DLTs were defined as TRAEs that met the following criteria: Hematological toxicities (grade 4 neutrophil count decreased or anemia lasting more than 7 days, or grade 3 or worse febrile neutropenia or platelet count decrease lasting more than 7 days); hepatic organ toxicities (grade 4 elevated blood BIL, AST or ALT, or grade 3 or worse AST, ALT or blood BIL elevation lasting more than 7 days); other grade 3 or worse major organ toxicities (except for grade 3 skin toxicity, nausea, vomiting, diarrhea or anorexia, or grade 3 fatigue that resolved to grade 1 or less within 7 days after treatment). A detailed definition of DLT is listed in the protocol. DLT monitoring was not continued formally during the dose-expansion phase in the ESCC cohort. The expansion phase was focused on further characterizing safety and preliminary efficacy at doses already determined to be tolerable. To mitigate safety risks during the expansion, we performed safety monitoring as follows: all patients were monitored closely for TEAEs, with predefined grading and response protocols; study investigators reviewed safety data on an ongoing basis to ensure rapid clinical response to any emerging toxicity patterns; protocol-defined dose modifications and treatment delays were implemented in the event of serious treatment-related toxicities.
Tumor assessments were performed at baseline and every 6 weeks after treatment initiation. Responses were categorized as CR, PR, SD or PD according to the RECIST v.1.1. An objective response was determined when patients had PR or CR as the best overall response. DCR was defined as the percentage of patients who achieved a CR, PR or SD. DOR was defined as the time from the first documentation of a confirmed response (CR or PR) to the first documentation of PD or death, whichever occurred first. The pharmacokinetic parameters included Cmax, Tmax, T1/2, AUC0–t, clearance and Ctrough (defined as the lowest drug concentration in blood system prior to the next administration). PFS was defined as the time from the treatment initiation to the documentation of PD or death, whichever occurred first. OS was defined as the time from treatment initiation until death from any cause.
Exploratory biomarker analysis
Given the dual-target nature of BL-B01D1, EGFR and HER3 protein expression were evaluated as exploratory biomarkers. Tumor samples were collected per protocol (archived or fresh tissue within 2 years). EGFR and HER3 proteins were visualized by IHC using antibodies targeting EGFR (D38B1) and HER3 (D22C5). The intensity of expression was classified and scored by a pathologist based on IHC staining depth:
Negative: no obvious DAB coloring in the field of view.
1+ intensity: cell membranes in at least 10% of the area showed a weak yellowish staining.
2+ intensity: cell membranes in at least 10% of the area showed clear yellow staining.
3+ intensity: cell membranes in at least 10% of the area showed clear brown staining.
If less than 10% of the area was positive, the intensity score was automatically reduced by 1. The Hscore was calculated as a numerical value, representing the weighted summation of staining percentage, intensity and the percentage of cells at each intensity.
Statistical analysis
In the dose-expansion phase, a prespecified study plan was implemented to guide cohort progression. Initially, approximately 20–30 patients were planned for enrollment in each dose group (2.0 mg kg−1 and 2.5 mg kg−1, D1D8 Q3W), based primarily on safety and efficacy considerations aligned with the United States Food and Drug Administration38 and National Medical Products Administration guidance for early-phase trials. Dose continuation decisions were based on predefined safety and efficacy criteria: if the incidence of grade ≥3 adverse events exceeded 40% in any group, or if the difference in ORR between groups exceeded 10%, the underperforming cohort would be discontinued. For the group meeting both safety and efficacy benchmarks, expansion to a target sample size of 40–60 patients was planned. These criteria were applied to ensure patient safety and support exploratory dose optimization.
Safety data were summarized descriptively. Point estimates of ORR and DCR were provided, along with a two-sided 95% CI using the Clopper–Pearson method. The Kaplan–Meier method was used to summarize time-to-event endpoints, such as PFS, DOR and OS. The 95% CIs for OS, PFS and DOR were calculated using the Brookmeyer–Crowley method. The Cox regression model was used to explore potential correlations between covariates and time-to-event endpoints with 95% CIs. Statistical analyses were conducted using SAS v.9.4, R v.4.4.1 and WinNonLin v.8.4.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of the present study are available within the paper and the Supplementary Information. All requests for further data sharing will be reviewed by the leading clinical site, Department of Gastrointestinal Oncology, Peking University Cancer Hospital and Institute, and the study collaborator, Sichuan Baili Pharmaceutical, to verify whether the request is subject to any intellectual property or confidentiality obligations. Requests for access to the individual participant level data from this study can be submitted via email to zhihaolupku@bjmu.edu.cn or shenlin@bjmu.edu.cn with detailed proposals for approval and will be responded to within 60 days. All data provided are anonymized to respect the privacy of patients who have participated in the trial, in line with applicable laws and regulations. Each participant’s right and privacy are key subjects to taken into consideration while sharing information. A signed data access agreement with the collaborator is required before accessing shared data.
Code availability
No custom code was used for statistical analysis in this study.
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263 (2024).
Morgan, E. et al. The global landscape of esophageal squamous cell carcinoma and esophageal adenocarcinoma incidence and mortality in 2020 and projections to 2040: new estimates from GLOBOCAN 2020. Gastroenterology 163, 649–658.e642 (2022).
Doki, Y. et al. Nivolumab combination therapy in advanced esophageal squamous-cell carcinoma. N. Engl. J. Med. 386, 449–462 (2022).
Sun, J. M. et al. Pembrolizumab plus chemotherapy versus chemotherapy alone for first-line treatment of advanced oesophageal cancer (KEYNOTE-590): a randomised, placebo-controlled, phase 3 study. Lancet 398, 759–771 (2021).
Lu, Z. et al. Sintilimab versus placebo in combination with chemotherapy as first line treatment for locally advanced or metastatic oesophageal squamous cell carcinoma (ORIENT-15): multicentre, randomised, double blind, phase 3 trial. Br. Med. J. 377, e068714 (2022).
Kato, K. et al. Nivolumab plus chemotherapy or ipilimumab versus chemotherapy in patients with advanced esophageal squamous cell carcinoma (CheckMate 648): 29-month follow-up from a randomized, open-label, phase III trial. Cancer Med 13, e7235 (2024).
Kim, T. K., Vandsemb, E. N., Herbst, R. S. & Chen, L. Adaptive immune resistance at the tumour site: mechanisms and therapeutic opportunities. Nat. Rev. Drug Discov. 21, 529–540 (2022).
Zhuo, N. et al. Characteristics and prognosis of acquired resistance to immune checkpoint inhibitors in gastrointestinal cancer. JAMA Netw. Open 5, e224637 (2022).
Schoenfeld, A. J. & Hellmann, M. D. Acquired resistance to immune checkpoint inhibitors. Cancer Cell 37, 443–455 (2020).
Huang, J. et al. Camrelizumab versus investigator’s choice of chemotherapy as second-line therapy for advanced or metastatic oesophageal squamous cell carcinoma (ESCORT): a multicentre, randomised, open-label, phase 3 study. Lancet Oncol. 21, 832–842 (2020).
Shen, L. et al. Tislelizumab versus chemotherapy as second-line treatment for advanced or metastatic esophageal squamous cell carcinoma (RATIONALE-302): a randomized phase III study. J. Clin. Oncol. 40, 3065–3076 (2022).
Arteaga, C. L. & Engelman, J. A. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 25, 282–303 (2014).
Kashyap, M. K. & Abdel-Rahman, O. Expression, regulation and targeting of receptor tyrosine kinases in esophageal squamous cell carcinoma. Mol. Cancer 17, 54 (2018).
Song, J. et al. Epidermal growth factor receptor and B7-H3 expression in esophageal squamous tissues correlate to patient prognosis. OncoTargets Ther. 9, 6257–6263 (2016).
Hanawa, M. et al. EGFR protein overexpression and gene amplification in squamous cell carcinomas of the esophagus. Int. J. Cancer 118, 1173–1180 (2006).
Gibault, L. et al. Diffuse EGFR staining is associated with reduced overall survival in locally advanced oesophageal squamous cell cancer. Br. J. Cancer 93, 107–115 (2005).
Moehler, M. et al. Cisplatin and 5-fluorouracil with or without epidermal growth factor receptor inhibition panitumumab for patients with non-resectable, advanced or metastatic oesophageal squamous cell cancer: a prospective, open-label, randomised phase III AIO/EORTC trial (POWER). Ann. Oncol. 31, 228–235 (2020).
Dutton, S. J. et al. Gefitinib for oesophageal cancer progressing after chemotherapy (COG): a phase 3, multicentre, double-blind, placebo-controlled randomised trial. Lancet Oncol. 15, 894–904 (2014).
Lorenzen, S. et al. Cetuximab plus cisplatin-5-fluorouracil versus cisplatin-5-fluorouracil alone in first-line metastatic squamous cell carcinoma of the esophagus: a randomized phase II study of the Arbeitsgemeinschaft Internistische Onkologie. Ann. Oncol. 20, 1667–1673 (2009).
Lu, Z. et al. Paclitaxel and cisplatin with or without cetuximab in metastatic esophageal squamous cell carcinoma: a randomized, multicenter phase II trial. Innovation (Camb.) 3, 100239 (2022).
Wang, L., Yuan, H., Li, Y. & Han, Y. The role of HER3 in gastric cancer. Biomed. Pharmacother. 68, 809–812 (2014).
Kountourakis, P. et al. Prognostic significance of HER3 and HER4 protein expression in colorectal adenocarcinomas. BMC Cancer 6, 46 (2006).
Renshaw, B., Khalili, J. S., Xiao, S. & Zhu, Y. Abstract 6309: Anti-tumor efficacy of SI-B001, a novel EGFR × HER3 bispecific antibody, against EGFR-driven epithelial tumors alone or in combination with paclitaxel and carboplatin. Cancer Res. 83, 6309 (2023).
Claus, J., Patel, G., Ng, T. & Parker, P. J. A role for the pseudokinase HER3 in the acquired resistance against EGFR- and HER2-directed targeted therapy. Biochem. Soc. Trans. 42, 831–836 (2014).
Hill, A. G. et al. Phase II study of the dual EGFR/HER3 inhibitor duligotuzumab (MEHD7945A) versus cetuximab in combination with FOLFIRI in second-line RAS wild-type metastatic colorectal cancer. Clin. Cancer Res. 24, 2276–2284 (2018).
Qiu, M. Z. et al. Evaluation of safety of treatment with anti–epidermal growth factor receptor antibody drug conjugate MRG003 in patients with advanced solid tumors: a phase 1 nonrandomized clinical trial. JAMA Oncol. 8, 1042–1046 (2022).
Yu, H. A. et al. HERTHENA-Lung01, a phase II trial of patritumab deruxtecan (HER3-DXd) in epidermal growth factor receptor–mutated non–small-cell lung cancer after epidermal growth factor receptor tyrosine kinase inhibitor therapy and platinum-based chemotherapy. J. Clin. Oncol. 41, 5363–5375 (2023).
Jänne, P. A. et al. Efficacy and safety of patritumab deruxtecan (HER3-DXd) in EGFR inhibitor-resistant, EGFR-mutated non-small cell lung cancer. Cancer Discov. 12, 74–89 (2022).
Xue, J. et al. Prediction of human pharmacokinetics and clinical effective dose of SI-B001, an EGFR/HER3 bi-specific monoclonal antibody. J. Pharm. Sci. 109, 3172–3180 (2020).
Wan, W. et al. Abstract 2642: BL-B01D1, a novel EGFR×HER3-targeting ADC, demonstrates robust anti-tumor efficacy in preclinical evaluation. Cancer Res. 83, 2642 (2023).
Ma, Y. et al. BL-B01D1, a first-in-class EGFR-HER3 bispecific antibody-drug conjugate, in patients with locally advanced or metastatic solid tumours: a first-in-human, open-label, multicentre, phase 1 study. Lancet Oncol. 25, 901–911 (2024).
Anandavadivelan, P. & Lagergren, P. Cachexia in patients with oesophageal cancer. Nat. Rev. Clin. Oncol. 13, 185–198 (2016).
Attar, A. et al. Malnutrition is high and underestimated during chemotherapy in gastrointestinal cancer: an AGEO prospective cross-sectional multicenter study. Nutr. Cancer 64, 535–542 (2012).
Meng, X. et al. Efficacy and safety of camrelizumab plus apatinib in patients with advanced esophageal squamous cell carcinoma previously treated with immune checkpoint inhibitors (CAP 02 Re-challenge): a single-arm, phase II study. Eur. J. Cancer 212, 114328 (2024).
Zhao, W. et al. Radiotherapy plus camrelizumab and irinotecan for oligometastatic esophageal squamous cell carcinoma patients after first-line immunotherapy plus chemotherapy failure: an open-label, single-arm, phase II trial. Radiother. Oncol. 184, 109679 (2023).
Harandi, A., Zaidi, A. S., Stocker, A. M. & Laber, D. A. Clinical efficacy and toxicity of anti-EGFR therapy in common cancers. J. Oncol. 2009, 567486 (2009).
Giovannini, M. et al. Clinical significance of skin toxicity due to EGFR-targeted therapies. J. Oncol. 2009, 849051 (2009).
Expansion Cohorts: Use in First-in-Human Clinical Trials to Expedite Development of Oncology Drugs and Biologics Guidance for Industry (United States Food and Drug Administration, 2022); www.fda.gov/regulatory-information/search-fda-guidance-documents/expansion-cohorts-use-first-human-clinical-trials-expedite-development-oncology-drugs-and-biologics
Acknowledgements
We thank all the participants in this trial and their families as well as the trial site investigators. We thank Y. Hou from Department of Biostatistics, Peking University for her assistance in revising the paper. This study was supported by Sichuan Baili Pharmaceutical, Chengdu, China.
Author information
Authors and Affiliations
Contributions
Z.L., L.S. and J.G. contributed to the conception and design of the study. C.L., D.L., Y.J., M.S., S. Gao, X.M., D.Z., J. Zhu, Y.C., C.Q., M.Z., P.Z., R.X., Z.P., J. Zhou, S. Ge, M.L., J.Y., Y.W., Z.W., J.L. and X.Z., contributed to patient enrollment and data acquisition. Y.Z., H.Z. and S.X. were responsible for data analysis. C.L., D.L., Y.Z., H.Z., S.X., Z.L., L.S. and J.G. contributed to data interpretation and medical review. C.L. and Z.L. drafted the paper. All authors contributed to the paper revision and approved the final version before submission. The authors affirm the accuracy and completeness of the data and adherence of the study to the protocol.
Corresponding authors
Ethics declarations
Competing interests
Y.Z., H.Z. and S.X. are employees of Sichuan Baili Pharmaceutical. The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Chen Hu, Florian Lordick and Steven Maron for their contribution to the peer review of this work. Primary Handling Editor: Ulrike Harjes, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Trial design.
The trial design of a phase 1b study in patients with ESCC populations. ESCC, esophageal squamous cell carcinoma; ECOG, Eastern Cooperative Oncology Group; RECIST, Response Evaluation Criteria in Solid Tumors; RP2D, recommended phase 2 dose; ORR, objective response rate; DCR, disease control rate; DOR, duration of response; PK, pharmacokinetic; ADA, anti-drug antibodies; PFS, progression-free survival; OS, overall survival.
Extended Data Fig. 2 Anti-tumor activity.
(a) Spider plot showing the change over time from baseline in the sum of the diameters of measurable tumors (n = 71). (b) Swimming plot showing the best response and duration for each patient (N = 82).
Extended Data Fig. 3 Subgroup analysis.
(a) Objective response rates by subgroup (n = 73), data are presented as ORR (95% CI). (b) Progression-free survival by subgroups (n = 82), data are presented as mPFS (95% CI). ORR, objective response rate; mPFS, median progression-free survival; ECOG, Eastern Cooperative Oncology Group.
Extended Data Fig. 4 BL-B01D1 concentration over time after a single dose in the two groups (n = 43).
Data are presented as mean values ± SD. Data are presented as mean values ± SD. ADC, antibody–drug conjugate; TAB, total antibody; TOX, Toxin.
Extended Data Fig. 5 Response based on EGFR or HER3 expression in 2.5 mg/kg dose group (n = 44).
(a) Differentiate efficacy in the waterfall plot based on EGFR expression. (b) Differentiate efficacy in waterfall plot based on HER3 expression. (c) Patients ordered by EGFR H-scores. (d) Patient ordered by HER3 H -score. (e) Scatter plot showed EGFR and HER3 H-scores on the x-axis and y-axis, respectively. Clinical responses were color-coded as partial response (PR), stable disease (SD), or progressive disease (PD). 1The following subjects overlap: confirmed best overall response of subjects 01085 and 06005 were SD.
Supplementary information
Supplementary Information
Supplementary Tables 1–5; Study protocol and Statistical analysis plan.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, C., Liu, D., Ji, Y. et al. A bispecific antibody–drug conjugate targeting EGFR and HER3 in metastatic esophageal squamous cell carcinoma: a phase 1b trial. Nat Med (2025). https://doi.org/10.1038/s41591-025-03792-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41591-025-03792-7
