
Abstract
At the first interim analysis of the phase 3 KEYNOTE-426 trial, first-line pembrolizumab plus axitinib showed superior overall survival (OS), progression-free survival (PFS) and objective response rate (ORR) over sunitinib for advanced renal cell carcinoma (RCC). To assess long-term durability of clinical outcomes and elucidate predictive biomarkers for RCC, we performed efficacy and prespecified exploratory biomarker analyses from KEYNOTE-426 with ≥5 years of follow-up. Pembrolizumab plus axitinib showed sustained benefits in OS (hazard ratio: 0.84; 95% confidence interval: 0.71–0.99), PFS (hazard ratio: 0.69; 95% confidence interval: 0.59–0.81) and ORR (60.6% versus 39.6%) compared to sunitinib. An 18-gene T-cell-inflamed gene expression profile (TcellinfGEP) was positively associated with OS (P = 0.002), PFS (P < 0.0001) and ORR (P < 0.0001) within the pembrolizumab plus axitinib arm. An angiogenesis signature was positively associated with OS (P = 0.004) within the pembrolizumab plus axitinib arm and with OS (P < 0.0001), PFS (P < 0.001) and ORR (P = 0.002) within the sunitinib arm. Across arms, programmed cell death ligand 1 combined positive score was only associated (negatively) with OS within the sunitinib arm (P = 0.025). Additionally, PBRM1 (polybromo-1) mutation had a positive association with ORR (P = 0.002) within the pembrolizumab plus axitinib arm. Within the sunitinib arm, OS was positively associated with VHL (von Hippel–Lindau tumor suppressor gene) (P = 0.040) and PBRM1 (P = 0.010) mutations and was negatively associated with BAP1 (BRCA1-associated protein 1) mutation (P = 0.019). Results showed a sustained clinical benefit with pembrolizumab plus axitinib over sunitinib and provide valuable information on biomarkers for immunotherapy-based treatment combinations in advanced RCC. Prospective clinical investigations are needed for biomarker-directed treatment for advanced RCC. ClinicalTrials.gov identifier: NCT02853331.
Similar content being viewed by others

Main
The combination of the programmed cell death protein 1 (PD-1) inhibitor pembrolizumab and the vascular endothelial growth factor receptor (VEGFR) tyrosine kinase inhibitor (TKI) axitinib is standard-of-care first-line treatment for patients with advanced clear cell RCC, as a result of outcomes from the open-label, randomized, phase 3 KEYNOTE-426 trial1,2,3,4,5. KEYNOTE-426 was the first trial of a PD-1 inhibitor and VEGFR-TKI combination in the first-line treatment setting and, therefore, has the longest follow-up duration of any combination of a PD-1 or programmed cell death ligand 1 (PD-L1) inhibitor and a TKI. Because the treatment duration of pembrolizumab is limited to 2 years, it is important to assess the long-term durability of clinical outcomes.
There is an unmet need for biomarkers that are predictive of patient outcomes after using available first-line treatment options in RCC6,7. Studies to evaluate predictive and prognostic biomarkers in metastatic RCC have been largely derived from phase 3 studies (for example, IMmotion151, JAVELIN Renal 101 and CheckMate 9ER) in which similar mechanisms (for example, PD-L1 inhibitor plus VEGF-TKI) but different drugs were evaluated as frontline therapy8,9,10,11,12,13,14,15,16,17,18,19,20,21,22. Thus, extrapolation of these data to define predictive biomarkers in frontline therapy for advanced RCC is potentially confounded. Nonetheless, the studies yielded relevant biological insight into the role of molecular features of RCC, including PD-L1 expression, interferon gamma (IFNγ) RNA signatures, specific DNA alterations (such as PBRM1 mutations), mRNA-based molecular clusters, circulating kidney injury molecule-1 (KIM-1) and serum glycopeptides8,9,10,11,14,17,19,20,21,22. Further investigation is warranted to identify tumor and (or) stromal biologic features that may define susceptible patient populations.
Here we report the final clinical data after 5 years of follow-up from KEYNOTE-426 and the results of a prespecified exploratory biomarker analysis that was conducted to determine whether molecular determinants relevant to the underlying disease biology are associated with clinical outcomes (ORR, PFS and OS) for pembrolizumab plus axitinib and for sunitinib in participants with advanced clear cell RCC.
Results
Participants
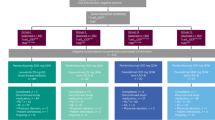
Between 24 October 2016 and 24 January 2018, 861 participants were randomly assigned to receive either pembrolizumab plus axitinib (n = 432) or sunitinib monotherapy (n = 429) (Fig. 1). As of the data cutoff date, the median follow-up (defined as time from randomization to the database cutoff date for this exploratory analysis) was 67.2 months (range, 60.0–75.0 months). Baseline demographics and characteristics are shown in Table 1. A total of 429 participants in the pembrolizumab plus axitinib arm and 425 participants in the sunitinib arm received at least one dose of the assigned study treatment. At the time of this analysis, 381 of 429 treated participants (88.8%) in the pembrolizumab plus axitinib arm and 406 of 425 treated participants (95.5%) in the sunitinib arm had permanently discontinued study treatment, most commonly due to radiographic progressive disease (pembrolizumab plus axitinib, n = 227 (52.9%); sunitinib, n = 260 (61.2%)) (Fig. 1). Thirty of 429 participants (7.0%) in the pembrolizumab plus axitinib arm and 19 of 425 participants (4.5%) in the sunitinib arm remained on treatment.

aCompleted pembrolizumab treatment after discontinuing axitinib.
Among participants who discontinued study treatment, 237 of 381 (62.2%) in the pembrolizumab plus axitinib arm and 300 of 406 (73.9%) in the sunitinib arm received subsequent systemic anticancer therapy, most commonly a VEGFR inhibitor in the pembrolizumab plus axitinib arm (206/237 (86.9%)) and a PD-L1 inhibitor in the sunitinib arm (240/300 (80.0%)) (Supplementary Table 1).
Efficacy outcomes
By the data cutoff date, 550 participants in the intention-to-treat population had died, including 270 of 432 participants (62.5%) in the pembrolizumab plus axitinib arm and 280 of 429 participants (65.3%) in the sunitinib arm. The median OS was 47.2 months in the pembrolizumab plus axitinib arm and 40.8 months in the sunitinib arm (hazard ratio: 0.84; 95% confidence interval: 0.71–0.99) (Fig. 2a). Median PFS was 15.7 months in the pembrolizumab plus axitinib arm and 11.1 months in the sunitinib arm (hazard ratio: 0.69; 95% confidence interval: 0.59–0.81) (Fig. 2b). Consistent OS and PFS benefits with pembrolizumab plus axitinib compared to sunitinib were seen across subgroups, including International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) risk favorable and intermediate-risk and poor-risk categories and PD-L1 combined positive score (CPS) cutoff values of <1 and ≥1 (Fig. 2c,d).

a, Kaplan–Meier estimates of OS. b, Kaplan–Meier estimates of PFS. c, OS by subgroups. d, PFS by subgroups. e, Kaplan–Meier estimates of DOR in participants with a confirmed objective response. In a, b and e, tick marks represent censored data. In c and d, shaded squares correspond to the hazard ratios (HRs), and the error bars (horizontal lines) correspond to the 95% confidence intervals (CIs).
Confirmed ORR was 60.6% (50 complete responses (11.6%), 212 partial responses (49.1%)) in the pembrolizumab plus axitinib arm and 39.6% (17 complete responses (4.0%), 153 partial responses (35.7%)) in the sunitinib arm (Extended Data Table 1). The median duration of response (DOR) was 23.6 months (range, 1.4+ months to 68.6+ months) in the pembrolizumab plus axitinib arm and 15.3 months (range, 2.3–68.3 months) in the sunitinib arm (Fig. 2e). The estimated percentage of participants with an objective response who would have an ongoing response at 60 months was 26.0% in the pembrolizumab plus axitinib arm and 14.4% in the sunitinib arm.
In a post hoc analysis of participants who completed 35 cycles of pembrolizumab (n = 120) (Supplementary Table 2), the median OS was not reached (95% confidence interval: 70.6 months to not reached), and the estimated 48-month and 60-month OS rates were 81.7% and 70.7%, respectively (Supplementary Fig. 1a). The median PFS was 37.4 months (95% confidence interval: 32.3–43.7 months), and the estimated 48-month and 60-month PFS rates were 38.3% and 32.8%, respectively (Supplementary Fig. 1b). The confirmed ORR was 85.0% (102/120; 22 complete responses (18.3%), 80 partial responses (66.7%)) (Supplementary Table 2).
Biomarker outcomes
Biomarker analysis population
Of 848 RNA sequencing samples, 797 passed quality control, resulting in a 94.0% success rate. Of the 775 whole-exome sequencing (WES) tumor samples with matched normal samples, 751 passed quality control, resulting in a 96.9% success rate. Of the samples that passed quality control, duplicates and samples from participants who did not receive study treatment were further excluded from the biomarker analysis population. Of 854 participants who received at least one dose of study treatment in the total KEYNOTE-426 population, 730 (85.5%) had evaluable RNA sequencing data, 698 (81.7%) had evaluable WES data and 816 (95.6%) had evaluable PD-L1 CPS data (Supplementary Table 3). Baseline characteristics in the evaluable RNA sequencing and WES analyses populations were well balanced between treatment arms and similar to those of the total study population (Table 1 and Supplementary Table 3). Consistent with the intention-to-treat population, OS and PFS favored pembrolizumab plus axitinib over sunitinib in the RNA sequencing and WES analyses populations (Supplementary Table 3).
TcellinfGEP, angiogenesis signature and PD-L1 CPS
Based on observed associations between biomarkers and clinical outcomes of pembrolizumab monotherapy in previous studies23,24,25,26,27,28, we examined whether an IFNγ-related 18-gene TcellinfGEP, the angiogenesis signature and PD-L1 CPS were separately associated with clinical outcomes of pembrolizumab plus axitinib or sunitinib. In the pembrolizumab plus axitinib arm, higher TcellinfGEP was associated with improved ORR (P < 0.0001), PFS (P < 0.0001) and OS (P = 0.002) (Table 2). In the sunitinib arm, no associations (P > 0.05) were observed between TcellinfGEP and clinical outcomes (Table 2 and Fig. 3a). For the angiogenesis signature, a positive association was observed only with OS (P = 0.004) in the pembrolizumab plus axitinib arm, and there was a positive association with ORR (P = 0.002), PFS (P < 0.001) and OS (P < 0.001) in the sunitinib arm (Table 2 and Fig. 3b). The significance of TcellinfGEP to clinical outcomes in the pembrolizumab plus axitinib arm and the significance of angiogenesis to clinical outcomes in the sunitinib arm remained the same in the joint TcellinfGEP and angiogenesis model (Supplementary Table 4). PD-L1 CPS as a continuous variable (square root scale) was negatively associated with OS for sunitinib (P = 0.025). No association was observed between continuous PD-L1 CPS and clinical outcomes for pembrolizumab plus axitinib (P > 0.05) (Table 2 and Fig. 3c), suggesting that PD-L1 expression (as measured by CPS) is not a predictive marker of outcomes with pembrolizumab plus axitinib in this disease setting. However, PD-L1 CPS showed a moderate correlation with TcellinfGEP (Spearman ρ = 0.46) (Extended Data Fig. 1), supportive of the respective roles of PD-L1 expression and TcellinfGEP in biologically defining an inflamed tumor microenvironment.

a, TcellinfGEP (pembrolizumab plus axitinib, n = 369; sunitinib, n = 361). b, Angiogenesis signature (pembrolizumab plus axitinib, n = 369; sunitinib, n = 361). c, PD-L1 CPS (pembrolizumab plus axitinib, n = 407; sunitinib, n = 409). The center line corresponds to the median, and the box is delineated by the first and third quartiles. Whiskers extend to any points within 1.5 times the interquartile range, with points lying beyond identified individually as potential outliers. P values are nominal (one-sided for pembrolizumab plus axitinib and two-sided for sunitinib) and were derived using a logistic regression model, with adjustment for IMDC risk category. Significance was prespecified at α = 0.05.
When assessing TcellinfGEP using a prespecified cutoff of the first tertile, improved OS and PFS for pembrolizumab plus axitinib compared to sunitinib was observed in the TcellinfGEPnon-low subgroup (OS hazard ratio: 0.77 (95% confidence interval: 0.61–0.96); PFS hazard ratio: 0.58 (95% confidence interval: 0.47–0.72)) (Fig. 4a,b). When assessing angiogenesis signature using a cutoff of ≥ or < the median, PFS favored pembrolizumab plus axitinib over sunitinib in the ≥ median subgroup (hazard ratio: 0.73 (95% confidence interval: 0.57–0.94)) (Fig. 4c,d). Notably, OS and PFS more strongly favored pembrolizumab plus axitinib over sunitinib in the < median subgroup (OS hazard ratio: 0.69 (95% confidence interval, 0.54–0.89); PFS hazard ratio: 0.62 (95% confidence interval: 0.48–0.79)) (Fig. 4c,d).

a, OS by TcellinfGEP cutoff. b, PFS by TcellinfGEP cutoff. c, OS by angiogenesis signature cutoff. d, PFS by angiogenesis signature cutoff. Tick marks represent censored data. CI, confidence interval; HR, hazard ratio.
Other gene expression signatures and molecular subtype
We evaluated other gene expression signatures and their association with clinical outcomes by hypothesis testing within each treatment arm. In the pembrolizumab plus axitinib arm, the monocytic myeloid-derived suppressor cell (mMDSC) signature was positively associated with ORR (P = 0.058), PFS (P = 0.039) and OS (P = 0.057) (Table 2). In the sunitinib arm, the hypoxia signature was positively associated with ORR and OS (P = 0.071 and P = 0.094, respectively); the MYC signature was negatively associated with PFS and OS (P = 0.017 and P < 0.001, respectively); and the proliferation signature was negatively associated with OS (P < 0.001). When evaluating the independence of the gene signatures with the angiogenesis signature, the mMDSC signature was significantly associated with improved ORR (P = 0.006), PFS (P = 0.002) and OS (P = 0.004) in the pembrolizumab plus axitinib arm (Supplementary Table 4). After adjusting for the angiogenesis signature in the sunitinib arm, the hypoxia signature was no longer significantly associated with any clinical outcome; the MYC signature was negatively associated with PFS (P = 0.039) and OS (P = 0.016); and the proliferation signature was negatively associated with OS (P = 0.043). These associations were weaker compared to those without adjustment, which can be partially explained by the correlations among the signatures with the angiogenesis signature. Among all samples analyzed, there was a modest positive correlation (ρ = 0.31) (Extended Data Fig. 1) between the hypoxia and angiogenesis signatures, whereas there was a modestly negative correlation for the MYC and proliferation signatures with the angiogenesis signature (ρ = 0.28 and ρ = 0.35, respectively) (Extended Data Fig. 1).
Given that TcellinfGEP was reported to be predictive of response to pembrolizumab monotherapy in other settings24,29, we adjusted these gene expression signatures for TcellinfGEP (in addition to IMDC risk category) to elucidate their additional predictive value for the pembrolizumab and axitinib combination (although adjustment was performed in both treatment arms). After adjusting for TcellinfGEP in the pembrolizumab plus axitinib arm, no associations between clinical outcomes and mMDSC signature were observed, and the proliferation signature was negatively associated with OS (P = 0.007) (Extended Data Table 2). Notably, mMDSC was strongly positively correlated with TcellinfGEP (ρ = 0.70) (Extended Data Fig. 1). The associations for other gene expression signatures within the sunitinib arm remained similar after adjusting for TcellinfGEP. The hypoxia signature was positively associated with ORR and OS (P = 0.065 and P = 0.095, respectively); the MYC signature was negatively associated with PFS and OS (P = 0.019 and P < 0.001, respectively); and the proliferation signature was negatively associated with OS (P < 0.001).
We additionally sought to assign tumor samples to molecular subtypes according to the transcriptomically defined clustering in the phase 3 IMmotion151 trial8. We observed that 18.4% of the treated participants in the RNA sequencing population were angiogenic–stromal, 15.1% were angiogenic, 21.6% were immune–proliferative, 14.9% were proliferative and 14.9% were stromal–proliferative (Supplementary Table 5 and Extended Data Fig. 2a); 15.1% of participants in the total RNA sequencing population could not be assigned to one of these five subtypes and constituted a sixth ‘other’ subtype. Next, we evaluated the distribution of these molecular subtypes across IMDC risk categories and by PD-L1 status and observed enrichment of the immune–proliferative subtype in the IMDC intermediate-risk and poor-risk groups and in participants with tumors of PD-L1 CPS ≥ 1 (Extended Data Fig. 2b,c); the angiogenic–stromal subtype was enriched in participants with tumors of PD-L1 CPS < 1.
Testing of the association of molecular subtype and clinical outcomes showed an association with OS (P = 0.010) in the sunitinib arm (Table 2). After prespecified adjustment for TcellinfGEP and the angiogenesis signature, no significant associations with clinical outcomes were observed in the pembrolizumab plus axitinib and sunitinib arms (P > 0.10) (Extended Data Table 2). Within the pembrolizumab plus axitinib arm, the ORR was lowest (50.0%) in the stromal–proliferative subtype and highest (75.6%) in the immune–proliferative subtype (Extended Data Fig. 3a). Within the sunitinib arm, the ORR was lowest (34.0%) in the proliferative subtype and highest (51.8%) in the angiogenic subtype.
Efficacy estimates by DNA mutational status
We examined the impact of mutations in genes with clinical relevance to RCC in other studies, including VHL, SETD2 (SET domain containing 2, histone lysine methyltransferase), PBRM1 and BAP1 using WES8,14,30. In the pembrolizumab plus axitinib arm, no significant associations between DNA mutations and PFS or OS were observed (Extended Data Table 3). The PBRM1 mutation was positively associated with the ORR within the pembrolizumab plus axitinib arm, with significantly higher rates in the PBRM1 mutant than in the wild-type subgroup (71.4% versus 52.3%; P = 0.002) (Extended Data Table 3 and Extended Data Fig. 3b). The ORR with the pembrolizumab plus axitinib was similar between the mutant and the wild-type subgroups for VHL, SETD2 and BAP1 (Extended Data Fig. 3b). In the sunitinib arm, VHL and PBRM1 mutations were associated with longer OS (P = 0.040 and P = 0.010, respectively), whereas BAP1 mutation was associated with shorter OS (P = 0.019) (Extended Data Table 3). The VHL, PBRM1 and SETD2 mutations were not associated with PFS or ORR in the sunitinib arm (P > 0.10). The ORR with sunitinib was similar between the mutant and the wild-type subgroups for VHL, PBRM1, SETD2 and BAP1 (Extended Data Fig. 3c).
When assessing efficacy by DNA mutational status, OS and PFS directionally favored pembrolizumab plus axitinib over sunitinib, with various hazard ratios in the mutant and wild-type subgroups for VHL, PBRM1, SETD2 and BAP1 (Extended Data Figs. 4–7).
Discussion
The final clinical follow-up analysis of KEYNOTE-426 showed sustained and durable clinical benefit of pembrolizumab plus axitinib compared to sunitinib2,31,32. In the prespecified exploratory biomarker analysis, significant associations between several genomic features and clinical outcomes with pembrolizumab plus axitinib or sunitinib were observed, which deepens understanding of RCC biology and potentially informs further advancement in treating patients with advanced clear cell RCC.
Higher TcellinfGEP was associated with improved ORR, PFS and OS for pembrolizumab plus axitinib, but there was no association with clinical outcomes for sunitinib. This association with pembrolizumab plus axitinib was not unexpected given that the TcellinfGEP comprises genes related to antigen presentation, adaptive immune resistance, cytolytic activity and chemokine expression23,29. The positive association between TcellinfGEP and outcomes in the combination arm is consistent with previous reports for pembrolizumab monotherapy in the pan-tumor and specific tumor settings, including clear cell RCC managed with pembrolizumab monotherapy (KEYNOTE-427 cohort A) and with reports that showed associations between T cell inflammation-related genes and clinical outcomes for avelumab plus axitinib (JAVELIN Renal 101 trial) and atezolizumab plus bevacizumab (IMmotion150/151 trial)23,24,25,26,27,28,29. However, in the phase 3 CLEAR/KEYNOTE-581 trial of participants with advanced clear cell RCC, TcellinfGEP was not associated with ORR and PFS for lenvatinib plus pembrolizumab but was associated with ORR for sunitinib33. In the phase 3 CheckMate 9ER trial of participants with advanced clear cell RCC, several gene expression signatures, including IFNγ, were not predictive of clinical outcomes of nivolumab plus cabozantinib19. The precise reasons for differences among the associations of immune signatures and outcomes across trials are not entirely clear but could include differences in the TKI partner, the number of evaluable samples and the duration of follow-up. The strength of the association between TcellinfGEP and response seems to be greater with the pembrolizumab and axitinib combination than with pembrolizumab monotherapy in specific tumor types (including clear cell RCC in the KEYNOTE-427 trial)23,25,27,28,29, suggesting a potential positive interaction between the TKI and the PD-1 inhibitor. Preclinical models have shown that TKIs exert immune-modulatory effects in the tumor microenvironment by enhancing tumor cell sensitivity to immune-cell-mediated lysis through an alteration in the tumor cell phenotype and by altering the frequency or function of immune cell subsets in the periphery or the tumor microenvironment, thus promoting more productive immune interactions34,35,36. The hypothesis of a positive interaction between axitinib and pembrolizumab is further supported by the lack of significant associations between TcellinfGEP and clinical outcomes for sunitinib, which suggests that the TcellinfGEP is not a predictor of response to sunitinib as monotherapy. However, axitinib and sunitinib are different TKIs.
The strong positive association between angiogenesis signature and all clinical outcomes for sunitinib is consistent with previous reports from the phase 3 COMPARZ (first-line pazopanib versus sunitinib) and IMmotion151 (first-line atezolizumab plus bevacizumab versus sunitinib) studies of participants with advanced clear cell RCC, supporting the present findings8,10,14. Because TKI monotherapy is not commonly used as first-line therapy, these findings have limited clinical applicability. Whether this association would be seen in a refractory setting in which TKIs are commonly used requires further investigation.
Previous analyses of the IMmotion151 trial identified seven molecular clusters related to differential clinical outcomes with first-line atezolizumab plus bevacizumab compared to sunitinib across molecular subtypes8,10. Survival outcomes were poorer for participants with tumors classified within the angiogenic cluster who were given atezolizumab plus bevacizumab than for those given sunitinib (OS hazard ratio: 1.32)10. In the present analysis, results of assessment of the associations within arms showed a positive association of the angiogenesis signature with OS, and the OS hazard ratio for pembrolizumab plus axitinib compared to sunitinib in the high (≥ median) angiogenesis subgroup was 0.94. An understanding of the different antiangiogenic effects among different TKIs and between TKIs and bevacizumab as related to the association with an angiogenic gene signature requires further investigation. In the phase 3 JAVELIN Renal 101 trial, participants with advanced or metastatic RCC in clusters 1 (angiogenic–stromal), 3 (complement–oxidation) and 4 (T-effector–proliferative) treated with avelumab plus axitinib tended to have improved PFS compared to sunitinib-treated participants11. In the present analysis of the KEYNOTE-426 trial, the highest ORR for pembrolizumab plus axitinib was observed in the immune–proliferative cluster, as expected given that this cluster comprises tumors that are angiogenesis poor but highly immunogenic, with highest infiltration in immune cell subsets (CD8+, CD4+ and regulatory T cells, B cells, macrophages and dendritic cells)8,10.
Genomic features, such as loss-of-function mutations in VHL, PBRM1, SETD2 and BAP1, have been evaluated to determine their association with clinical outcomes of systemic therapies in patients with advanced clear cell RCC; however, data are often conflicting8,12,13,14,15,16,17,18,30. The relationship between clinical outcomes and PBRM1 DNA alterations has attracted attention in the past. Although the pressent dataset showed that PBRM1 mutation tended to be associated with improved outcomes, PRRM1 mutation does not seem to be a reliable biomarker considering the totality of the data.
Although PD-L1 CPS has been positively associated with clinical outcomes of pembrolizumab monotherapy or pembrolizumab-based combination treatment in advanced clear cell RCC and other tumor types25,28, the lack of a significant association between PD-L1 CPS and clinical outcomes in the present analyses suggests that PD-L1 expression (as measured by CPS) is not a predictive marker of outcomes of pembrolizumab plus axitinib in this disease setting and should not be used in this clinical setting. Similarly, PD-L1 expression (tumor proportion score ≥ 1%) was not associated with clinical outcomes of nivolumab plus cabozantinib in the CheckMate 9ER trial19. Notably, TcellinfGEP, which was positively associated with clinical outcomes within the pembrolizumab plus axitinib arm, was moderately correlated with PD-L1 CPS (this correlation is not unexpected given that the TcellinfGEP includes mRNA expressions for PD-L1)29. These data suggest the respective roles of TcellinfGEP and PD-L1 CPS in biologically defining an inflamed tumor microenvironment, but their independent contribution as predictive biomarkers of immunotherapy may be therapy specific and tumor type specific24,26,29.
These data have both clinical and biomarker relevance in the treatment choice for patients with advanced clear cell RCC. First, with longer follow-up of patients in the KEYNOTE-426 trial and other phase 3 trials of a VEGF-TKI plus PD-1 inhibitor (CheckMate 9ER (cabozantinib plus nivolumab) and CLEAR/KEYNOTE-581 (lenvatinib plus pembrolizumab)), as well as more mature data for the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) inhibitor ipilimumab plus nivolumab (CheckMate 214), all four treatment approaches remain reasonable, supported by a significant survival advantage37,38,39. Second, employing the IMDC risk score to select therapy appears increasingly flawed; it remains a useful prognostic tool based on clinical characteristics rather than tissue-based biomarkers. Biomarker data from this and previous trials show some consistency but also many instances of inconsistency8,9,10,11,14,19,20. Although the signatures evaluated in this trial look promising, particularly for sunitinib, and other studies have shown potential clinical utility of molecular subsets and emerging biomarkers (for example, serum glycopeptides and circulating KIM-1)8,10,11,20,21,22, it is not currently possible to define a biomarker to select for combination regimens with either a specific VEGF-TKI plus PD-1 inhibitor or CTLA-4 inhibitor plus PD-1 inhibitor. Moving forward, prospective biomarker trials are ongoing, including those testing for PD-L1 expression and RNA signatures30,40,41.
A strength of the biomarker analysis is that the populations included a high proportion of the treated population in each arm; therefore, inferences drawn from these respective datasets are largely representative of the KEYNOTE-426 trial population. However, the prespecified exploratory biomarker analysis from the KEYNOTE-426 trial is limited by the small sample sizes of some of the subgroups and the lack of statistical power and (or) multiplicity adjustments for association analysis of some biomarkers, hindering definitive conclusions. Additionally, given the complex interplay of biological processes involved in the RCC tumor microenvironment42, the evaluation of each signature or gene individually most likely does not capture the potential joint effects of the biomarkers on clinical outcomes within each treatment arm. Furthermore, inter-trial differences in definitions for PD-L1 expression, different algorithms for clustering patient samples into molecular subtypes and different gene expression signatures evaluated limit comparative interpretations of the biomarker data. Lastly, because a VEGF-TKI was present in both treatment arms, the relative contribution of VEGF-TKI in the survival outcomes cannot be determined from a clinical and biomarker standpoint.
In conclusion, results of the present analysis showed sustained OS, PFS and ORR benefit of the use of pembrolizumab plus axitinib compared to sunitinib monotherapy. An extensive biomarker analysis adds to the increasing amount of information on biomarkers in patients treated with immunotherapy-based combinations. Although the analysis showed potential clinical utility of some RNA signatures in identifying patients who are likely to benefit the most from each treatment, additional correlative data and further prospective clinical investigations are needed to inform biomarker-directed treatment of patients with advanced or metastatic RCC who are being considered for combination treatment with antiangiogenic and PD-1 inhibitor therapies. Pembrolizumab plus axitinib is a first-line treatment option for patients with advanced RCC regardless of biomarker subtypes.
Methods
Inclusion and ethics
KEYNOTE-426 (NCT02853331) is a randomized, open-label, phase 3 trial conducted across 129 medical centers globally. The trial was conducted in accordance with the principles of Good Clinical Practice and was approved by the appropriate institutional review boards and regulatory agencies. Written informed consent was provided by all participants before enrollment.
Trial design, participants and treatments
Details of the trial design and eligibility criteria were published previously2,31. In brief, eligible participants were adults with newly diagnosed stage IV or recurrent clear cell RCC who had not previously received systemic therapy for advanced disease. Participants had a Karnofsky Performance Scale score of 70% or higher at baseline, one or more measurable lesions per Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1) as assessed by the investigator and a tumor sample available for biomarker assessment. Sex of participants was determined based on self-report.
Participants were randomly assigned in a 1:1 ratio to receive pembrolizumab 200 mg intravenously once every 3 weeks for up to 35 cycles (~2 years) plus axitinib 5 mg by mouth twice daily continuously or sunitinib 50 mg by mouth once daily for 4 weeks on and 2 weeks off, continuously. Randomization was stratified according to the IMDC risk group (favorable versus intermediate versus poor risk) and by geographic region (North America versus Western Europe versus rest of the world). Treatment was continued until disease progression, unacceptable toxicity or physician or participant decision to discontinue. In the pembrolizumab plus axitinib arm, if one drug was discontinued because of toxicity, the other drug could be continued.
Outcomes
The dual primary endpoints of OS and PFS per RECIST v1.1 by blinded independent central review (BICR) and key secondary endpoint of ORR per RECIST v1.1 by BICR were met at the first interim analysis2. Therefore, the subsequent analyses of efficacy are exploratory.
The prespecified objectives of the exploratory biomarker analysis, defined in a statistical analysis plan, were as follows:
-
(1)
To assess whether an IFNγ-related 18-gene TcellinfGEP and 10 other signatures (angiogenesis, glycolysis, granulocytic myeloid-derived suppressor cells (gMDSCs), hypoxia, mMDSCs, MYC, proliferation, RAS, stroma/EMT/TGFβ and WNT)24 are individually associated with clinical outcomes (ORR, OS and PFS) of pembrolizumab plus axitinib or sunitinib;
-
(2)
To assess whether prespecified molecular subtypes as categorical variables are separately associated with clinical outcomes of pembrolizumab plus axitinib or of sunitinib;
-
(3)
To assess whether continuous PD-L1 CPS is separately associated with clinical outcomes of pembrolizumab plus axitinib or of sunitinib; and
-
(4)
To assess whether mutation status of key RCC driver genes (VHL, PBRM1, SETD2 and BAP1), as determined by WES, are separately associated with clinical outcomes of pembrolizumab plus axitinib or of sunitinib.
Estimation of PFS and OS hazard ratios for pembrolizumab plus axitinib compared to sunitinib was also performed by TcellinfGEP subgroups based on a prespecified cutoff of the first tertile23; by angiogenesis signature subgroups defined by a prespecified cutoff of the median; and by mutational status of VHL, PBRM1, SETD2 and BAP1.
Assessments
Details on the assessment of efficacy outcomes were published previously and follow standard guidance by the US Food and Drug Administration (https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-trial-endpoints-approval-cancer-drugs-and-biologics)2,31,43. For the biomarker analysis, formalin-fixed, paraffin-embedded (FFPE) pretreatment tumor tissue samples collected at screening were used. PD-L1 expression was centrally determined using the PD-L1 IHC 22C3 pharmDx (Agilent Technologies). CPS was calculated as the number of PD-L1-staining cells (tumor cells, lymphocytes and macrophages) divided by the total number of viable tumor cells, multiplied by 100.
RNA sequencing was performed on an Illumina HiSeq by use of the TruSeq Access protocol. Raw reads were processed using a customized data analysis pipeline in OmicSoft Array Suite version 9 (Qiagen)24. In brief, the raw sequence reads were filtered for quality and subsequently aligned to the reference genome Human.B37.3 using OmicSoft Sequence Aligner44. After reference alignment, gene expression levels (raw read counts and fragments per kilobase of exon per million mapped fragments) were quantified using the RNA-Seq by Expectation Maximization (RSEM) algorithm45 with the gene model Ensembl.R75. The TcellinfGEP is composed of 18 inflammatory genes related to antigen presentation, adaptive immune resistance, cytolytic activity and chemokine expression, including CCL5, CD27, CD274 (PD-L1), CD276 (B7-H3), CD8A, CMKLR1, CXCL9, CXCR6, HLA–DQA1, HLA–DRB1, HLA-E, IDO1, LAG3, NKG7, PDCD1LG2 (PD-L2), PSMB10, STAT1 and TIGIT23,29. The TcellinfGEP score was calculated as the weighted sum of normalized expression values for the 18 genes. The TcellinfGEP was established on the NanoString platform (NanoString Technologies), was evaluated across the pembrolizumab clinical development program and was predictive of response to pembrolizumab in both pan-tumor and histology-specific settings23,24,25,27,28,29. The 10 other signature scores (angiogenesis, glycolysis, gMDSCs, hypoxia, mMDSCs, MYC, proliferation, RAS, stroma/EMT/TGFβ and WNT) were calculated as the average of the genes (on the logarithmic scale) in each signature gene set, as previously described24,29.
Profiled tumor RNA sequencing samples were assigned to molecular subtypes according to the transcriptomically defined clustering in the phase 3 IMmotion151 trial of atezolizumab plus bevacizumab as first-line therapy compared to sunitinib in participants with advanced or metastatic RCC8. The molecular subtypes were assigned as follows. First, tumors with an angiogenesis consensus signature score above the upper tertile were assigned to the angiogenic group. Second, among tumors assigned to the angiogenic group, those that had a stroma/EMT/TGFβ consensus signature score above the median (evaluated over all samples) were assigned to the angiogenic–stromal group. Third, among the remaining samples, those that had a TcellinfGEP score above the 75th percentile (evaluated over all samples) were assigned to the immune–proliferative group. Fourth, among the remaining (yet unassigned to the other groups) samples, those with a low proliferation consensus signature score (defined for which the proliferation score was below the lower tertile (evaluated over the not-already-assigned samples)) were assigned to the ‘other’ group. Finally, among the remaining samples, those for which a stroma/EMT/TGFβ score was higher than the median (evaluated over the remaining samples) were assigned to the stromal–proliferative group, whereas the other half was assigned to the proliferative group.
WES was performed on FFPE sections of pretreatment tumor samples and on matched normal (blood cell) samples using ACE Cancer Exome technology (Personalis), with average coverage of 200× (range, 13–475)23,46. WES reads were aligned to the Genome Reference Consortium Human Build 37 assembly by use of the Burrows–Wheeler Aligner MEM algorithm, followed by preprocessing steps that included duplicate marking, indel realignment and base recalibration using Picard (version 1.114; Broad Institute) and generation of analysis-ready binary alignment map files using Genome Analysis Toolkit (version 2; Broad Institute) analysis software. Thereafter, somatic single-nucleotide variant (SNV) calls were generated by comparing binary alignment map files from tumor and matched normal samples using default parameters from the MuTect method47. MuTect-called SNVs that were present in the Single Nucleotide Polymorphism Database (version 141; National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov/snp/) but not in the Catalogue of Somatic Mutations in Cancer (version 68; http://cancer.sanger.ac.uk) were filtered out48. SNVs with mutant reads of fewer than four in tumor samples were also eliminated. MuTect2 was further used to comprehensively characterize insertion or deletion–spliced mutations.
Statistical analysis
We assessed efficacy in the intention-to-treat population, which included all randomly assigned participants, and followed guidelines published previously2,31. Because the trial outcome was previously defined as positive and the present analysis is exploratory, no formal hypothesis testing was performed in the present analysis. In the biomarker analysis population, we included all participants who received at least one dose of study treatment and had available PD-L1, RNA sequencing or WES data that passed quality control.
We used the Kaplan–Meier method to estimate OS, PFS and DOR in each treatment arm. To estimate the magnitude of the treatment difference (that is, hazard ratio) between the treatment arms, we used a stratified Cox proportional hazards model with the Efron method for handling ties. The stratification factors used for randomization were applied to the stratified Cox model. The stratified Miettinen and Nurminen method, with weights proportional to the stratum size, was used for comparison of ORR between the treatment arms. For ORR, 95% confidence intervals were based on the binomial exact confidence interval method for binomial data. Additionally, we assessed OS and PFS in protocol-prespecified subgroups based on participantsʼ baseline characteristics, including IMDC risk category and PD-L1 status. Post hoc analysis of efficacy was also performed for participants who completed 35 cycles of pembrolizumab. No formal hypothesis testing was conducted for the follow-up analysis of efficacy.
The biomarker analysis followed a statistical analysis plan written before merging of the clinical data with biomarker assessment, specifying where statistical testing would be used and what biomarker cutoffs defined the subgroups for treatment arm comparisons. The association between each biomarker and the clinical outcomes with pembrolizumab plus axitinib or sunitinib was assessed using logistic regression for ORR or a Cox proportional hazards regression model for OS and PFS. All models were adjusted by IMDC risk category, as prespecified in the statistical analysis plan. Statistical significance for associations between biomarkers and clinical outcomes (ORR, OS and PFS) was prespecified at α < 0.05 (without multiplicity adjustment) for TcellinfGEP, the angiogenesis signature and PD-L1 CPS separately; at α < 0.10 after multiplicity adjustment (Hochberg step-up procedure; tested as one family of 10 hypotheses (before TcellinfGEP adjustment) or nine hypotheses (after TcellinfGEP adjustment) within each treatment arm) for the other signatures and molecular subtypes; and at α < 0.10 after multiplicity adjustment for DNA mutations, as prespecified in the statistical analysis plan. The direction of the hypothesis tests (that is, non-zero association (two-tailed test), positive association or negative association (one-tailed test)) for the TcellinfGEP and other signatures was informed by an internal evaluation of published data from other trials, including the phase 3 JAVELIN Renal 101 trial of avelumab plus axitinib compared to sunitinib in participants with previously untreated advanced RCC9,11. The Spearman correlation was used to evaluate the relationship between pairs of RNA signatures and (or) biomarkers. Using prespecified cutoffs for the TcellinfGEP (≥ first tertile (TcellinfGEPnon-low) and < first tertile (TcellinfGEPlow) as previously defined and validated using pan-tumor clinical data23,49) and angiogenesis signature (≥ median and < median; the choice of median as the cutoff was for illustrative purposes) and mutational status (mutant versus wild-type) for VHL, PBRM1, SETD2 and BAP1, we performed descriptive subgroup analyses to estimate OS and PFS benefits of pembrolizumab plus axitinib compared to sunitinib and to assess the relative prognostic and predictive effects of the biomarkers. Statistical analyses were performed in SAS version 9.4 and R version 4.2.1 software. The data cutoff for this analysis was 23 January 2023.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Merck Sharp & Dohme (MSD), a subsidiary of Merck & Co., is committed to providing qualified scientific researchers access to anonymized data and clinical study reports from the company’s clinical trials for the purpose of conducting legitimate scientific research. MSD is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The MSD data-sharing website (https://externaldatasharing-msd.com/) outlines the process and requirements for submitting a data request. Applications will be promptly assessed for completeness and policy compliance. Feasible requests will be reviewed by a committee of MSD subject matter experts to assess the scientific validity of the request and the qualifications of the requestors. In line with data privacy legislation, submitters of approved requests must enter into a standard data-sharing agreement with MSD before data access is granted. Data will be made available for request after product approval in the United States and the European Union or after product development is discontinued. Certain circumstances may prevent MSD from sharing requested data, including country-specific or region-specific regulations. If the request is declined, it will be communicated to the investigator. Access to genetic or exploratory biomarker data requires a detailed, hypothesis-driven statistical analysis plan that is collaboratively developed by the requestor and MSD subject matter experts; after approval of the statistical analysis plan and execution of a data-sharing agreement, MSD will either perform the proposed analyses and share the results with the requestor or will construct biomarker covariates and add them to a file with clinical data that is uploaded to an analysis portal so that the requestor can perform the proposed analyses.
References
Keytruda (pembrolizumab injection), for intravenous use. Package insert. (Merck & Co., Inc., 2025).
Rini, B. I. et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N. Engl. J. Med. 380, 1116–1127 (2019).
Ljungberg, B. et al. EAU guidelines on renal cell carcinoma. European Association of Urology https://d56bochluxqnz.cloudfront.net/documents/full-guideline/EAU-Guidelines-on-Renal-Cell-Carcinoma-2023.pdf (2023).
Motzer, R. J. et al. NCCN Guidelines® Insights: Kidney Cancer, Version 2.2024. J. Natl Compr. Cancer Netw. 22, 4–16 (2024).
Powles, T. et al. Renal cell carcinoma: ESMO clinical practice guideline for diagnosis, treatment and follow-up. Ann. Oncol. 35, 692–706 (2024).
Raimondi, A. et al. Predictive biomarkers of response to immunotherapy in metastatic renal cell cancer. Front. Oncol. 10, 1644 (2020).
Lyskjær, I. et al. Management of renal cell carcinoma: promising biomarkers and the challenges to reach the clinic. Clin. Cancer Res. 30, 663–672 (2024).
Motzer, R. J. et al. Molecular subsets in renal cancer determine outcome to checkpoint and angiogenesis blockade. Cancer Cell 38, 803–817 (2020).
Motzer, R. J. et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat. Med. 26, 1733–1741 (2020).
Motzer, R. J. et al. Final overall survival and molecular analysis in IMmotion151, a phase 3 trial comparing atezolizumab plus bevacizumab vs sunitinib in patients with previously untreated metastatic renal cell carcinoma. JAMA Oncol. 8, 275–280 (2022).
Saliby, R. M. et al. Impact of renal cell carcinoma molecular subtypes on immunotherapy and targeted therapy outcomes. Cancer Cell. 42, 732–735 (2024).
Signoretti, S. et al. Renal cell carcinoma in the era of precision medicine: from molecular pathology to tissue-based biomarkers. J. Clin. Oncol. 36, JCO2018792259 (2018).
Hakimi, A. A. et al. A pan-cancer analysis of PBAF complex mutations and their association with immunotherapy response. Nat. Commun. 11, 4168 (2020).
Hakimi, A. A. et al. Transcriptomic profiling of the tumor microenvironment reveals distinct subgroups of clear cell renal cell cancer: data from a randomized phase III trial. Cancer Discov. 9, 510–525 (2019).
McDermott, D. F. et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 24, 749–757 (2018).
Miao, D. et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359, 801–806 (2018).
Santos, M. et al. PBRM1 and KDM5C cooperate to define high-angiogenesis tumors and increased antiangiogenic response in renal cancer. Am. J. Cancer Res. 13, 2116–2125 (2023).
Braun, D. A. et al. Clinical validation of PBRM1 alterations as a marker of immune checkpoint inhibitor response in renal cell carcinoma. JAMA Oncol. 5, 1631–1633 (2019).
Choueiri, T. K. et al. Biomarker analysis from the phase 3 CheckMate 9ER trial of nivolumab + cabozantinib v sunitinib for advanced renal cell carcinoma (aRCC). J. Clin. Oncol. 41, 608 (2023).
Braun, D. A. et al. 1694MO Novel serum glycoproteomic biomarkers predict response to nivolumab plus cabozantinib (NIVO+CABO) versus sunitinib (SUN) in advanced RCC (aRCC): analysis from CheckMate 9ER. Ann. Oncol. 35, S1012–S1013 (2024).
Machaalani, M. et al. KIM-1 as a circulating biomarker in metastatic RCC: post-hoc analysis of JAVELIN Renal 101. J. Clin. Oncol. 43, 594 (2025).
Xu, W. et al. Evaluation of circulating kidney injury marker-1 (KIM-1) as a prognostic and predictive biomarker in advanced renal cell carcinoma (aRCC): post-hoc analysis of CheckMate 214. J. Clin. Oncol. 43, 437 (2025).
Cristescu, R. et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362, eaar3593 (2018).
Cristescu, R. et al. Transcriptomic determinants of response to pembrolizumab monotherapy across solid tumor types. Clin. Cancer Res. 28, 1680–1689 (2022).
Bellmunt, J. et al. Putative biomarkers of clinical benefit with pembrolizumab in advanced urothelial cancer: results from the KEYNOTE-045 and KEYNOTE-052 landmark trials. Clin. Cancer Res. 28, 2050–2060 (2022).
Ott, P. A. et al. T-cell-inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J. Clin. Oncol. 37, 318–327 (2019).
Shitara, K. et al. Association between gene expression signatures and clinical outcomes of pembrolizumab versus paclitaxel in advanced gastric cancer: exploratory analysis from the randomized, controlled, phase III KEYNOTE-061 trial. J. Immunother. Cancer 11, e006920 (2023).
McDermott, D. F. et al. Association of gene expression with clinical outcomes in patients with renal cell carcinoma treated with pembrolizumab in KEYNOTE-427. J. Clin. Oncol. 38, 5024 (2020).
Ayers, M. et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 127, 2930–2940 (2017).
Saliby, R. M. et al. Update on biomarkers in renal cell carcinoma. Am. Soc. Clin. Oncol. Educ. Book 44, e430734 (2024).
Powles, T. et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 21, 1563–1573 (2020).
Plimack, E. R. et al. Pembrolizumab plus axitinib versus sunitinib as first-line treatment of advanced renal cell carcinoma: 43-month follow-up of the phase 3 KEYNOTE-426 study. Eur. Urol. 84, 449–454 (2023).
Motzer, R. J. et al. Biomarker analyses from the phase III randomized CLEAR trial: lenvatinib plus pembrolizumab versus sunitinib in advanced renal cell carcinoma. Ann. Oncol. 36, 375–386 (2024).
Lu, C. et al. Understanding the dynamics of TKI-induced changes in the tumor immune microenvironment for improved therapeutic effect. J. Immunother. Cancer 12, e009165 (2024).
Nuti, M. et al. Immunomodulatory effects of tyrosine kinase inhibitors (TKIs) in renal cell carcinoma (RCC) patients. J. Clin. Oncol. 35, e14506 (2017).
Aparicio, L. M. A. et al. Tyrosine kinase inhibitors reprogramming immunity in renal cell carcinoma: rethinking cancer immunotherapy. Clin. Transl. Oncol. 19, 1175–1182 (2017).
Motzer, R. J. et al. Lenvatinib plus pembrolizumab versus sunitinib in first-line treatment of advanced renal cell carcinoma: final prespecified overall survival analysis of CLEAR, a phase III study. J. Clin. Oncol. 42, 1222–1228 (2024).
Tannir, N. M. et al. Nivolumab plus ipilimumab versus sunitinib for first-line treatment of advanced renal cell carcinoma: extended 8-year follow-up results of efficacy and safety from the phase 3 CheckMate 214 trial. Ann. Oncol. 35, 1026–1038 (2024).
Motzer, R. J. et al. Nivolumab plus cabozantinib (N+C) vs sunitinib (S) for previously untreated advanced renal cell carcinoma (aRCC): final follow-up results from the CheckMate 9ER trial. J. Clin. Oncol. 43, 439 (2025).
Reddy, A. et al. Biomarker-driven prospective clinical trial in renal cell carcinoma: developing machine learning models to allocate patients to treatment arms using RNA sequencing. J. Clin. Oncol. 41, 4525 (2023).
Chouieri, T. SAMETA: a phase III study of savolitinib + durvalumab vs sunitinib and durvalumab monotherapy in patients with MET-driven, unresectable, locally advanced/metastatic papillary renal cell carcinoma. Oncologist 28, S11–S12 (2023).
Monjaras-Avila, C. U. et al. The tumor immune microenvironment in clear cell renal cell carcinoma. Int. J. Mol. Sci. 24, 7946 (2023).
US Department of Health and Human Services. Guidance for industry: clinical trial endpoints for the approval of cancer drugs and biologics (2018).
Hu, J., Ge, H., Newman, M. & Liu, K. OSA: a fast and accurate alignment tool for RNAseq. Bioinformatics 28, 1933–1934 (2012).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
Cristescu, R. et al. Tumor mutational burden predicts the efficacy of pembrolizumab monotherapy: a pan-tumor retrospective analysis of participants with advanced solid tumors. J. Immunother. Cancer 10, e003091 (2022).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Bamford, S. et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 91, 355–358 (2004).
Ayers, M. et al. Molecular profiling of cohorts of tumor samples to guide clinical development of pembrolizumab as monotherapy. Clin. Cancer Res. 25, 1564–1573 (2019).
Acknowledgements
We thank the participants, their families and caregivers for their involvement in this trial as well as all investigators and site personnel. Medical writing and (or) editorial assistance was provided by O. T. Ezeokoli and R. Steger of ApotheCom. This assistance was funded by Merck Sharp & Dohme, a subsidiary of Merck & Co. This work was supported by Merck Sharp & Dohme. The sponsor played a role in the design and conduct of the study; the collection, management, analysis and interpretation of the data; and the preparation, review and approval of the paper.
Author information
Authors and Affiliations
Contributions
B.I.R., E.R.P., D.S., S.W., A.L., R.F.P., J. Burgents and T.P. conceptualized the study. B.I.R., E.R.P., V.S., R.G., T.W., D.N., F.P., B.A., B.M., I.V., S.J.d.A., R.S.M., J. Bedke, S.W. and T.P. contributed to the acquisition of the data. B.I.R., E.R.P., R.G., T.W., F.P., B.A., B.M., J. Bedke, S.W., J.M. and A.V. curated the data. B.I.R., D.N., R.G., T.W., D.S., B.M., S.J.d.A., D.B., S.W., J.M., Y.Z., A.V. and T.P. analyzed and interpreted the data. B.I.R., E.R.P., T.W., D.S., S.W., Y.Z., A.L., A.V., R.F.P., J. Burgents and T.P. contributed to the study methodology. A.V. provided software. D.S., S.T. and J.M. administered the project. B.A. and J. Bedke contributed to funding acquisition. B.A., B.M. and J. Bedke provided resources. B.I.R., E.R.P., T.W., B.A., D.S., J. Bedke, S.T., A.L., R.F.P. and J. Burgents supervised the study. B.I.R., V.S., T.W., F.P., B.A., D.S., B.M., S.J.d.A. and J. Bedke validated the results. B.I.R., B.A., D.S., J. Bedke, Y.Z. and A.V. contributed to data visualization. B.I.R., D.S., A.V. and T.P. wrote the original draft of the paper. All authors reviewed or edited the paper for important intellectual content and approved the final draft for publication.
Corresponding author
Ethics declarations
Competing interests
B.I.R. reports receiving writing support for the present paper from Merck Sharp & Dohme (MSD); payment to his institution for clinical trial research from MSD; and consulting fees from MSD. E.R.P. reports receiving honoraria for scientific advisory from 23andMe, AbbVie, Adaptimmune, Astellas, AstraZeneca, Aura Biosciences, Bristol Myers Squibb, Daiichi Sankyo, Eisai, EMD Serono, Exelixis, IMV, Merck, Pfizer, Seagen, Seattle Genetics, Signatera, Synthekine, 23andMe and UroGen and continuing medical education talks for Cancer Network Readout 360, Clinical Care Options, CUA, Everyday Health Med Page Today, GU Oncology Now, Mashup Media, Mayo Clinic Hematology–Oncology Reviews, MJH Oncology, OncLive, PlatformQ, Research to Practice, Targeted Oncology, TouchME and VJ Oncology. V.S. reports receiving honoraria for presentations by Pfizer and support for attending meetings and (or) travel from Bionorica. R.G. reports funding for the present paper from MSD; grants or contracts to him and his institution from Astellas, AstraZeneca, Bristol Myers Squibb, Bayer, Ipsen, Janssen, Pfizer and Roche; payment or honoraria for lectures, presentations, speakersʼ bureaus, paper writing or educational events from Astellas, AstraZeneca, Bristol Myers Squibb, Bayer, Ipsen, Janssen, Pfizer and Roche; support for attending meetings and (or) travel from Astellas, AstraZeneca, Bristol Myers Squibb, Bayer, Ipsen, Janssen, Pfizer and Roche; and participation on a data safety monitoring board or advisory board for Astellas, AstraZeneca, Bristol Myers Squibb, Bayer, Ipsen, Janssen, Pfizer and Roche. T.W. reports a research grant paid to his institution from MSD; honoraria for lectures, presentations, speakersʼ bureaus, paper writing or educational events from Bristol Myers Squibb, EUSA Pharma, Ipsen and Pfizer; travel support from Bristol Myers Squibb, EUSA Pharma and Ipsen; and advisory board fees from Bristol Myers Squibb, Eisai, EUSA Pharma, Ipsen and MSD. D.N. reports honoraria for lectures from Astellas, AstraZeneca, Eisai, Pfizer and Roche and honoraria for participation on advisory boards from AstraZeneca, Pfizer and Roche. F.P. reports grants or contracts for research from Astellas, Janssen, Merck & Co., Novartis, Pfizer, Tersera and Tolmar; consulting fees from Astellas, Bayer, Janssen, Novartis, Tersera and Tolmar; payment or honoraria for lectures, presentations, speakersʼ bureaus, paper writing or educational events from Astellas, Bayer, Janssen, Novartis, Tersera and Tolmar; support for attending meetings and (or) travel from Novartis, Tersera and Tolmar; and leadership or fiduciary role for the National Cancer Institute Prostate Cancer Task Force and the Canadian Cancer Trials Group Genito-Urinary Clinical Trial Group. B.A. reports support for the present paper from MSD; grants or contracts from Astellas, AstraZeneca, Bayer, Bristol Myers Squibb, Eisai, Ipsen, Janssen, Merck & Co., Pfizer and Roche; consulting fees from Astellas, AstraZeneca, Bayer, Eisai, Ipsen, Janssen, Merck & Co., MSD, Pfizer and Roche; payment or honoraria for lectures, presentations, speakersʼ bureaus, paper writing or educational events from Astellas, AstraZeneca, Bayer, Eisai, Ipsen, Janssen, Merck & Co., MSD, Pfizer and Roche; support for attending meetings and (or) travel from Astellas, AstraZeneca, Bayer, Eisai, Ipsen, Janssen, Merck & Co. and Roche; and participation on a data safety monitoring board or advisory board for Astellas, AstraZeneca, Bayer, Eisai, Ipsen, Janssen, Merck & Co., Pfizer and Roche. D.S. reports support for the present paper from MSD; grants to his institution from Bristol Myers Squibb, Merck & Co. and Novartis; consulting fees from Bristol Myers Squibb, Eisai, Ipsen, Merck & Co. and Pfizer; and payment or honoraria for lectures, presentations, speakersʼ bureaus, paper writing or educational events from Bristol Myers Squibb, Merck & Co. and Pfizer. B.M. reports receiving honoraria for participation on advisory boards from Astellas, AstraZeneca, Bristol Myers Squibb, Eli Lilly, Eisai, Merck Serono, MSD, Novartis, Pfizer, Pierre Fabre, Roche and Servier; honoraria for lectures from Astellas, AstraZeneca, Bristol Myers Squibb, Eli Lilly, Eisai, Merck Serono, MSD, Novartis, Pfizer, Pierre Fabre, Roche and Servier; and support for attending meetings and (or) travel from AstraZeneca, Bristol Myers Squibb, Merck Serono and MSD. D.B. reports grants to her institution from Bristol Myers Squibb and Pfizer; grants or contracts to her institution for clinical trials from Aveo, Bristol Myers Squibb, Exelixis, MSD, Roche and Pfizer; consulting fees for consulting or advisory board from Bristol Myers Squibb, Ipsen, MSD and Pfizer; and support for traveling to meetings from Ipsen, MSD and Pfizer. R.S.M. reports payment or honoraria for lectures, presentations, speakersʼ bureaus, paper writing or educational events from Astellas, Bristol Myers Squibb, Ipsen, Janssen and MSD and support for attending meetings and (or) travel from Bayer, Novartis and Pfizer. J. Bedke reports payment or honoraria for lectures, presentations, speakersʼ bureaus, paper writing or educational events from Apogepha, Astellas, Bristol Myers Squibb, Eisai, F. Hoffman-La Roche, Gilead Sciences, Ipsen Innovation, Janssen, Merck KGaA, MSD, Pfizer and Seagen. S.T. reports payment or honoraria for lectures, presentations, speakersʼ bureaus, paper writing or educational events from Merck Biopharma, MSD, Pfizer and Takeda. S.W., J.M., Y.Z., A.L., A.V., R.F.P. and J. Burgents are employees of MSD, a subsidiary of Merck & Co., and own stock in Merck & Co. T.P. reports grants or contracts from AstraZeneca, Roche, Bristol Myers Squibb, Exelixis, Ipsen, MSD, Novartis, Pfizer, Seattle Genetics, Merck Serono, Astellas, Johnson & Johnson and Eisai; consulting fees and honoraria from AstraZeneca, Bristol Myers Squibb, Exelixis, Incyte, Ipsen, MSD, Novartis, Pfizer, Seattle Genetics, Merck Serono, Astellas, Johnson & Johnson, Eisai, Roche, Mashup and Gilead Sciences; support for attending meetings and (or) travel from Roche, Pfizer, MSD, AstraZeneca, Ipsen, Gilead Sciences, Astellas and MSD; participation on a data safety monitoring board or advisory board for AstraZeneca, Roche, Bristol Myers Squibb, Exelixis, Ipsen, MSD, Novartis, Pfizer, Seattle Genetics, Merck Serono, Astellas, Johnson & Johnson and Eisai; and other financial interests from Novartis, Pfizer, AstraZeneca, Roche/Genentech, Eisai and Merck. I.V. and S.J.d.A. declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Ganessan Kichenadasse and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Ulrike Harjes, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Correlogram plot of PD-L1 CPS, RNA sequencing signatures, and single genes.
CPS, combined positive score; EMT, epithelial-to-mesenchymal transition; gMDSC, granulocytic myeloid-derived suppressor cell; mMDSC, monocytic myeloid-derived suppressor cell; PD-L1, programmed cell death ligand 1; TcellinfGEP, T-cell–inflamed gene expression profile; TGF-β, transforming growth factor β.
Extended Data Fig. 2 Clustering and distribution of molecular subtypes.
a, Molecular subtype clustering. b, Distribution by IMDC risk categories. c, Distribution by tumor PD-L1 CPS. Molecular subtypes are based on transcriptomically defined clustering patterns identified in the IMmotion151 phase 3 study8. “Other” includes samples that could not be assigned to the angiogenic/stromal, angiogenic, immune/proliferative, proliferative, or stromal/proliferative subtype. CPS, combined positive score; EMT, epithelial-to-mesenchymal transition; gMDSC, granulocytic myeloid-derived suppressor cell; HIF-2α, hypoxia-inducible factor- α; IMDC, International Metastatic Renal Cell Carcinoma Database Consortium; mMDSC, monocytic myeloid-derived suppressor cell; PD-L1, programmed cell death ligand 1; TcellinfGEP, T-cell–inflamed gene expression profile; TGF-β, transforming growth factor β.
Extended Data Fig. 3 ORR by molecular subtypes and mutational status.
a, ORR per transcriptomically defined clustering pattern identified in the IMmotion151 phase 3 study8 (pembrolizumab plus axitinib, n = 369; sunitinib, n = 361). b, ORR by mutational status in the pembrolizumab plus axitinib arm (n = 347). c, ORR by mutational status in the sunitinib arm (n = 351). In panel a, “Other” includes samples that could not be assigned to angiogenic/stromal, angiogenic, immune/proliferative, proliferative, or stromal/proliferative subtype. In panels a-c, bars represent ORR, derived by dividing the number of participants with a complete or partial response by the total number of patients in that subgroup, multiplied by 100. Error bars indicate 95% confidence intervals. P values shown in panels b and c are multiplicity-adjusted (one-sided for pembrolizumab plus axitinib and two-sided for sunitinib) and were derived using logistic regression model, with adjustment for IMDC risk category. Significance was prespecified at α = 0.10. BAP1, BRCA1-associated protein 1 gene; IMDC, International Metastatic Renal Cell Carcinoma Database Consortium; NS, not significant; ORR, objective response rate; PBRM1, polybromo-1 gene; SETD2, SET domain containing 2, histone lysine methyltransferase gene; VHL, von Lindau-Hippel tumor suppressor gene.
Extended Data Fig. 4 Kaplan-Meier estimates of survival by VHL mutational status.
a, Overall survival. b, Progression-free survival. HR was estimated using a Cox proportional hazards model, with adjustment for IMDC risk category. Tick marks represent censored data. HR, hazard ratio; IMDC, International Metastatic Renal Cell Carcinoma Database Consortium; OS, overall survival; PFS, progression-free survival; VHL, von Lindau-Hippel tumor suppressor gene; WT, wild type.
Extended Data Fig. 5 Kaplan-Meier estimates of survival by PBRM1 mutational status.
a, Overall survival. b, Progression-free survival. HR was estimated using the Cox proportional hazards model, with adjustment for IMDC risk category. HR, hazard ratio; IMDC, International Metastatic Renal Cell Carcinoma Database Consortium; NE, not evaluable; OS, overall survival; PBRM1, polybromo-1 gene; PFS, progression-free survival; WT, wild type.
Extended Data Fig. 6 Kaplan-Meier estimates of survival by SETD2 mutational status.
a, Overall survival. b, Progression-free survival. HR was estimated using the Cox proportional hazards model, with adjustment for IMDC risk category. HR, hazard ratio; IMDC, International Metastatic Renal Cell Carcinoma Database Consortium; OS, overall survival; PFS, progression-free survival; SETD2, SET domain containing 2, histone lysine methyltransferase gene; WT, wild type.
Extended Data Fig. 7 Kaplan-Meier estimates of survival by BAP1 mutational status.
a, Overall survival. b, Progression-free survival. HR was estimated using the Cox proportional hazards model, with adjustment for IMDC risk category. BAP1, BRCA1-associated protein 1 gene; HR, hazard ratio; IMDC, International Metastatic Renal Cell Carcinoma Database Consortium; OS, overall survival; PFS, progression-free survival; WT, wild type.
Supplementary information
Supplementary Information
Supplementary Tables 1–5 and Supplementary Fig. 1
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rini, B.I., Plimack, E.R., Stus, V. et al. Pembrolizumab plus axitinib versus sunitinib for advanced clear cell renal cell carcinoma: 5-year survival and biomarker analyses of the phase 3 KEYNOTE-426 trial. Nat Med (2025). https://doi.org/10.1038/s41591-025-03867-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41591-025-03867-5
