
Abstract
Although genomic sequencing presents groundbreaking newborn screening (NBS) opportunities, critical feasibility and utility questions remain. Here we present initial results from the Early Check program—an observational study assessing the feasibility and clinical utility of genomic NBS in North Carolina. Recruitment was statewide through mailed letters with electronic consent. Genome sequencing with analysis of 169 high actionability genes (plus 29 optional lower actionability genes) was performed using residual NBS dried blood spots. In 8 months, 1,979 newborns were screened, with 50 (2.5%) screen positives. Negative results were returned electronically, positive results by genetic counselors. Twenty-eight results (55%) were true positives, all received anticipatory guidance, surveillance and management recommendations, and referral to specialists as appropriate. We report technical feasibility and preliminary clinical utility finding, along with interpretation and follow-up challenges that hinder public health implementation. We propose standardized terminology to facilitate cross-study comparisons and accurate characterization of genomic NBS outcomes.
Similar content being viewed by others


Main
Newborn screening (NBS) programs in the United States are established, state-based public health programs designed to identify health conditions in newborns for which early treatment substantially improves outcomes. Advances in genomic sequencing technology and a rapidly expanding treatment landscape offer opportunities to greatly expand NBS programs. Despite its promise, introducing genomic sequencing to NBS without adequate preparation could disrupt the current screening infrastructure and endanger this profoundly successful public health offering1. Thus, research programs worldwide are generating data to inform future decision making and support effective implementation2. Results from initial small cohorts have been published3,4,5,6.
Before genomic NBS (gNBS) can be incorporated into public health, key issues related to feasibility, acceptability, utility and the ethics of sequencing-based population-wide screening must be addressed. Numerous unresolved questions include whether and how to obtain parental consent, store genomic data and revise criteria to determine what genes to screen7,8. Furthermore, the utility of using existing diagnostic variant data to interpret population-based screening results must be explored, as penetrance in unselected populations is largely unknown. Clinicians will need tools to aid in anticipating phenotypes based on genotypes and develop appropriate follow-up to avoid overmedicalization. Cost effectiveness must also be demonstrated9. Diverse cohorts of participants are needed to address potential healthcare disparities that gNBS could exacerbate10.
Early Check is an NBS research program that has offered supplemental NBS to infants born in North Carolina since October 201811,12,13,14. Led by researchers at RTI International in partnership with the University of North Carolina at Chapel Hill (UNC), GeneDx and the North Carolina State Laboratory of Public Health (NCSLPH), Early Check transitioned to gNBS in September 2023. Here we report Early Check results for the first 1,979 newborns screened by gNBS.
Spanning the entire state of North Carolina, Early Check is positioned uniquely to address critical questions about incorporating technological and therapeutic innovations into NBS. The overall objective of the program is to inform whether, when and how gNBS could be implemented universally using existing NBS infrastructure or a parallel public health system.
Study aims for the first participants reported here include to (1) characterize study enrollment and panel selection choices, (2) assess feasibility of using state-collected NBS cards for gNBS, (3) return screen-positive results and measure workforce efforts, (4) characterize screen-positive outcomes, (5) compare gNBS to standard NBS results and (6) develop nomenclature to categorize gNBS molecular diagnoses and report on related interpretation challenges.
Results
Enrollment rates
Between 28 September 2023 and 10 June 2024 (~8.5 months), a total of 2,125 newborns were enrolled. Recruitment letters were mailed to mothers of 42,434 newborns. We estimate approximately five enrollments for every 100 letters mailed (~5%). During the same period, 816 postpartum mothers were approached at UNC Hospital. Most (71%) were willing to learn about Early Check, and 183 (32%) enrolled their newborn during the encounter; we cannot estimate how many enrolled after their engagement with the recruiter. Few parents completed the decliners survey (n = 16). Reasons for not enrolling endorsed by three or more parents included concerns with DNA storage, worry about my child’s wellbeing, worry about my child’s privacy and not interested in research studies.
Participant characteristics
Compared to birthing mothers in North Carolina, self-report race/ethnicity data in Early Check indicated over-enrollment of white participants (58.8% versus 50.8%) (Table 1). Representation was lower among participants with both parents identifying exclusively as Black or Hispanic. However, significantly more Early Check participants reported two or more race/ethnicity categories than reported by the North Carolina State Center for Health Statistics (22.1% versus 2.9%).
Panel selection
Panel 1 included 178 gene–condition pairs (169 unique genes) with high actionability during the first 2 years of life, whereas Panel 2 included an additional 29 gene–condition pairs. All enrolled newborns were screened for Panel 1 conditions. Additionally, 82.9% of parents opted for Panel 2. In comparison to mothers of white babies, mothers of babies reported as African American/Black (odds ratio (OR) = 0.37; P < .0001), Asian (OR = 0.61; P < .05), Hispanic/Latino or Spanish (OR = 0.43; P < 0.001) and Unknown/Not Reported (OR = 0.30; P < .01) were significantly less likely to choose Panel 2 screening.
Frequency of successful screening
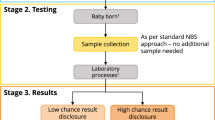
Of 2,125 enrolled newborns, screening was completed for 1,979 (93%). Successful screening required a series of steps. First, the Early Check team identified and ‘matched’ the state NBS card with the consenting parent. Second, the team confirmed sufficient residual dried blood spot (DBS) material to punch. Third, the laboratory confirmed adequate DNA extracted for genome sequencing (GS). Participant attrition at each step is shown in Fig. 1. All consents that did not match an NBS card had discrepant information (for example, mother’s name did not match) and the consenting parent did not respond to requests for identity verification. One participant was administratively withdrawn after learning that the father provided consent on behalf of the mother, without the mother’s permission. Parents of newborns whose samples failed at any step were notified that screening was not completed. No significant differences in race/ethnicity were observed between newborns who were and were not sequenced successfully.

Enrolled: signed the consent. Matched: consent identifiers matched to an NBS card. Sampled: sufficient DBS sample remained on the NBS card to punch. Sequenced: successful sequencing from DBS punches. Published: participants reported in this paper.
Turnaround time
Median time from consent to return of results was 35 days (range 16–104 days) for negative results and 38 days (range 24–82 days) for positive results. The median time from consent to shipping samples to GeneDx was 11 days (range 1–63 days). The average time from sample arrival at GeneDx to completion of the results report was 13 days (range 6–40 days). The median time from completion of the results report, transfer of the report between institutions and availability of report in the Early Check portal was 5 days (range 1–58 days).
Screen-positive results
Fifty newborns (2.5%) received a positive screening result. One newborn had two positive results (SCN1A and MITF). Most positive results were from Panel 1 (n = 47), with four from Panel 2 (two SCN1A and two MECP2; Supplementary Table 1). The most common screen-positive results were G6PD deficiency and a pathogenic (P) variant (c.952G>A) in MITF associated with increased risk for melanoma15. Variants in G6PD accounted for nearly half of all screen positives, with a positive rate of 1.2%. The MITF gene was included in Panel 1 due to its association with Waardenburg syndrome, not melanoma risk16; therefore, the c.952G>A variant is considered an off-target result (phenotypic false positive). Excluding the 11 newborns whose only finding was the off-target c.952G>A variant in MITF, the overall positive screening rate was 2.0%. Excluding both MITF and G6PD screen positives, the overall positive screening rate was 0.8%.
Return of results
Several metrics were collected for each screen-positive result (Table 2). At the time of screening result disclosure, two newborns already had symptoms or a clinical diagnosis of the condition (COL2A1—Stickler syndrome, TMPRSS3—congenital hearing loss). Four parents were aware of a positive family history (F8—Hemophilia A, COL2A1—Stickler syndrome) or their carrier status (SLC26A4—Pendred syndrome, G6PD—G6PD deficiency) before enrollment. Screen-negative results (n = 1,929; 97.5%) were returned electronically.
Confirmatory molecular testing
Of 50 newborns with screen-positive results, 37 parents (74%) agreed to follow-up testing of their newborn and 32 (64%) provided newborn buccal samples for confirmatory molecular testing. One newborn had undergone clinical genetic testing for COL2A1—Stickler syndrome due to family history, so further confirmatory testing was unnecessary. Samples for follow-up testing were collected at home for 28 newborns and at UNC for four newborns. In all completed cases, the variant(s) identified on screening were confirmed. At publication, 17 participants (34%) had not completed molecular confirmation, mostly for nonurgent conditions (G6PD (n = 12); MITF (n = 3)).
Additional follow-up testing
Additional testing beyond molecular confirmation was recommended for seven newborns and completed for six (Table 2). Hearing evaluation confirmed the initial screen-positive result for two newborns (SLC26A4—Pendred syndrome, TMPRSS3—hearing loss). Other orthogonal testing modalities did not confirm any diagnoses. Concurrent biochemical testing for the newborn with IDUA variants demonstrated normal enzyme activity; urine glycosaminoglycan was not completed due to an unsatisfactory specimen. For three others, follow-up testing was normal or inconclusive; these newborns were deemed at risk to develop symptoms later in life (COL4A5—Alport syndrome, SCNN1B—pseudohypoaldosteronism, DUOX2—thyroid dyshormonogenesis).
Penetrance in infancy
Most infants with screen-positive results did not have signs or symptoms of the condition within the first few months of life. Three exhibited symptoms consistent with their molecular diagnoses: SLC26A4—Pendred syndrome, TMPRSS3—hearing loss and COL2A1—Stickler syndrome (Supplementary Table 1). Six infants had symptoms that were potentially associated with their molecular diagnoses. Five with G6PD deficiency had a history of neonatal jaundice, and one with a likely pathogenic (LP) variant in SCN1A had seizure-like episodes, although a normal EEG.
Comparison with standard state NBS
Three screen-positive results could have resulted in abnormal NBS due to their clear association with hearing loss (SLC26A4—Pendred syndrome, COL2A1—Stickler syndrome, TMPRSS3—hearing loss). However, only the newborn with variants in TMPRSS3 failed the NBS hearing screen. The newborns with SLC26A4 and COL2A1 variants were confirmed to have Pendred syndrome and Stickler syndrome, respectively, but did not have congenital hearing loss, consistent with natural history studies that indicate variable expression at birth17,18.
A newborn with a screen-positive result (DUOX2—thyroid dyshormonogenesis) had negative NBS despite having a homozygous P frameshift variant. Follow-up studies revealed normal thyroid function, and this result remains categorized as a ‘probable’ molecular diagnosis. Continued follow-up will be necessary due to the incomplete penetrance and variable expressivity described in this condition19.
Standard NBS results were reviewed for all 1,979 newborns screened by Early Check. Three newborns with confirmed diagnoses through standard state NBS were not detected by Early Check, indicating potential false negatives on gNBS. First was a newborn with sickle cell disease. Although initial gNBS was reported to the parents as negative, reinspection of the GS data revealed a homozygous P variant p.E7V in the HBB gene, and a revised, positive screening report was issued.
The second false negative was a newborn with late-onset Pompe disease. Standard NBS follow-up identified one P variant and one variant of uncertain significance (VUS) in the GAA gene. Since Early Check does not report VUS, this participant was assessed as a carrier and the result was not reported as screen positive. The third was a newborn with congenital hypothyroidism, which is most commonly not monogenic. Early Check did not screen for all possible monogenic causes. Thus, it is unclear whether this result represents a false negative result within the context of detectable monogenic forms of congenital hypothyroidism.
Molecular diagnosis and result classification
We applied study-developed rubrics (Table 3 and Fig. 2) to classify our screen-positive results (Table 4). Barriers to ‘definite’ molecular diagnoses included detection of LP (versus P) variants, inability to determine phase and potential variant misclassification. True positive or probable true positive was assigned to 28 results (55%). Four results (8%) were possible true positive/possible false positive and require longer follow-up.

A genomic screening test must first have an established set of criteria that define a ‘positive’ or ‘negative’ screen (1), typically variant-level thresholds (for example, pathogenic/likely pathogenic versus VUS) and zygosity requirements for any given monogenic condition. Variant confirmation in a new sample using an orthogonal method (2) can identify rare cases of sample swap (estimated at 1:1,000 for clinical laboratory workflows) or artifactual variant calls (probability depending on the type of variant). Both types of laboratory errors could be considered ‘technical’ false positives that will happen at a somewhat predictable frequency. Family studies to determine de novo occurrence or to phase putative biallelic variants (3) may reveal that two variants reported as a positive screen are actually in cis and therefore not a disease-causing genotype, considered an ‘allelic’ false positive or, alternatively, may reveal inheritance of a heterozygous variant from an unaffected parent, potentially causing the clinical laboratory to downgrade the variant, considered a ‘classification’ false positive. A molecular diagnosis is thereby established (4) with varying degrees of confidence (Table 3). With general population screening, many people will initially be asymptomatic. Subsequent clinical correlation results in accumulation of laboratory findings, clinical features and family history (5). People with a definite molecular diagnosis are considered a true positive even if they never develop signs or symptoms of the condition (nonpenetrant). If clinical findings consistent with the putative molecular diagnosis emerge, then it can be confirmed as a true positive with penetrance established (though expressivity may vary). Sometimes, the phenotypic consequence of the variant(s) identified may end up being different than the intent of the screening program, such as hypomorphic variants with milder phenotypes, or variants having a different molecular mechanism that cause a phenotype not intended as part of the screen. These could be considered ‘phenotypic’ false positives, even though such findings may ultimately have clinical relevance. People with probable, likely or possible molecular diagnoses who remain asymptomatic over time make up a category in which the screening result could represent a false positive or a true positive (with incomplete penetrance and/or variable expressivity). Longitudinal follow-up and management of these people will be needed, thus creating additional burden with unclear benefit. Rarely, a person with a negative screen may develop symptoms of a monogenic disease over time, indicating a potential false negative (6). As with ‘technical’ false positives, a sample swap or low confidence variant call could result in a false negative. Similarly, a single heterozygous variant in a gene associated with a recessive condition could result in an ‘allelic’ false negative if the other variant was not detected, or a ‘classification’ false negative if the other variant was a VUS and therefore not reported. The category of VUS in general presents a challenge for screening in both recessive and dominant conditions, potentially resulting in ‘classification’ false positives and/or false negatives, depending on whether this category of variant is returned in a screening program. In some cases, disease manifestations may be due to locus heterogeneity (where the gene responsible for symptoms was not included in the screen) or because symptoms are caused by a nonmonogenic etiology; these would be ‘phenotypic’ false negatives. Each of these potential scenarios is unlikely in genomic screening due to the rarity of the screened conditions, but clinicians must remain alert to the possibility of a false negative as they are for any condition included in standard public health NBS. If a person remains asymptomatic over time, a negative becomes increasingly likely to be a true negative. FN, false negative; FP, false positive; TN, true negative; TP, true positive.
Phenotypic false positives (n = 12, 24%) arose from detection of variants unrelated to the intended actionable condition, such as the MITF variant associated with melanoma risk. These diagnoses have clinical implications for participating newborns but were off-target findings. Parental testing revealed two maternally inherited variants in IDUA for one newborn, ruling out the initial molecular diagnosis of mucopolysaccharidosis type I. This result was classified as an allelic false positive. Four additional allelic false positives were due to detection of single heterozygous variants in genes associated with both autosomal dominant and autosomal recessive phenotypes and were more consistent with carrier status for the recessive condition.
Using our rubric, the sickle cell result that was initially reported as negative was classified as a technical false negative due to an oversight during a manual analysis step. This false negative may be correctable in the future by increasing automation support of sample analysis to increase throughput and reduce opportunities for error. The late-onset Pompe diagnosis detected on standard NBS but not gNBS is a classification false negative resulting from the stringency of the variant threshold used for reporting.
Clinical utility
The Early Check program demonstrated clinical utility by identifying actionable diagnoses for several families. One participant passed newborn hearing screening but was identified to have two P variants in SLC26A4. Auditory brainstem response performed at 15 weeks showed normal hearing sloping to mild/moderate sensorineural hearing loss. Repeat auditory brainstem response at 6 months showed bilateral mild sloping to severe mixed hearing loss. Brain MRI confirmed a diagnosis of Pendred syndrome. She received hearing aids at 8 months and is receiving speech therapy with good response to therapy goals. At 18 months she verbalizes 30–40 words and uses American sign language. Hearing loss continues to progress, with unilateral profound hearing loss. Assessment for cochlear implant is underway.
Two infants received actionable diagnoses of Stickler syndrome and TMPRSS3—hearing loss that were also identified clinically. Hearing loss in the infant with pathogenic variants in TMPRSS3 was initially attributed to congenital CMV until the molecular diagnosis was discovered. Parents of infants identified at risk for future morbidity received surveillance and management recommendations, along with connection to specialists, as needed.
Several screen-positive results from Panel 2 resulted in clinical actions, although were associated with greater uncertainty. One newborn with a de novo, previously reported, P variant in the MECP2 gene was diagnosed with Rett syndrome and referred to a multidisciplinary Rett Syndrome Clinic for management. Neurological and developmental assessments at 8 months were normal. The care team discussed the availability of trofinetide for children 2 years and older with the family; the family has expressed interest in interventional clinical trials for this or similar medications. Return appointment with the Rett Clinic team is planned for the second year of life to monitor development. One infant with an LP variant in SCN1A developed seizure-like episodes and was seen by neurology but subsequently lost to follow-up. Parents of the newborn with an LP variant in SCN1A that was downgraded by the sequencing laboratory to a VUS were given anticipatory guidance on seizures and are following with the pediatrician.
Discussion
By demonstrating strategies that are potentially scalable for population-level implementation, Early Check is a model for future public health adoption. Early Check conducts statewide recruitment, online consent and return of results, and sequencing from NBS-collected DBS. This approach is supported by a partnership with a high-throughput commercial sequencing laboratory. Although the feasibility of using GS to identify monogenic disease risks in newborns has been demonstrated, the ability to conduct gNBS at scale has yet to be shown in a true public health context. Early exploratory gNBS projects conducted by NC NEXUS and BabySeq, as part of the Newborn Sequencing in Genomic Medicine and Public Health consortium, focused on small cohorts (<200 newborns) using hospital-based recruitment and traditional consent approaches5,6. These studies did not use DBS samples and relied on time-intensive and logistically restrictive methods for return of results, including in-person visits with physicians and genetic counselors. Recently, results from larger gNBS studies that are being conducted concurrently with Early Check have been reported3,4. The GUARDIAN study recruits from six New York hospitals and uses NBS-collected DBS for gNBS4. BabyDetect recruits in one maternity ward in Belgium and collects a sample for targeted sequencing performed by the NBS reference laboratory3. To provide data on feasibility of statewide implementation in the United States, Early Check used statewide recruitment to successfully enroll, sequence and return results for nearly 2,000 newborns. Notably, 97.5% of parents enrolled their newborns and received results without direct interaction with study staff. However, this volume is just a small fraction of total births in the state. Substantial implementation challenges remain, particularly in scaling the return and follow-up of screen-positive results, turnaround time and addressing the cost of sequencing and variant interpretation, which are critical for broader public health adoption.
In the initial cohort of 1,979 newborns screened by Early Check, 2.5% had a positive screening result. If implemented at scale in North Carolina, which has approximately 120,000 annual births, this rate would represent 3,000 newborns with a screen-positive result each year. Screen-positive frequencies in other gNBS studies range from 1.8% to 7.9%, with higher positive screening rates observed in studies screening for larger numbers of genes3,4,5,6,20. However, one empirical study estimated the global birth prevalence of rare monogenic conditions to be ~9 of 1,000 (0.9%)21, suggesting that many of these molecular findings may not result in clinical diagnoses. Without long-term follow-up data, it remains unclear how many newborns with positive screening results will develop symptoms. Most newborns with screen-positive results in Early Check were asymptomatic during the short-term follow-up period. This finding highlights a key challenge in population-wide screening: the identification of P variants with reduced penetrance or misclassified pathogenicity22. Genes included in screening must be selected carefully, prioritizing those with highly penetrant variants and definitive confirmatory testing to avoid unnecessary surveillance for large numbers of newborns, associated parental worry and strain on the healthcare system. For example, G6PD deficiency accounted for nearly half of all screen-positive results in Early Check as well as other contemporaneous studies4, required considerable follow-up resources, and lacks robust evidence supporting the utility of screening in reducing morbidity. Thoughtful discussion among key interest holders is needed to determine whether G6PD deficiency should remain a candidate for NBS.
To standardize the interpretation and reporting of molecular diagnoses, Early Check developed reproducible classification systems that may be valuable resources for the broader community (Table 3 and Fig. 2). Consistent terminology is critical for cross-study comparisons and accurate characterization of gNBS outcomes. Current literature often lacks the granularity needed to distinguish the certainty or nuance of screen-positive results4. Screen positives are not a uniform category, especially among asymptomatic newborns, and reporting them in such simplistic terms fails to capture the complexity of genomic results. Likewise, false positives and false negatives are not homogenous categories23. Therefore, a deeper delineation of different types of false positive and false negative results is needed. For example, there could be technical (for example, sequencing errors) or allelic (for example, variants in cis) explanations for false positive or false negative results. For policy decision-makers less familiar with clinical genetics, rubrics such as ours will be critical for evaluating and communicating the implications of gNBS results.
Previous studies described challenges reporting all P and LP variants in screened genes consistent with condition inheritance patterns3, which was also experienced in this study. Although many rare monogenic diseases follow a simple ‘one gene to one disease’ relationship, some genes have been implicated in more than one distinct disease entity, often differentiated by a combination of molecular mechanism, inheritance pattern and/or phenotypic features. For example, the TECTA gene has been implicated in both autosomal dominant (DFNA12) and autosomal recessive (DFNB21) forms of nonsyndromic hearing loss, which are thought to have dominant negative and loss of function mechanisms, respectively. The heterozygous frameshift variant identified in a newborn in our study is expected to result in loss of function and was inherited from the reportedly unaffected father. This result is therefore most consistent with carrier status and not with autosomal dominant TECTA-related hearing loss24,25. Furthermore, sequencing genes associated with more than one phenotype, such as MITF, may identify variants associated with phenotypes that may be considered off-target findings26. Predetermination of specific reportable variants, based on the associated phenotype, penetrance and expressivity, could reduce the number of carriers and off-target results.
Comparison of Early Check results with state NBS results identified three potential false negatives, which is consistent with several previous studies that demonstrated reduced clinical sensitivity of sequencing compared to standard biochemical NBS. The NC NEXUS study included blinded analysis of infants with known positive NBS results and failed to identify two newborns due to a single heterozygous P variant in one and a homozygous VUS in the other6. Another study found the overall sensitivity of exome sequencing was 88%, in comparison to 99% for tandem mass spectrometry27. Reduced sensitivity was attributed largely to the presence of variant types not detectable by exome sequencing (for example, copy number variations) and challenges in variant interpretation. Bodian et al. sequenced a cohort of 1,696 presumed healthy newborns and found generally good coverage of the genes associated with NBS conditions, yet three out of five newborns with state-screened conditions were missed by gNBS20. A recent study examined the sensitivity of sequencing to identify 635 newborns with inborn errors of metabolism, reporting an overall sensitivity of 80.3%. Notably, people self-identifying as Black were found to have significantly reduced sensitivity28. These findings underscore that gNBS cannot currently replace traditional NBS and presents additional challenges to avoid healthcare disparities. However, integrating appropriate workflows that combine tandem mass spectrometry with sequencing results could reduce both false negatives and false positives, potentially enhancing the overall effectiveness.
Despite many automated processes in Early Check, interpretation, return and follow-up of screen-positive results remained labor intensive, requiring genetics expertise. Unexpected genetic findings were not unusual and would be challenging for nongeneticists to interpret. More than half of newborns with screen-positive results required several attempts to reach the consenting parent by phone and, in some cases, the initial phone call lasted more than an hour. Contacting primary care providers to relay results also demanded considerable effort. The addition of sequencing to NBS would require additional specialized staffing at all stages of screening and follow-up, which may be difficult to accommodate due to budget limitations and the already existing shortage of clinical genetics providers29. Streamlined follow-up workflows would be necessary to make public health implementation feasible.
Cost, turnaround time and throughput are additional challenges to public health implementation. Although sequencing costs have decreased dramatically over the last two decades, they are still prohibitively high for universal screening30. Early Check’s median time from consent to positive result reporting of 38 days was substantially longer than the NBS standard of return within 5–7 days of life31. Due to Early Check being a research study, there are administrative procedures that are not part of standard NBS, such as matching consents to NBS cards, that add time to the process. Although rapid GS can reduce turnaround times to 5 days or fewer, this reduction in time is accompanied by a substantial increase in cost32.
Finally, this study highlights logistical implications for DBS sample collection. As NBS programs expand to include more conditions, the demand for greater sample volumes will grow, necessitating adjustments to DBS collection protocols. Some states are already considering expanding the number of DBS collected due to sample volume challenges. In a public health NBS setting, unsatisfactory samples would require collection of another DBS sample for a repeat attempt at sequencing, which again has workforce and cost implications.
There are limitations to the present study. Based on available data, demographic characteristics of newborns enrolled in Early Check were not representative of birthing mothers in the state. Furthermore, since this is a consented study, parents who are unsupportive of gNBS or uncomfortable participating in research are probably not represented. Early Check uses letter-based recruitment to achieve targeted enrollment numbers, which is unlikely to be an effective model for public health implementation. Our uptake from letters was approximately 5%, which exceeds other reports of letter-based recruitment in the literature (1–2%)33, but is not effective for universal access. Identification of false negatives is limited to those conditions detected on standard NBS; thus, we cannot determine an overall false negative rate. Longer-term follow-up is needed to distinguish false positives from true positives, as well as to identify false negatives.
Advances in rare disease therapeutics continue to expand the number of newborns with conditions that would benefit from a diagnosis before, or at the earliest stages of, phenotypic manifestation34. However, many questions remain about the feasibility, acceptability and utility of gNBS35. The study results presented here add to the growing body of evidence informing future policy decisions. Early Check stands apart from other gNBS studies due to its statewide recruitment in partnership with the state laboratory of public health, thus modeling potential public health implementation.
Emerging data from Early Check and other studies indicate the technical feasibility of gNBS using DBS. Improvement in bioinformatic pipelines and variant classification are anticipated, alongside reduced costs of short-read sequencing30 and increased adoption of advanced sequencing technologies like long-read sequencing36 in the coming years. Several other implementation challenges, such as the need for repeat sample collection in a small subset of children, are familiar to public health NBS programs. Studies like Early Check are essential for developing, testing and optimizing appropriate education and consent approaches, efficient return of results, streamlined follow-up procedures and family-centered supportive services. Further research is needed to demonstrate whether gNBS improves clinical outcomes—a critical question to answer before universal adoption can be considered.
Methods
Preimplementation planning for the transition of Early Check11,16 to gNBS began in 2021. Development of the protocol was informed by extensive formative research and guidance from an external advisory committee. The study protocol and electronic consent were reviewed and approved by the UNC Institutional Review Board (IRB no. 18-0009), with registration on Clinicaltrials.gov (NCT03655223). Parents use a self-directed web-based portal (https://portal.earlycheck.org), available in both English and Spanish, to learn more about the study and enroll their newborns. Mothers give permission for the newborn to participate unless the mother is not the legal guardian, in which case another guardian can enroll the newborn. GS of the first 2,000 newborns was provided in kind by GeneDx and Illumina. This publication reports that initial cohort. Surveys and interviews to assess the psychosocial impact of study participation on parents, and surveys to assess healthcare provider perceptions of clinical utility, are ongoing and will be reported separately.
Panel design
The gene panel design process was published previously16. Gene–condition pairs were evaluated using systematic, actionability based, frameworks and divided into two panels. Panel 1 included gene–condition pairs with high actionability and early age of action (≤2 years age at intervention initiation), comparable to core conditions on the Recommended Uniform Screening Panel. Panel 2 offers additional screening for gene–condition pairs that do not meet the high thresholds for Panel 1 inclusion but have emerging potential for high actionability, such as those with promising clinical trials. The Early Check Gene Panel Working Group included four clinical geneticists, one pediatrician practicing in primary care, five genetic counselors, one PhD social scientist/genetic counselor, one PhD genetic epidemiologist, one PhD translational genomics researcher and one clinical psychologist (Panel 2 meetings only). A previously developed framework for evaluating gene–condition pairs for newborn sequencing, the Age-based Semi Quantitative Metric (ASQM)38 was the starting point for development of the Panel 1 framework for Early Check. The ASQM assesses five criteria of clinical actionability on a scale of 0–3: severity and likelihood of the manifested condition, efficacy and acceptability of the intervention, and knowledge about the gene–condition association and intervention. Increasing ASQM scores, with a maximum of 15, suggest greater actionability. We prioritized gene–condition pairs with total scores of 13 or higher for Panel 1; however, those with scores of ≥9 could be considered. Gene–condition pairs that received most positive votes were approved for inclusion; unanimous agreement was not required. All currently Recommended Uniform Screening Panel conditions for which sequencing is informative were included on Early Check Panel 1, regardless of ASQM score. Although ASQM scores were calculated for Panel 2 gene–condition pairs, no minimum ASQM score was set for inclusion in Panel 2. At launch, Panel 1 included 178 gene–condition pairs (169 unique genes) with high actionability during the first 2 years of life (Supplementary Table 2), while Panel 2 (Supplementary Table 3) included an additional 29 gene–condition pairs.
Participants
For this pilot, the goal was to recruit a representative sample of 2,000 participants across the state. Eligible participants were newborns up to 31 days old who had undergone standard NBS in North Carolina and lived in North Carolina or South Carolina. The consenting parent also needed to provide an email address to enroll. The primary recruitment methods were letters addressed from the NCSLPH informing birthing parents about the voluntary, free Early Check program and in-person recruitment on the labor and delivery floor at UNC Hospital33. The online Early Check consent portal was opened for enrollment on 28 September 2023, enabling English- and Spanish-speaking birthing parents or legal guardians to enroll their newborns after birth39. There was no option for a paper consent, all participating newborns were enrolled through the online consent portal. Parents who were approached by an in-person recruiter and declined enrollment were asked to complete a decliners survey. All enrolled newborns were screened for conditions on Panel 1; consenting parents could decide to accept or decline optional screening for Panel 2 conditions and/or lifetime risk for type 1 diabetes determined by a genetic risk score40. Through the Early Check portal, the consenting parent provided the newborn’s sex and the birthing parent’s date of birth as well as self-reported the race/ethnicity of both parents. The race/ethnicity of the newborn was recorded as the combined self-reported race/ethnicity of both parents. Basic descriptive statistics were conducted to describe demographic characteristics of the study population. Logistic regression estimated the likelihood (ORs) of different racial/ethnic groups opting into Panel 2 screening. SAS Institute Inc. (2020) SAS Enterprise Guide v.8.3 was used for analyses.
Genome sequencing and variant classification
Screening was performed on the residual state-collected DBS after consent was obtained. For participants with insufficient DBS samples, it was not feasible to collect repeat DBS through the study. Eight deidentified 3.2 mm DBS punches were sent to GeneDx—a Clinical Laboratory Improvement Amendments (CLIA)-certified and College of American Pathologists (CAP)-accredited sequencing laboratory—for DNA extraction and sequencing. Sequencing was performed using PCR-free GS libraries prepared using Illumina DNA PCR-Free Prep, Tagmentation kit following the manufacturer’s protocol (Illumina Inc.). Massively parallel (NextGen) sequencing was performed on NovaSeq 6000 at 2 × 150. Alignment and variant calling for sequencing variants and most deletions greater than 1 kb in size were performed using the Dragen commercial platform41 and additional sequencing technology and variant interpretation protocol42. The general assertion criteria for variant classification are publicly available on the GeneDx ClinVar submission page (http://www.ncbi.nlm.nih.gov/clinvar/submitters/26957/).
Only the genes on Panel 1 and optional Panel 2 were interrogated and reported. Variants were classified according to criteria defined jointly by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology43. Reportable variants constituting screen-positive results included P and LP variants when consistent with the inheritance pattern. VUSs were not reported. GeneDx issued a clinical report for each participating newborn.
Molecular diagnosis and result classification
A rubric was developed to describe the molecular interpretation of screen-positive results, depending on the inheritance pattern, variant classification and confirmation status (Table 3). Results were also classified as true positive, possible true positive/possible false positive, false positive or false negative after a series of evaluations, as shown in Fig. 2. True positive results included ‘Definite’ molecular diagnoses that were either penetrant or nonpenetrant. ‘Probable’, ‘Likely’ and ‘Possible’ molecular diagnoses were also considered true positives if clinical correlation was consistent with the diagnosis (for example, the newborn was penetrant for a phenotypic feature of the condition). Categories of false positive/false negatives include ‘Technical’, ‘Allelic’, ‘Classification’ and ‘Phenotypic’. Basic statistics were calculated to determine the percentage of enrolled participants with positive screening results and classification into screen-positive result categories.
Return of results
Negative results were returned in the Early Check portal44. Positive results were called out by a study genetic counselor. Phenotypic false positive results were returned like all other screen-positive results. The genetic counselor documented pregnancy history, neonatal history, family history and any current medical concerns in a REDCap follow-up tracker at the time screen-positive results were conveyed to the parent or guardian45. When screening results were considered urgent, or at the request of the parents, a study clinical geneticist or genetic counselor discussed results with the newborn’s primary care provider. If the condition was potentially detectable through standard NBS, the parent was asked to sign a Health Insurance Portability and Accountability Act authorization for the release of state NBS results to the study team. Median days were calculated to measure the time for return of results. As per the consent, unexpected results that may affect the health of the newborn (for example, sex discrepancies) will be returned to the parents.
Follow-up testing
This publication includes follow-up data through 8 May 2025. To rule out any chance of a DBS sample swap, all parents of newborns with positive screening results were offered molecular confirmation of the reported variant(s) on a buccal sample from the newborn (ORAcollect, OCD-100). Targeted variant testing of the parent(s) and other family members was offered as needed to aid interpretation of the newborn result. Additional follow-up testing was offered as appropriate, depending on the screen-positive result. Parents signed an additional consent form for the newborn and themselves (if applicable) to participate in follow-up activities. Cascade testing in family members was not offered through Early Check unless testing a relative would aid interpretation of the newborn result. Our team was available to help identify clinical services for other family members interested in genetic testing.
Newborns with urgent screen-positive results, or those who needed blood or urine collected for follow-up testing, were seen by a clinical geneticist and genetic counselor at the Clinical Translational Research Center at UNC Hospital. Nonurgent confirmatory molecular testing was performed on buccal samples from collection kits mailed to the family. Molecular confirmation, orthogonal testing, genetic counseling and consultation with a clinical geneticist were offered at no cost to participating newborns and their parents. Frequencies were used to examine participation in follow-up or confirmatory testing.
Comparison with standard state NBS
Sample IDs of newborns enrolled in Early Check were shared with the NCSLPH. Newborns with confirmed diagnoses in the cohort were identified by the NCSLPH and a deidentified list of these diagnoses was shared with the RTI study team.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data described in this paper, including the gene list, screen-positive genotypes and phenotypes, are provided in Supplementary Information. Deidentified data that support the results reported in this article will be made available to suitably qualified researchers through any requests that comply with ethical standards to the corresponding author. Data must be requested within 12 months after the paper has been published, and the proposed use of the data must be IRB approved. Upon approval, deidentified data will be provided by the corresponding author to the applicants within 2 months. Sequencing data files are not available publicly for privacy, ethical and data protection reasons. Clinically interpreted variants from GeneDx are deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar) under organization ID 26957 (https://www.ncbi.nlm.nih.gov/clinvar/submitters/26957).
Code availability
Custom code developed for data preprocessing and modeling is available from the corresponding author to suitably qualified researchers through any requests that comply with ethical standards. The code is not publicly available due to institutional privacy policies but will be shared for academic purposes under a data use agreement.
References
Bick, D. et al. Newborn screening by genomic sequencing: opportunities and challenges. Int. J. Neonatal Screen. 8, 40 (2022).
Minten, T. et al. Data-driven consideration of genetic disorders for global genomic newborn screening programs. Genet. Med. 27, 101443 (2025).
Boemer, F. et al. Population-based, first-tier genomic newborn screening in the maternity ward. Nat. Med. 31, 1339–1350 (2025).
Ziegler, A. et al. Expanded newborn screening using genome sequencing for early actionable conditions. JAMA 333, 232–240 (2024).
Ceyhan-Birsoy, O. et al. Interpretation of genomic sequencing results in healthy and ill newborns: results from the BabySeq project. Am. J. Hum. Genet. 104, 76–93 (2019).
Roman, T. S. et al. Genomic sequencing for newborn screening: results of the NC NEXUS project. Am. J. Hum. Genet. 107, 596–611 (2020).
Downie, L. et al. Gene selection for genomic newborn screening: moving toward consensus? Genet. Med. 26, 101077 (2024).
Knoppers, B. M., Bonilha, A. E., Laberge, A. M., Ahmed, A. & Newson, A. J. Genomic sequencing in newborn screening: balancing consent with the right of the asymptomatic at-risk child to be found. Eur. J. Hum. Genet. 33, 182–188 (2024).
Baple, E. L. et al. Exploring the benefits, harms and costs of genomic newborn screening for rare diseases. Nat. Med. 30, 1823–1825 (2024).
Sobotka, S. A. & Ross, L. F. Newborn screening for neurodevelopmental disorders may exacerbate health disparities. Pediatrics 152, e2023061727 (2023).
Bailey, D. B. Jr et al. Early Check: translational science at the intersection of public health and newborn screening. BMC Pediatr. 19, 238 (2019).
Kucera, K. S. et al. A voluntary statewide newborn screening pilot for spinal muscular atrophy: results from Early Check. Int J. Neonatal Screen. 7, 20 (2021).
Kucera, K. S. et al. Two years of newborn screening for Duchenne muscular dystrophy as a part of the statewide Early Check research program in North Carolina. Genet. Med. 26, 101009 (2024).
Okoniewski, K. C. et al. Early identification of fragile X syndrome through expanded newborn screening. Brain Sci. 9, 4 (2019).
Guhan, S. M. et al. Cancer risks associated with the germline MITF(E318K) variant. Sci. Rep. 10, 17051 (2020).
Cope, H. L. et al. A systematic framework for selecting gene-condition pairs for inclusion in newborn sequencing panels: Early Check implementation. Genet. Med. 26, 101290 (2024).
Mey, K., Bille, M., Rye Rasmussen, S. H., Tranebjaerg, L. & Caye-Thomasen, P. The natural history of hearing loss in Pendred syndrome and non-syndromic enlarged vestibular aqueduct. Otol. Neurotol. 40, e178–e185 (2019).
Acke, F. R., Dhooge, I. J., Malfait, F. & De Leenheer, E. M. Hearing impairment in Stickler syndrome: a systematic review. Orphanet J. Rare Dis. 7, 84 (2012).
Peters, C. et al. DUOX2/DUOXA2 mutations frequently cause congenital hypothyroidism that evades detection on newborn screening in the United Kingdom. Thyroid 29, 790–801 (2019).
Bodian, D. L. et al. Utility of whole-genome sequencing for detection of newborn screening disorders in a population cohort of 1,696 neonates. Genet. Med. 18, 221–230 (2016).
Blencowe, H. et al. Rare single gene disorders: estimating baseline prevalence and outcomes worldwide. J. Community Genet. 9, 397–406 (2018).
Shieh, J. T. C. Genomic sequencing expansion and incomplete penetrance. Pediatrics 143, S22–S26 (2019).
Berg, J. S. The path to genomic screening—far from simple, but the journey has begun. JAMA 333, 210–212 (2024).
Mustapha, M. et al. An alpha-tectorin gene defect causes a newly identified autosomal recessive form of sensorineural pre-lingual non-syndromic deafness, DFNB21. Hum. Mol. Genet. 8, 409–412 (1999).
Verhoeven, K. et al. Mutations in the human alpha-tectorin gene cause autosomal dominant non-syndromic hearing impairment. Nat. Genet. 19, 60–62 (1998).
Yokoyama, S. et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature 480, 99–103 (2011).
Adhikari, A. N. et al. The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat. Med. 26, 1392–1397 (2020).
Bick, S. L. et al. Estimating the sensitivity of genomic newborn screening for treatable inherited metabolic disorders. Genet. Med. 27, 101284 (2024).
Dragojlovic, N. et al. The composition and capacity of the clinical genetics workforce in high-income countries: a scoping review. Genet. Med. 22, 1437–1449 (2020).
Wetterstrand, K. A. DNA Sequencing Costs: Data (NHGRI Genome Sequencing Program, accessed 1 December 2024); https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data
Sontag, M. K. et al. Newborn screening timeliness quality improvement initiative: Impact of national recommendations and data repository. PLoS ONE 15, e0231050 (2020).
Kingsmore, S. F. et al. A genome sequencing system for universal newborn screening, diagnosis, and precision medicine for severe genetic diseases. Am. J. Hum. Genet. 109, 1605–1619 (2022).
Paquin, R. S. et al. Outreach to new mothers through direct mail and email: recruitment in the Early Check research study. Clin. Transl. Sci. 14, 880–889 (2021).
Kaufmann, P., Pariser, A. R. & Austin, C. From scientific discovery to treatments for rare diseases—the view from the National Center for Advancing Translational Sciences—Office of Rare Diseases Research. Orphanet J. Rare Dis. 13, 196 (2018).
National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Health Sciences Policy; Roundtable on Genomics and Precision Health. The Promise and Perils of Next-Generation DNA Sequencing at Birth: Proceedings of a Workshop-in Brief The National Academies Collection: Reports funded by National Institutes of Health (eds Beachy, S. H. et al.) (National Academies Press, 2023).
Cohen, A. S. A. et al. Genomic answers for children: dynamic analyses of >1000 pediatric rare disease genomes. Genet. Med. 24, 1336–1348 (2022).
Division of Public Health NC State Center for Health Services. CY2023 North Carolina resident births by county of residence and race/ethnicity of mother. NCDHHS https://schs.dph.ncdhhs.gov/data/provisional/Birth/2023/CY2023PB7ResidentBirthsbyCountybyMaternalRace_Ethnicity.html (2023).
Milko, L. V. et al. An age-based framework for evaluating genome-scale sequencing results in newborn screening. J. Pediatr. 209, 68–76 (2019).
Peay, H. L. et al. Education and consent for population-based DNA screening: a mixed-methods evaluation of the Early Check newborn screening pilot study. Front. Genet. 13, 891592 (2022).
Sharp, S. A. et al. Development and standardization of an improved type 1 diabetes genetic risk score for use in newborn screening and incident diagnosis. Diabetes Care 42, 200–207 (2019).
Behera, S. et al. Comprehensive genome analysis and variant detection at scale using DRAGEN. Nat. Biotechnol. 43, 1177–1191 (2024).
Retterer, K. et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18, 696–704 (2016).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Cope, H. et al. Use of a web-based portal to return normal individual research results in Early Check: exploring user behaviors and attitudes. Clin. Genet. 103, 672–680 (2023).
Harris, P. A. et al. The REDCap consortium: building an international community of software platform partners. J. Biomed. Inf. 95, 103208 (2019).
Acknowledgements
We gratefully acknowledge our Early Check research participants, their parents and our Early Check Monogenic External Advisory Committee (https://earlycheck.org/what-is-early-check/who-we-are/). We acknowledge the critical support of the Early Check collaborating organizations involved in this study: NCSLPH, UNC, Illumina and GeneDx. The findings and conclusions in this publication are those of the authors and do not necessarily represent the views of the North Carolina Department of Health and Human Services, Division of Public Health. This work was initially supported by funding from Janssen Pharmaceuticals (July 2021 to July 2022). Study implementation is supported by Breakthrough T1D (formerly JDRF International), the Leona M. and Harry B. Helmsley Charitable Trust, Travere Therapeutics and Orchard Therapeutics. GS reagents were provided by Illumina, and GS including variant interpretation was provided by GeneDx. The project described was supported by the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health, through grant award no. UM1TR004406. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The development of the Early Check research platform was also supported by grants from the John Merck Fund and the National Center for Advancing Translational Sciences of the National Institutes of Health (Award no. UL1TR002489).
Author information
Authors and Affiliations
Consortia
Contributions
H.L.C., J.S.B., J.A.S., K.S.K., L.V.M., A.C.W., M.R., D.B.B.J., S.M.S., E.R.J., C.M.P. and H.L.P. conceived and designed the study. J.S.B. conceived and designed the result classification rubrics, with contributions from H.L.C., C.M.P., E.R.J., J.A.S. and H.L.P. H.L.C., E.R.J., J.A.S., K.S.K., H.E.F., A.Y.G., A.N.F., B.A.M., B.W., R.R.M., C.M.P. and H.L.P. contributed to the day-to-day conduct of the Early Check gNBS study. H.E.F., A.Y.G., J.A.S., A.N.F. and H.L.C. recorded and analyzed data. R.S.Z., S.F.S., A.B., A.J.G., K.G.L., K.G.M., K.M., C.K., K.S.H. and P.K. participated in gene list curation at GeneDx and test development/validation, processed participant samples, generated and analyzed genomic data and produced genetic test reports. H.L.C. wrote the initial draft of the paper. All authors contributed and approved the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
R.S.Z., S.F.S., A.B., A.J.G., K.G.L., K.G.M., K.M., C.K., K.S.H. and P.K. were employees of GeneDx, LLC at the time of this study and may have held company stock. All RTI authors (H.L.C., K.S.K., H.E.F., A.Y.G., A.N.F., B.A.M., B.W., R.R.M., D.B.B.J., A.C.W., M.R. and H.L.P.) were supported in this research by Janssen Pharmaceuticals (July 2021 to July 2022), Breakthrough T1D (formerly JDRF International), the Leona M. and Harry B. Helmsley Charitable Trust, Travere Therapeutics and Orchard Therapeutics. UNC authors (E.R.J., J.A.S. and C.M.P.) were supported in this research by Breakthrough T1D (formerly JDRF International). The other authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks François Boemer and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Anna Maria Ranzoni, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
List of consortium members and their affiliations.
Supplementary Table 1
Panel 1 and Panel 2 screen positives.
Supplementary Tables 2 and 3
Table 2. Gene–condition pairs on Early Check Panel 1 (28 September 2023–10 June 2024). Table 3. Gene–condition pairs on Early Check Panel 2 (28 September 2023–10 June 2024).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cope, H.L., Jalazo, E.R., Berg, J.S. et al. Feasibility and clinical utility of expanded genomic newborn screening in the Early Check program. Nat Med (2025). https://doi.org/10.1038/s41591-025-03945-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41591-025-03945-8
