
Abstract
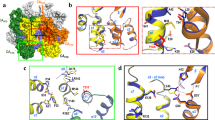
HIV-1 capsid plays multiple key roles in viral replication, and inhibition of capsid assembly is an attractive target for therapeutic intervention. Here, we report the atomic-resolution structure of capsid protein (CA) tubes, determined by magic-angle spinning NMR and data-guided molecular dynamics simulations. Functionally important regions, including the NTD β-hairpin, the cyclophilin A-binding loop, residues in the hexamer central pore, and the NTD-CTD linker region, are well defined. The structure of individual CA chains, their arrangement in the pseudo-hexameric units of the tube and the inter-hexamer interfaces are consistent with those in intact capsids and substantially different from the organization in crystal structures, which feature flat hexamers. The inherent curvature in the CA tubes is controlled by conformational variability of residues in the linker region and of dimer and trimer interfaces. The present structure reveals atomic-level detail in capsid architecture and provides important guidance for the design of novel capsid inhibitors.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


Data availability
The coordinates of the atomic structures have been deposited in the Protein Data Bank under accession codes PDB 6WAP for the single CA chain and PDB 6X63 for the CA tube. MAS-NMR chemical shifts, dihedral constraints and distance constraints have been deposited in the Biological Magnetic Resonance Data Bank under accession code 30741.
References
Ganser, B. K., Li, S., Klishko, V. Y., Finch, J. T. & Sundquist, W. I. Assembly and analysis of conical models for the HIV-1 core. Science 283, 80–83 (1999).
Briggs, J. A. et al. The stoichiometry of Gag protein in HIV-1. Nat. Struct. Mol. Biol. 11, 672–675 (2004).
Pornillos, O., Ganser-Pornillos, B. K. & Yeager, M. Atomic-level modelling of the HIV capsid. Nature 469, 424–428 (2011).
Zhao, G. P. et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497, 643–646 (2013).
Campbell, E. M. & Hope, T. J. HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 13, 471–483 (2015).
Ambrose, Z. & Aiken, C. HIV-1 uncoating: connection to nuclear entry and regulation by host proteins. Virology 454–455, 371–379 (2014).
Luban, J., Bossolt, K. L., Franke, E. K., Kalpana, G. V. & Goff, S. P. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73, 1067–1078 (1993).
Goujon, C. et al. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502, 559–562 (2013).
Liu, Z. et al. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 14, 398–410 (2013).
Stremlau, M. et al. The cytoplasmic body component TRIM5α restricts HIV-1 infection in Old World monkeys. Nature 427, 848–853 (2004).
Lukic, Z., Dharan, A., Fricke, T., Diaz-Griffero, F. & Campbell, E. M. HIV-1 uncoating is facilitated by dynein and kinesin 1. J. Virol. 88, 13613–13625 (2014).
Malikov, V. et al. HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat. Commun. 6, 6660 (2015).
Brass, A. L. et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science 319, 921–926 (2008).
Konig, R. et al. Global analysis of host–pathogen interactions that regulate early-stage HIV-1 replication. Cell 135, 49–60 (2008).
Ocwieja, K. E. et al. HIV integration targeting: a pathway involving Transportin-3 and the nuclear pore protein RanBP2. PLoS Pathog. 7, e1001313 (2011).
Rasaiyaah, J. et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 503, 402–405 (2013).
Perilla, J. R. & Gronenborn, A. M. Molecular architecture of the retroviral capsid. Trends Biochem. Sci. 41, 410–420 (2016).
Ganser-Pornillos, B. K., Cheng, A. & Yeager, M. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell 131, 70–79 (2007).
López, C. S. et al. Determinants of the HIV-1 core assembly pathway. Virology 417, 137–146 (2011).
Gamble, T. R. et al. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87, 1285–1294 (1996).
Byeon, I. J. L. et al. Structural convergence between Cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 139, 780–790 (2009).
Gres, A. T. et al. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 349, 99–103 (2015).
Jacques, D. A. et al. HIV-1 uses dynamic capsid pores to import nucleotides and fuel encapsidated DNA synthesis. Nature 536, 349–353 (2016).
Han, Y. et al. Magic angle spinning NMR reveals sequence-dependent structural plasticity, dynamics and the spacer peptide 1 conformation in HIV-1 capsid protein assemblies. J. Am. Chem. Soc. 135, 17793–17803 (2013).
Lu, M. et al. Dynamic allostery governs cyclophilin A-HIV capsid interplay. Proc. Natl Acad. Sci. USA 112, 14617–14622 (2015).
Mattei, S., Glass, B., Hagen, W. J., Krausslich, H. G. & Briggs, J. A. The structure and flexibility of conical HIV-1 capsids determined within intact virions. Science 354, 1434–1437 (2016).
Russell, R. W. et al. Accuracy and precision of protein structures determined by magic angle spinning NMR spectroscopy: for some ‘with a little help from a friend’. J. Biomol. NMR 73, 333–346 (2019).
Gupta, R. et al. Dynamic nuclear polarization magic-angle spinning nuclear magnetic resonance combined with molecular dynamics simulations permits detection of order and disorder in viral assemblies. J. Phys. Chem. B 123, 5048–5058 (2019).
Byeon, I. J. L. et al. Motions on the millisecond time scale and multiple conformations of HIV-1 capsid protein: implications for structural polymorphism of CA assemblies. J. Am. Chem. Soc. 134, 6455–6466 (2012).
Zhang, H. et al. HIV-1 capsid function is regulated by dynamics: quantitative atomic-resolution insights by integrating magic-angle-spinning NMR, QM/MM and MD. J. Am. Chem. Soc. 138, 14066–14075 (2016).
Campos-Olivas, R. & Summers, M. F. Backbone dynamics of the N-terminal domain of the HIV-1 capsid protein and comparison with the G94D mutant conferring cyclosporin resistance/dependence. Biochemistry 38, 10262–10271 (1999).
Fritz, M. et al. Determination of accurate backbone chemical shift tensors in microcrystalline proteins by integrating MAS NMR and QM/MM. Phys. Chem. Chem. Phys. 20, 9543–9553 (2018).
Dick, R. A. et al. Inositol phosphates are assembly co-factors for HIV-1. Nature 560, 509–512 (2018).
Bayro, M. J. & Tycko, R. Structure of the dimerization interface in the mature HIV-1 capsid protein lattice from solid state NMR of tubular assemblies. J. Am. Chem. Soc. 138, 8538–8546 (2016).
Han, Y. et al. Solid-state NMR studies of HIV-1 capsid protein assemblies. J. Am. Chem. Soc. 132, 1976–1987 (2010).
Hou, G. J., Yan, S., Trebosc, J., Amoureux, J. P. & Polenova, T. Broadband homonuclear correlation spectroscopy driven by combined R2(n)(v) sequences under fast magic angle spinning for NMR structural analysis of organic and biological solids. J. Magn. Reson. 232, 18–30 (2013).
Brauniger, T., Wormald, P. & Hodgkinson, P. Improved proton decoupling in NMR spectroscopy of crystalline solids using the SPINAL-64 sequence. Monatsh. Chem. 133, 1549–1554 (2002).
Fung, B. M., Khitrin, A. K. & Ermolaev, K. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 142, 97–101 (2000).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Goddard, T. D. & Kneller, D. G. SPARKY 3 (Univ. California, 2004).
Stevens, T. J. et al. A software framework for analysing solid-state MAS NMR data. J. Biomol. NMR 51, 437–447 (2011).
Schwieters, C. D., Kuszewski, J. J., Tjandra, N. & Clore, G. M. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 (2003).
Schwieters, C. D., Kuszewski, J. J. & Clore, G. M. Using Xplor-NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 48, 47–62 (2006).
Schwieters, C. D., Bermejo, G. A. & Clore, G. M. Xplor-NIH for molecular structure determination from NMR and other data sources. Protein Sci. 27, 26–40 (2018).
Bermejo, G. A., Clore, G. M. & Schwieters, C. D. Smooth statistical torsion angle potential derived from a large conformational database via adaptive kernel density estimation improves the quality of NMR protein structures. Protein Sci. 21, 1824–1836 (2012).
Schwieters, C. D. & Clore, G. M. A pseudopotential for improving the packing of ellipsoidal protein structures determined from NMR data. J. Phys. Chem. B 112, 6070–6073 (2008).
Schwieters, C. D., Bermejo, G. A. & Clore, G. M. A three-dimensional potential of mean force to improve backbone and sidechain hydrogen bond geometry in Xplor-NIH protein structure determination. Protein Sci. 29, 100–110 (2020).
Shen, Y. & Bax, A. Protein structural information derived from NMR chemical shift with the neural network program TALOS-N. Methods Mol. Biol. 1260, 17–32 (2015).
Perilla, J. R. et al. CryoEM structure refinement by integrating NMR chemical shifts with molecular dynamics simulations. J. Phys. Chem. B 121, 3853–3863 (2017).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Pettersen, E. F. et al. UCSF chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Gong, Z., Schwieters, C. D. & Tang, C. Conjoined use of EM and NMR in RNA structure refinement. PLoS ONE https://doi.org/10.1371/journal.pone.0120445 (2015).
Phillips, J. C. et al. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 (2005).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Olsson, M. H. M., Søndergaard, C. R., Rostkowski, M. & Jensen, J. H. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537 (2011).
Søndergaard, C. R., Olsson, M. H. M., Rostkowski, M. & Jensen, J. H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 7, 2284–2295 (2011).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Gullingsrud, J., Saam, J. & Phillips, J. psfgen User’s Guide (Theoretical and Computational Biophysics Group, University of Illinois and Beckman Institute, 2006).
Stone, J. E., Vandivort, K. L. & Schulten, K. in Advances in Visual Computing. ISVC 2011. Lecture Notes in Computer Science in Lecture Notes in Computer Science Vol. 6939 (eds. Bebis, G. et al.) 1–12 (2011).
Fiorin, G., Klein, M. L. & Hénin, J. Using collective variables to drive molecular dynamics simulations. Mol. Phys. 111, 3345–3362 (2013).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Skeel, R. D. & Biesiadecki, J. J. Symplectic integration with variable stepsize. Ann. Numer. Math. 1, 191–198 (1994).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Bussi, G., Zykova-Timan, T. & Parrinello, M. Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J. Chem. Phys. 130, 074101 (2009).
Andersen, H. C. Rattle: a ‘velocity’ version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 52, 24–34 (1983).
Kräutler, V., van Gunsteren, W. F. & Hünenberger, P. H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 22, 501–508 (2001).
The PyMOL Molecular Graphics System v.2.0 (Schrödinger, 2000).
Stone, J. E. An Efficient Library for Parallel Ray Tracing and Animation. Masters thesis, Univ. Missouri-Rolla (1998).
Rousseeuw, P. J. Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math. 20, 53–65 (1987).
Acknowledgements
This work was supported by the National Institutes of Health (NIH; grant no. P50AI1504817). We acknowledge the support of the National Science Foundation (NSF; grant no. CHE0959496) for acquisition of the 850-MHz NMR spectrometer, the NIH (grant no. P30GM110758) for support for the core instrumentation infrastructure at the University of Delaware and also grant no. S10OD012213 for acquisition of the 750-MHz NMR spectrometer at the University of Pittsburgh. This work was partially supported by the Intramural Research Program of the Center for Information Technology at the NIH. This research used resources from the Oak Ridge Leadership Computing Facility (OLCF) at Oak Ridge National Laboratory, which is supported by the Office of Science of the Department of Energy under contract no. DE-AC05-00OR22725. We acknowledge a Director’s Discretionary Award on the Summit supercomputer from the OLCF.
Author information
Authors and Affiliations
Contributions
T.P. and A.M.G. conceived the project and guided the work. J.R.P. designed and guided the MD simulations and structure calculations of the CA tube. M.L. prepared the samples, performed NMR experiments and analyzed the experimental data. R.W.R. and M.L. performed the structure calculations of the CA hexamer unit. C.M.Q. assisted in the structure calculations of the hexamer unit. A.J.B. conducted the MD simulations and structure calculation of the CA tube. M.L., R.W.R. and A.J.B. prepared figures for the manuscript. R.W.R. and A.J.B. wrote scripts for analysis of calculation results and visualization of the hexamer unit and tube, respectively. C.D.S. provided critical input in the NIH-Xplor-based structure calculations. C.M.Q., G.H. and H.Z. took part in the design or analysis of NMR experiments. T.P. and A.M.G. took the lead in writing the manuscript. All authors discussed the results and contributed to manuscript preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Inês Chen was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Summary of MAS-derived NMR distances.
a, Inter-residue contact plot. b, Intrachain distances (gray dotted lines), mapped onto the ribbon diagram structure of a single CA chain. NTD helices are colored purple, the β-hairpin yellow, loops gray, and the CTD helices cyan.
Extended Data Fig. 2 Initial MAS NMR structure of full-length CA protein.
Ensemble of 10 lowest-energy structures for the single-chain. Best-fit superpositions for the NTD (a) and CTD (b), respectively, are shown. The color coding is identical to that in Extended Data Fig. 1.
Extended Data Fig. 3 Protocol for the structure determination of a CA hexamer using MAS NMR and cryo-EM data.
Ensembles of the 10 lowest-energy structures at each step are depicted in ribbon representation, with NTD helices colored purple, the β-hairpin yellow, loops gray, and CTD helices cyan.
Extended Data Fig. 4 Structural clusters of HIV-1 CA tube.
a–f, Plots of k-medoid clusters4 for different structural units in the final tube structure by Silhouette analysis5. g–l, Conformations from clustering analyses and the number of members within each cluster. g, Representative structures (medoids) of dimer interface clusters. h, Alignment of the two trimer interface medoids yielded by clustering. i, CypA loops grouped and colored by conformational cluster. j, Two structural clusters from analysis of monomeric β-hairpins. k, Nine clusters from analysis of hexameric β-hairpins. l, Flexible linkers grouped into four conformational clusters.
Extended Data Fig. 5 Water occupancy in the MD simulation of the HIV-1 CA tube.
a, Water occupancy map, averaged over 80 ns of the trajectory. For each frame in the trajectory, a binary mask is used to indicate the presence of water at every point in space. The average of these masks for a trajectory is the water occupancy map, and each point in the latter describes the fractional occupancy of water at a specific location. 3-D visualizations, isosurfaces, are generated according to the value at each point in the occupancy map and are shown at varying occupancy cutoffs. b, Histogram of dimer interfaces involved in water-mediated contacts, the latter determined by proximity of S149, E175 and W184 side chain atoms within 4.0 Å of the water occupancy map at the specified occupancy cutoff values. c, Illustration of a dimer interface with the side chains of S149, E175 and W184 in stick representation and an isosurface of the water occupancy map (red; > 0.70 fractional occupancy).
Extended Data Fig. 6 Analysis of trimer interfaces in the HIV-1 CA tube.
a, A hexamer of hexamers, highlighting the two trimer interfaces: one involving chains A, D and F (blue) and the other between chains B, C and E (orange), shown from two perspectives. b, Histogram of the shortest L205 Cα–Cα distance at each trimer interface in the CA tube, for the orange and blue interfaces. The histogram is drawn as a frequency polygon, where vertices represent the center of each bin along the x-axis. c, Best-fit superposition of the two medoids from clustering analysis of the trimer interface (blue, red) and the cryo-electron tomography (cryo-ET) structure (accession codes: EMDB 3475, PDB ID 5MD7)26.
Extended Data Fig. 7 Molecular dynamics simulation setup for the HIV-1 CA tube.
a, The 13.9-million atom system comprises water molecules (transparent), Na+ and Cl- ions (yellow and blue, respectively), and protein (white). View of the dimer (b) and trimer (c) interface docked into the 8.6 Å cryo-EM envelope4 (accession code EMD-5582). d, Fourier Shell Correlation (FSC) between the 8.6 Å CA tube density and simulated 8.6 Å densities of the CA tube before (blue) and after (red) data-guided MD refinement. The dashed lines in the plot correspond to FSC = 0.50 and FSC = 0.143, for resolutions of 10.0 Å and 8.3 Å, respectively. e, Scaling analysis of the MD simulations of the current system on the Oak Ridge National Laboratory’s Summit supercomputer.
Extended Data Fig. 8 Superposition of synthetic peak positions, back calculated from the MAS NMR structure of a hexamer unit and the CORD spectrum of [1,6-13C]-glucose,U-15N-CA tubular assemblies.
The spectrum was collected at 14.1 T, with a MAS frequency of 14 kHz, and a CORD mixing time of 500 ms. Cross peak positions were calculated for the lowest-energy structure of the hexameric unit, using the experimental 13C chemical shifts, and all intrachain and interchain 13C-13C contacts, corresponding to distances up to 7 Å. Peak positions corresponding to intrachain contacts are colored red and blue for distances up to 5 Å and those between 5 Å and 7 Å, respectively. Peak positions corresponding to interchain contacts are colored in green. Peak positions due to isotope scrambling are colored yellow.
Extended Data Fig. 9 Superposition of synthetic peak positions, back calculated from the MAS NMR structure of a hexamer unit, and the CORD spectrum of U-13C,15N-CA tubular assemblies.
The spectrum was collected at 14.1 T, a MAS frequency of 14 kHz, and a CORD mixing time of 500 ms. Cross peak positions were calculated for the lowest-energy structure of the hexameric unit, using the experimental 13C chemical shifts, and all intrachain and interchain 13C-13C contacts corresponding to distances up to 7 Å. Peak positions corresponding to intrachain contacts are colored red and blue for distances up to 5 Å and those between 5 Å and 7 Å, respectively. Peak positions corresponding to interchain contacts are colored in green.
Extended Data Fig. 10 Interchain contacts (up to 7 Å) identified in the CORD spectra extracted from simulated cross peak positions.
Interchain correlations are mapped on two neighboring CA chains in the lowest-energy structure of the hexameric unit. Intermolecular correlations are shown as green dashed lines and the associated residues are depicted in orange stick representation.
Supplementary information
Supplementary Information
Supplementary Figs. 1–4 and Tables 1–8.
Rights and permissions
About this article

Cite this article
Lu, M., Russell, R.W., Bryer, A.J. et al. Atomic-resolution structure of HIV-1 capsid tubes by magic-angle spinning NMR. Nat Struct Mol Biol 27, 863–869 (2020). https://doi.org/10.1038/s41594-020-0489-2
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41594-020-0489-2
This article is cited by
-
Structural basis of protein condensation on microtubules underlying branching microtubule nucleation
Nature Communications (2023)
-
Structural basis of HIV-1 maturation inhibitor binding and activity
Nature Communications (2023)
-
Early events in amyloid-β self-assembly probed by time-resolved solid state NMR and light scattering
Nature Communications (2023)
-
5D solid-state NMR spectroscopy for facilitated resonance assignment
Journal of Biomolecular NMR (2023)
-
Magic-angle-spinning NMR structure of the kinesin-1 motor domain assembled with microtubules reveals the elusive neck linker orientation
Nature Communications (2022)
