
Abstract
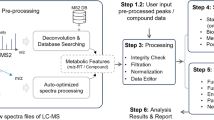
Liquid chromatography coupled with high-resolution mass spectrometry (LC–HRMS) has become a workhorse in global metabolomics studies with growing applications across biomedical and environmental sciences. However, outstanding bioinformatics challenges in terms of data processing, statistical analysis and functional interpretation remain critical barriers to the wider adoption of this technology. To help the user community overcome these barriers, we have made major updates to the well-established MetaboAnalyst platform (www.metaboanalyst.ca). This protocol extends the previous 2011 Nature Protocol by providing stepwise instructions on how to use MetaboAnalyst 5.0 to: optimize parameters for LC–HRMS spectra processing; obtain functional insights from peak list data; integrate metabolomics data with transcriptomics data or combine multiple metabolomics datasets; conduct exploratory statistical analysis with complex metadata. Parameter optimization may take ~2 h to complete depending on the server load, and the remaining three stages may be executed in ~60 min.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout









Similar content being viewed by others


Data availability
All example datasets used in the protocol are integrated as example datasets in their respective modules and are also available for download from the ‘Format’ page of MetaboAnalyst (https://www.metaboanalyst.ca/MetaboAnalyst/docs/Format.xhtml). There are no restrictions on their use.
Code availability
MetaboAnalyst is freely accessible as a web-based application. The underlying R code is freely available at GitHub as the MetaboAnalystR (https://github.com/xia-lab/MetaboAnalystR) and OptiLCMS (https://github.com/xia-lab/OptiLCMS) packages under the GNU General Public License version 2 or later.
References
Alseekh, S. et al. Mass spectrometry-based metabolomics: a guide for annotation, quantification and best reporting practices. Nat. Methods 18, 747–756 (2021).
Alseekh, S. & Fernie, A. R. Metabolomics 20 years on: what have we learned and what hurdles remain? Plant J. 94, 933–942 (2018).
Doerr, A. Global metabolomics. Nat. Methods 14, 32–32 (2017).
Cajka, T. & Fiehn, O. Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics. Anal. Chem. 88, 524–545 (2016).
Vermeulen, R., Schymanski, E. L., Barabasi, A. L. & Miller, G. W. The exposome and health: where chemistry meets biology. Science 367, 392–396 (2020).
Beger, R. D. et al. Metabolomics enables precision medicine: “A White Paper, Community Perspective”. Metabolomics 12, 149 (2016).
Want, E. J. et al. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 8, 17–32 (2013).
Zhou, G. et al. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 47, W234–W241 (2019).
Chong, J., Liu, P., Zhou, G. & Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 15, 799–821 (2020).
Pang, Z. et al. MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 49, W388–W396 (2021).
Xia, J., Psychogios, N., Young, N. & Wishart, D. S. MetaboAnalyst: a web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 37, W652–W660 (2009).
Xia, J., Mandal, R., Sinelnikov, I. V., Broadhurst, D. & Wishart, D. S. MetaboAnalyst 2.0—a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 40, W127–W133 (2012).
Xia, J., Sinelnikov, I. V., Han, B. & Wishart, D. S. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucleic Acids Res. 43, W251–W257 (2015).
Chong, J. et al. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 46, W486–W494 (2018).
Stanstrup, J. et al. The metaRbolomics Toolbox in Bioconductor and beyond. Metabolites https://doi.org/10.3390/metabo9100200 (2019).
Gardinassi, L. G., Xia, J., Safo, S. E. & Li, S. Bioinformatics tools for the interpretation of metabolomics data. Curr. Pharmacol. Rep. 3, 374–383 (2017).
Chang, H. Y. et al. A practical guide to metabolomics software development. Anal. Chem. 93, 1912–1923 (2021).
Tautenhahn, R., Patti, G. J., Rinehart, D. & Siuzdak, G. XCMS Online: a web-based platform to process untargeted metabolomic data. Anal. Chem. 84, 5035–5039 (2012).
Giacomoni, F. et al. Workflow4Metabolomics: a collaborative research infrastructure for computational metabolomics. Bioinformatics 31, 1493–1495 (2015).
Yang, Q. et al. NOREVA: enhanced normalization and evaluation of time-course and multi-class metabolomic data. Nucleic Acids Res. 48, W436–W448 (2020).
Chambers, M. C. et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 (2012).
Du, X. X., Smirnov, A., Pluskal, T., Jia, W. & Sumner, S. Metabolomics data preprocessing using ADAP and MZmine 2. Computational Methods Data Anal. Metabolomics 2104, 25–48 (2020).
Tsugawa, H. et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 38, 1159–1163 (2020).
Tsugawa, H. et al. Hydrogen rearrangement rules: computational MS/MS fragmentation and structure elucidation using MS-FINDER software. Anal. Chem. 88, 7946–7958 (2016).
Rost, H. L. et al. OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 13, 741–748 (2016).
Danczak, R. E. et al. Using metacommunity ecology to understand environmental metabolomes. Nat. Commun. 11, 6369 (2020).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Xia, J. & Wishart, D. S. Web-based inference of biological patterns, functions and pathways from metabolomic data using MetaboAnalyst. Nat. Protoc. 6, 743–760 (2011).
Xia, J. & Wishart, D. S. Metabolomic data processing, analysis, and interpretation using MetaboAnalyst. Curr. Protoc. Bioinformatics https://doi.org/10.1002/0471250953.bi1410s34 (2011).
Xia, J. & Wishart, D. S. Using MetaboAnalyst 3.0 for comprehensive metabolomics data analysis. Curr. Protoc. Bioinforma. 55, 14 10 11–14 10 91 (2016).
Xia, J., Broadhurst, D. I., Wilson, M. & Wishart, D. S. Translational biomarker discovery in clinical metabolomics: an introductory tutorial. Metabolomics 9, 280–299 (2013).
Chong, J., Wishart, D. S. & Xia, J. Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinforma. 68, e86 (2019).
Chong, J. & Xia, J. Using MetaboAnalyst 4.0 for metabolomics data analysis, interpretation, and integration with other omics data. Methods Mol. Biol. 2104, 337–360 (2020).
Tautenhahn, R., Bottcher, C. & Neumann, S. Highly sensitive feature detection for high resolution LC/MS. BMC Bioinforma. 9, 504 (2008).
Yu, T., Park, Y., Johnson, J. M. & Jones, D. P. apLCMS—adaptive processing of high-resolution LC/MS data. Bioinformatics 25, 1930–1936 (2009).
Kenar, E. et al. Automated label-free quantification of metabolites from liquid chromatography-mass spectrometry data. Mol. Cell Proteom. 13, 348–359 (2014).
Pang, Z., Chong, J., Li, S. & Xia, J. MetaboAnalystR 3.0: toward an optimized workflow for global metabolomics. Metabolites https://doi.org/10.3390/metabo10050186 (2020).
Chaleckis, R., Meister, I., Zhang, P. & Wheelock, C. E. Challenges, progress and promises of metabolite annotation for LC–MS-based metabolomics. Curr. Opin. Biotechnol. 55, 44–50 (2019).
Sindelar, M. & Patti, G. J. Chemical discovery in the era of metabolomics. J. Am. Chem. Soc. 142, 9097–9105 (2020).
Kuhl, C., Tautenhahn, R., Bottcher, C., Larson, T. R. & Neumann, S. CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 84, 283–289 (2012).
Senan, O. et al. CliqueMS: a computational tool for annotating in-source metabolite ions from LC-MS untargeted metabolomics data based on a coelution similarity network. Bioinformatics 35, 4089–4097 (2019).
Kind, T. & Fiehn, O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics https://doi.org/10.1186/1471-2105-8-105 (2007).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Xia, J. & Wishart, D. S. MSEA: a web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 38, W71–W77 (2010).
Tarca, A. L. et al. A novel signaling pathway impact analysis. Bioinformatics 25, 75–82 (2009).
Xia, J. & Wishart, D. S. MetPA: a web-based metabolomics tool for pathway analysis and visualization. Bioinformatics 26, 2342–2344 (2010).
Li, S. et al. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 9, e1003123 (2013).
Xia, J. et al. INMEX—a web-based tool for integrative meta-analysis of expression data. Nucleic Acids Res. 41, W63–W70 (2013).
Zhou, G. & Xia, J. OmicsNet: a web-based tool for creation and visual analysis of biological networks in 3D space. Nucleic Acids Res. 46, W514–W522 (2018).
Schriml, L. M. et al. COVID-19 pandemic reveals the peril of ignoring metadata standards. Sci. Data 7, 188 (2020).
Kahan, B. C., Jairath, V., Dore, C. J. & Morris, T. P. The risks and rewards of covariate adjustment in randomized trials: an assessment of 12 outcomes from 8 studies. Trials 15, 139 (2014).
Chong, J. & Xia, J. MetaboAnalystR: an R package for flexible and reproducible analysis of metabolomics data. Bioinformatics 34, 4313–4314 (2018).
Chong, J., Yamamoto, M. & Xia, J. MetaboAnalystR 2.0: from raw spectra to biological insights. Metabolites https://doi.org/10.3390/metabo9030057 (2019).
Gardinassi, L. G. et al. Integrative metabolomics and transcriptomics signatures of clinical tolerance to Plasmodium vivax reveal activation of innate cell immunity and T cell signaling. Redox Biol. 17, 158–170 (2018).
Pang, Z., Zhou, G., Chong, J. & Xia, J. Comprehensive meta-analysis of COVID-19 global metabolomics datasets. Metabolites https://doi.org/10.3390/metabo11010044 (2021).
Walker, D. I. et al. High-resolution metabolomics of occupational exposure to trichloroethylene. Int. J. Epidemiol. 45, 1517–1527 (2016).
Gatto, L., Gibb, S. & Rainer, J. MSnbase, efficient and elegant R-based processing and visualization of raw mass spectrometry data. J. Proteome Res. 20, 1063–1069 (2021).
Conley, C. J. et al. Massifquant: open-source Kalman filter-based XC-MS isotope trace feature detection. Bioinformatics 30, 2636–2643 (2014).
Wishart, D. S. et al. HMDB 5.0: the Human Metabolome Database for 2022. Nucleic Acids Res. 50, D622–D631 (2022).
Vaughan, A. M. et al. Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cell Microbiol. 11, 506–520 (2009).
Cumnock, K. et al. Host energy source is important for disease tolerance to malaria. Curr. Biol. 28, 1635–1642 e1633 (2018).
Smith, C. A., Want, E. J., O’Maille, G., Abagyan, R. & Siuzdak, G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78, 779–787 (2006).
Prince, J. T. & Marcotte, E. M. Chromatographic alignment of ESI–LC–MS proteomics data sets by ordered bijective interpolated warping. Anal. Chem. 78, 6140–6152 (2006).
Libiseller, G. et al. IPO: a tool for automated optimization of XCMS parameters. BMC Bioinforma. 16, 118 (2015).
McLean, C. & Kujawinski, E. B. AutoTuner: high fidelity and robust parameter selection for metabolomics data processing. Anal. Chem. 92, 5724–5732 (2020).
Zhou, G., Ewald, J. & Xia, J. OmicsAnalyst: a comprehensive web-based platform for visual analytics of multi-omics data. Nucleic Acids Res. 49, W476–W482 (2021).
Rappoport, N. & Shamir, R. Multi-omic and multi-view clustering algorithms: review and cancer benchmark. Nucleic Acids Res. 47, 1044 (2019).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Acknowledgements
We thank Genome Canada, Génome Québec, US National Institutes of Health (U01 CA235493), Natural Sciences and Engineering Research Council of Canada (NSERC) and Canada Research Chairs (CRC) Program for funding support.
Author information
Authors and Affiliations
Contributions
Z.P., J.E., N.B. and J.X. prepared the manuscript. Z.P., G.Z., J.E., L.C., O.H. and J.X. contributed to the development and testing of the MetaboAnalyst. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Protocols thanks Julia Kuligowski, Zhenzhen Xu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations
Related links
Key references using this protocol
Pang, Z. et al. Metabolites 10, 186 (2020): https://doi.org/10.3390/metabo10050186
Pang, Z. et al. Metabolites 11, 44 (2021): https://doi.org/10.3390/metabo11010044
Pang, Z. et al. Nucleic Acids Res. 49, W388–W396 (2021): https://doi.org/10.1093/nar/gkab382
Key data used in this protocol
Gardinassi, L. G. et al. Redox Biol. 17, 158–170 (2018): https://doi.org/10.1016/j.redox.2018.04.011
Walker, D. I. et al. Int. J. Epidemiol. 45, 1517–1527 (2016): https://doi.org/10.1093/ije/dyw218
This protocol is an extension to: Nat. Protoc. 6, 743–760 (2011): https://doi.org/10.1038/nprot.2011.319.
Supplementary information
Supplementary Information
Supplementary Figs. 1 and 2 and Supplementary Table 1.
Rights and permissions
About this article

Cite this article
Pang, Z., Zhou, G., Ewald, J. et al. Using MetaboAnalyst 5.0 for LC–HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nat Protoc 17, 1735–1761 (2022). https://doi.org/10.1038/s41596-022-00710-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41596-022-00710-w
This article is cited by
-
Selective neuronal restoration of progranulin does not prevent the frontotemporal dementia like-phenotype of progranulin knockout mice
Journal of Neuroinflammation (2026)
-
Metabolomic and transcriptomic analyses identify metabolic alterations and immune suppression in ovarian cancer
Scientific Reports (2026)
-
An integrated drug repositioning analysis identifies rosiglitazone as a treatment for sarcopenia
Communications Biology (2026)
-
Comparison of human metabolome changes identified in a placebo-controlled amphetamine administration study versus those using forensic toxicology routine data
Scientific Reports (2026)
-
Fontan associated protein-losing enteropathy is linked to distinct metabolic and hepatic alterations
Scientific Reports (2026)
